

Abstract
Exome sequencing of families of related individuals has been highly successful in identifying genetic polymorphisms responsible for Mendelian disorders. Here, we demonstrate the value of the reverse approach, where we use exome sequencing of a sample of unrelated individuals to analyze allele frequencies of known causal mutations for Mendelian diseases. We sequenced the exomes of 100 individuals representing the three major genetic subgroups of the Qatari population (Q1 Bedouin, Q2 Persian-South Asian, Q3 African) and identified 37 variants in 33 genes with effects on 36 clinically significant Mendelian diseases. These include variants not present in 1000 Genomes and variants at high frequency when compared to 1000 Genomes populations. Several of these Mendelian variants were only segregating in one Qatari subpopulation, where the observed subpopulation specificity trends were confirmed in an independent population of 386 Qataris. Pre-marital genetic screening in Qatar tests for only 4 out of the 37, such that this study provides a set of Mendelian disease variants with potential impact on the epidemiological profile of the population that could be incorporated into the testing program if further experimental and clinical characterization confirms high penetrance.
Keywords: Mendelian, consanguinity, exome sequencing, OMIM, diagnostic biomarkers
Introduction
The nation of Qatar, residing in a peninsula on the northeast coast of the Arabian peninsula, sits at the crossroads of human migration out of Africa with human habitation dating over 50,000 years (Oppenheimer, 2012). The current Qatari population is comprised of approximately 300,000 nationals within a resident population of 1.8 million (Qatar Statistics Authority, 2010). The Qataris are descendants of nomadic tribes with European, Persian and Southern African influences that reflect the complex migration history of the region (Omberg et al., 2012). Consistent with this history, the one major genomic study of the Qatari population conducted to date using DNA microarrays found that the population can be divided into 3 distinct genetic groups: Bedouin (Q1), Persian-South Asian (Q2) and African (Q3) (Hunter-Zinck et al., 2010).
Despite the importance of the Qatari people in the history of human evolution and the importance of this nation in the region and globally, there have been relatively few applications of genome-wide microarray genotyping (Hunter-Zinck et al., 2010; Omberg et al., 2012) or high-throughput next-generation sequencing to study this population (Rodriguez-Flores et al., 2012), particularly in comparison to populations that have been sampled as part of major genomics consortiums such as the Human Genome Diversity Project (HGDP) (Cann et al., 2002), HapMap (1000 Genomes Project Consortium, 2010; ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/), or the 1000 Genomes (1000G) Project (1000 Genomes Project Consortium et al., 2012). As a consequence, there is limited information concerning the genomes of the Qatari people. For example, there is little known about what Mendelian disease variants are segregating in Qatari genomes and the contribution of these to the health profile of the modern Qatari population, a situation which is mirrored in other native populations of the Arabian Peninsula and in other understudied regions of the world.
In this study, we demonstrate the power of applying next-generation exome sequencing in populations such as the Qatari, who have been understudied from a genomics perspective. Specifically, to gain further insight into the exome genetic variation of the population of Qatari nationals and their genome-based risk for inherited disorders, we carried out massive parallel exome sequencing of a sample of 100 Qatari genomes. By applying a detailed annotation analysis, we identified 37 polymorphisms that we infer are likely to be responsible for Mendelian diseases in the Qatari population. Using 1000G, the largest and most diverse resource of human exome variation (1000 Genomes Project Consortium et al., 2012) available in populations with some slight degree of genetic relatedness to Qataris (Hunter-Zinck et al., 2010; Omberg et al., 2012; Rodriguez-Flores et al., 2012), we assessed whether any of the 37 disease variant alleles identified are at higher frequency in Qatar when benchmarked vs this worldwide sample. Of the variants we identified, 10 were not represented in the populations of the 1000 Genomes consortium and an additional 2 were found to be at significantly higher frequency when comparing to the 1000 Genomes sample. We also found that 2 of the Mendelian polymorphisms were segregating at disease allele frequency >1% only within one of the genetic subpopulations (Q1, Q2, or Q3), where these subpopulations tend to be good predictors of marriage patterns among Qatari (Sandridge et al., 2010).
Methods
Ethics Statement
Human subjects were recruited and written informed consent obtained at Hamad Medical Corporation (HMC), Doha, Qatar under protocols approved by the Medical Research Center & Research Committee and the Institutional Review Board of Weill Cornell Medical College in Qatar.
Inclusion Criteria
The goal of the study was to assess genetic variation in a population that could be clearly demarcated by population genetic criteria and also reflects a unit relevant for the current population in terms of intra-marrying frequency. As selection criteria, we therefore required that subjects be third generation Qataris where all ancestors were Qatari citizens born in Qatar, as assessed by questionnaires. Recent immigrants or residents of Qatar who traced their recent ancestry to other geographic regions were excluded. A previous population genetic study utilizing genotyping microarrays that used these criteria (Pritchard et al., 2000; Hunter-Zinck et al., 2010; Rodriguez-Flores et al., 2012) found this approach produced a sample clearly definable by principal component analysis (PCA) when compared to other worldwide populations. Individuals selected using this criteria fell into three clearly definable subpopulations: Q1-Bedouin, Q2-Persian-South Asian, and Q3-African ancestry (Hunter-Zinck et al., 2010; Omberg et al., 2012) that reflect the historical migration patterns in the region (Omberg et al., 2012) where studies indicate there tend to be strong patterns of intra-marrying within population subgroups and that these individuals tend not to marry outside of this population as a whole (Sandridge et al., 2010). We used a panel of 48 SNPs genotyped by TaqMan (Life Technologies, Carlsbad, CA) sufficient for classification of Qataris in one of these 3 groups based on >70% ancestry (Figure 1A) in one cluster in a STRUCTURE analysis with k=3 to identify individuals that could unambiguously be placed in one of these three groups (Rodriguez-Flores et al., 2012).
Figure 1.
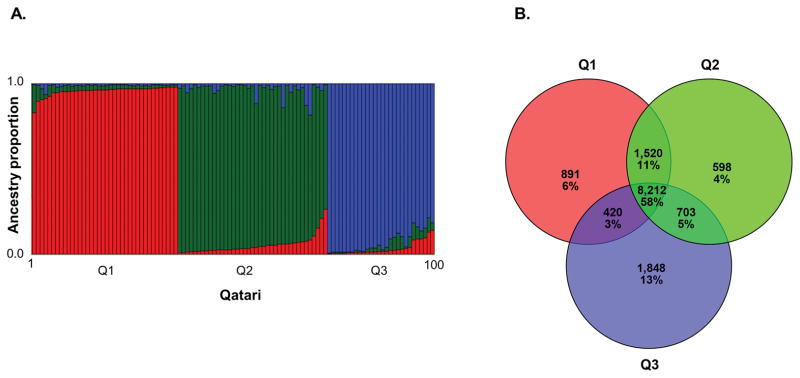
Qatari subpopulation structure and exome major alleles. A. Plot showing the results of a STRUCTURE analysis used for the selection of the 100 Qatari in the study (Pritchard et al., 2000). Each individual was genotyped for 48 SNPs by TaqMan where this panel was designed to quantify the proportion of Bedouin (Q1), Persian-South Asian (Q2), and African (Q3) ancestry in a Qatari individual. Each pair of columns represents the proportion of Red = Q1, Green = Q2, Blue = Q3 ancestry for an individual. All individuals in the study had >70% ancestry in one of Q1, Q2 or Q3 population clusters. B. A Venn diagram showing the overlap between three sets of Bedouin (Q1), Persian-South Asian (Q2), and African (Q3) subpopulation major allele alternate variants (total 14,192 variants). For each subpopulation, the number of sites where the alternate allele is the major allele was counted, and the list of major alternate allele sites was compared between subpopulations.
Subjects with no known familial relationships satisfying the Qatari ancestry criteria and with unambiguous assignment to a Qatari subpopulation were selected from a group visiting the health clinics at Hamad Hospital, Doha, Qatar, for a routine diabetes screening; the prevalence of type-2 diabetes in Qatar is extremely high (20%) (http://www.idf.org/diabetesatlas). Selection of the study sample attempted to produce a relatively even distribution of males and females (32 M and 68 F), a relatively even distribution across each of the three Qatari subpopulations (36 Q1, 38 Q2, and 26 Q3), and an even distribution of those with and without type 2 diabetes (51 with and 49 without). The final set of subjects in the sample matching these criteria was selected from medical record information by researchers at Weill Cornell Medical College in New York City, who did not have any direct interactions with the subjects.
Variants Discovered in Qatar
In order to characterize the spectrum of genetic variation in Qataris, 100 exomes were sequenced by the Beijing Genomics Institute to a median depth of 67x using paired-end 90 bp Illumina reads on a HiSeq 2000 (BGI Americas, Cambridge, MA). Reads were mapped to human reference genome GRCh37 using BWA 0.5.9 (Li and Durbin, 2010) with parameters (quality threshold for read trimming 15, maximum number of fraction gap opens 1, indels disallowed within 15 bp of end of read, long gaps disabled, alignment seed length 32 with a maximum of 2 mismatches in seed, and maximum distance between paired end read mapping of 2,000 bp). Each exome was verified to have ≥10x depth at >80% of exome target sites (38 Mb Agilent enrichment platform) with reads mapped in a proper pair of mapping quality >10 and base quality >17. Mapped reads were prepared for genotyping using the “Best practices for variant detection v3” GATK (http://www.broadinstitute.org/gatk/) pipeline, including removal of PCR-duplicate reads, realignment across known indels, and base quality score recalibration (DePristo et al., 2011). The number of exome bases covered at ≥1x and ≥10x was determined using SAMTOOLS (Li et al., 2009).
Genotypes were called in each individual exome at all exome sites with sufficient coverage to call a genotype (ref/ref, ref/alt, or alt/alt) using GATK v1.1-24 in “emit all confident sites” mode. The 100 individual VCF files were then filtered and merged into a single population VCF file using a Perl script. First, a site list of variants discovered in at least one exome at ≥10x depth, <180x depth, and with quality >50 was generated. Second, the 100 individual VCF files were combined to include a genotype (ref/ref, ref/alt, alt/alt) for each exome. Genotypes with insufficient coverage, excess coverage, or low quality were marked missing. The allele balance for homozygous and heterozygous genotypes was then examined using a binomial model. For homozygous sites, a maximum of 1 alternate base was allowed. For heterozygous sites, a minimum of 2 alternate bases was required, and the binomial p value of both the reference and alternate allele count was required to be >10−3, given the expected count for both reference and alternate to be 50% of the depth (5 and 5 for 10x depth). Variants not meeting the heterozygous and homozygous criteria were filtered out. Next, a Chi-square test of Hardy-Weinberg Equilibrium was assessed for each variant, and variants with p<10−5 were filtered out. Finally, a 90% callability filter was applied, removing sites where high-confidence genotypes were obtained for less than 90% of the exomes. A VCF file containing genotypes for this final variant set is available online at: http://mezeylab.cb.bscb.cornell.edu/Software.aspx.
Coding Variant Allele Frequency
The variants identified in the 100 Qatari exomes were assigned to genes and functionally classified using gene models from the ENSEMBL (Flicek et al., 2012) database of genes and transcripts (build 65) by SNPEFF (Cingolani et al., 2012). The population frequency distribution of 95,840 high-confidence coding variants with known function on the 22 autosomal chromosomes and chromosome X was calculated for the Qatari population while accounting for gender at sites on the X chromosome (Schaffner, 2004). Variants on the X chromosome were filtered using gender-aware criteria (1000 Genomes Project Consortium et al., 2012). Female X chromosome variants were treated as described for autosomes above, while male X chromosome variants were treated as homozygous haploid variants. Variants were binned as the “variant is the major allele”, where the alternate allele was the major allele in Qataris and “variant in the minor allele,” where the alternate allele was the minor allele in Qataris. The minor allele site frequency spectrum of coding variants was generated by binning variants by minor allele frequency in 1% bins from 0 to 0.5 and plotted on a log scale.
Comparison of Qatari Sub-population Allele Frequency
In order to define the “Qatari exome”, variants were binned for the Qatari and for the Q1, Q2 and Q3 subpopulations as the “variant is the major allele”, where the alternate allele was the major allele in Qataris and “variant is the minor allele,” where the alternate allele was the minor allele in Qataris. For each subpopulation, a major allele reference exome was generated, where the reference allele was replaced with the major allele in Q1, Q2, or Q3.
In order to identify variants that distinguish the Q1, Q2 and Q3 Qatari sub-populations, the allele frequency was compared for 95,840 coding variants using two methods, Fst and allele frequency difference (Akey et al., 2002; Holsinger and Weir, 2009). The Fixation Index (Fst) was used as a measure of population differentiation and was calculated using the unbiased estimation approach comparing observed mean square errors within and between subpopulations described elsewhere (Akey et al., 2002). These two statistics were calculated for comparison of Q1 vs Q2; Q1 vs Q3; and Q2 vs Q3.
Variants Linked to Mendelian Disorders
To identify coding variants linked to Mendelian disorders, a functional classification was used where in cases of multiple transcripts the most severe was selected in order to maximize the likelihood of identifying a variant present in the OMIM or HGMD database. Of these coding variants, 251 were previously reported genetic disorder variants in the OMIM database (http://omim.org) or HGMD database GenomeTrax webserver (http://www.biobase-international.com/product/genome-trax. This original list was reduced by a literature review manual curation down to 37 variants in 33 genes where both the variants and the genes were clearly linked to 36 recessive, dominant or X-linked Mendelian disorders by excluding variants under the following categories: greater than 5% disease allele frequency in Qatari; disease allele unclear; association/risk/susceptibility variant; compound with other variants in cases; observed in controls, molecular basis not known for phenotype; insufficient evidence to determine causality; reclassified by OMIM as variant of unknown significance; conflicting reports in literature; drug metabolism variant not linked to disease; genotype of cases not specified; modifier not causative; polymorphism with no known functional impact [referred to by OMIM as a “polymorphism” (http://www.omim.org/help/faq)]; somatic mutation in cancer; gene not expressed in disease tissue; observed in heterozygous state in cases; and SNPEFF and OMIM (or HGMD) disagree on function. This final list was considered to have the strongest evidence for including true Mendelian disease-causative variants identified in the sample given available information. These 37 variants observed in 100 Qatari exomes were compared to the 14 SNP Mendelian disease variants directly genotyped in the panel of genetic tests conducted in the Laboratory Medicine and Pathology, Hamad Medical Corporation, Doha, Qatar, which is used to screen Qatari couples before marriage (Supp. Table S1).
In addition, to determine if prior reports of the 36 diseases linked to the 33 genes were previously observed in Qatar or other Arab populations, the Center of Arab Genomics Studies (CAGS) database (http://www.cags.org.ae) was queried using the MIM ID number for each gene (http://omim.org). The CAGS database lists 113 genetic disorders and 27 associated gene loci in the Arab population of Qatar (Tadmouri, 2012), in both text and online database formats, with each disease and locus indexed by its MIM number. The list was compiled from articles in Pub-Med and the WHO Index Medicus for the Eastern Mediterranean.
The analysis identified additional variants observed in the Qatari population that are present within disease causing genes, although the function of the variants is not yet known. These variants have the potential to be disease causing. Although it was not the focus of this study, the list of 3,293 genes present in either the OMIM or HGMD databases was compared to the list of coding mutations with potential effect on protein function, including nonsynonymous, missense, splice site donor, splice site acceptor, frameshift, start loss, codon insertion, stop lost, and codon change plus codon insertion.
Comparison of Variant Frequencies in 1000 Genomes Populations
In order to identify variants unique to or at elevated allele frequency in the Qatari, we assessed the frequency of each of the 37 variants in the 1000 Genomes (1000G) Project Phase 1 v3 release of genotypes for 14 populations (1000G: ASW, CEU, CHB, CLM, FIN, GBR, GIH, IBS, JPT, LWK, MXL, PUR, TSI, YRI) (1000 Genomes Project Consortium et al., 2012). For variants segregating with at least 2 observed alleles that were also observed in at least one 1000 Genomes population, we used a one-sided binomial test to compare the frequency of the Qatari sample as a whole to the entire 1000 Genomes sample, using the latter as the expected frequency. A Bonferroni-corrected threshold was used to assess significance. For variants found to be significant, we considered the non-zero Q1, Q2, or Q3 subpopulation frequencies of these variants and again used a one-sided binomial test to check if these subpopulation frequencies were significantly higher when compared to each of the 1000 Genomes populations that were segregating for the variant, where again the 1000 Genomes population frequencies were used as the expected frequency for these tests.
TaqMan Genotyping
Potential disease-causative mutations were confirmed by allelic discrimination TaqMan (Life Technologies) assays. To accomplish this, an additional 386 Qatari genomes were genotyped, including n=217 Q1, n=154 Q2, and n=15 Q3. Pre-designed or custom allelic discrimination reagents were used in TaqMan genotyping assays using vendor’s protocols in the ABI 7500 Sequence Detection System (Life Technologies). End point reads were confirmed with inspection of fluorophore-specific amplification plots when necessary.
Results
Variants Discovered in Qatar
To characterize the spectrum of genetic variation in Qatari, 100 exomes were sequenced to a median depth of 67x using paired-end 90 bp Illumina reads on a HiSeq 2000. A total of 2.2 Gb of sequence data was collected for each exome, consisting of reads mapped to human reference genome GRCh37 using BWA 0.5.9 with reads of mapping quality >10 and base quality >17 (Table 1). The resulting median depth was 67x in the target exome of 37.4 Mb, with a median of 36.5 Mb covered with at least one read (97.3% of target exome). All exomes met a quality threshold of >80% of target sites at ≥10x depth, a median of 33.3 Mb covered at this depth. Coverage depth in the X and Y chromosomes was lower and differed between sexes. For females, the X chromosome mean depth was 76x, with 89.1% of target sites at ≥10x depth. For males, the X chromosome mean depth was 43x, with 76.6% of target sites at ≥10x depth, lower than in females. The male Y chromosome mean depth was 49x, with 53.2% of target sites at ≥10x depth.
Table 1.
Variants Identified in 100 Qatari Exomes1
| Statistic | Chromosomes
|
||||
|---|---|---|---|---|---|
| Total (%) | Autosomal | Female X | Male X | Male Y | |
| Per individual2 | |||||
| Alleles | 200 | 200 | 136 | 32 | 32 |
| Samples | 100 | 100 | 68 | 32 | 32 |
| Total collected bases (Gb) | 2.2 | 2.1 | 0.10 | 0.054 | 0.0021 |
| Mean mapped depth on target (X) | 67 | 67 | 76 | 43 | 49 |
| Target exome (Mb) | 37.5 | 35.9 | 1.53 | 1.53 | 0.077 |
| Megabases accessed (% of target exome) | 36.5 (97.3) | 35.1 (97.7) | 1.46 (95.4) | 1.45 (94.8) | 0.046 (60.2) |
| Megabases at ≥10x (% of target exome) | 33 (87.2) | 31 (87.4) | 1.4 (89.1) | 1.2 (76.6) | 0.041 (53.2) |
| Mean variants per individual (% novel) | 17,487 (2) | 17,392 (2) | 114 (11) | 57 (4) | 0 (*) |
| Mean variant SNPs per individual (% novel) | 17,399 (2) | 17,304 (2) | 113 (11) | 57 (4) | 0 (0) |
| Mean Ti:Tv per individual (novel) | 3.04 (2.57) | 3.04 (2.57) | 3.16 (3.41) | 3.15 (2.46) | * (*) |
| Mean variant indel sites per individual (% novel) | 88 (15) | 88 (15) | 0.75 (59) | 0.06 (0) | 0 (*) |
| Population3 | |||||
| Number of variants (% novel) | 132,303 (23) | 129,938 (23) | 2,349 (29) | 756 (8) | 0 (*) |
| Number of SNPs (% novel) | 131,036 (23) | 128,693 (23) | 2,327 (29) | 755 (8) | 0 (*) |
| Number of indels (% novel) | 1,267 (53) | 1,245 (53) | 22 (72) | 1 (0) | 0 (*) |
| Ti:Tv SNPs (novel) | 2.97 (2.60) | 2.97 (2.60) | 2.98 (2.82) | 2.95 (3.07) | 0 (*) |
100 exomes were sequenced to >10x depth at >80% of target exome sites (37.4MB) using paired-end 90 bp Illumina reads on a HiSeq 2000. Reads were mapped to human reference genome GRCh37 using BWA 0.5.9 (Li and Durbin, 2010), and variants were called from reads of mapping quality >10 and bases with quality >17 using GATK (DePristo et al., 2011). Variants were filtered to remove deviations from Hardy-Weinberg equilibrium, sites with low coverage, sites with less than 90% callability, and sites with an allele balance likely to be a false positive. Top section shows a summary per individual, and the bottom section shows a summary for the Qatari population. Results are stratified by chromosome, with autosomal, X and Y chromosome variants reported separately. X chromosome results are shown separate for males and females.
From top-to-bottom is shown the number of Qatari alleles (2n), number of Qatari individuals; total gigabases mapped to reference within target exons; mean depth of exome sites covered with ≥1 read; total target exome size in megabases; size and percentage of target sites at coverage ≥1x and ≥10x; mean number and % novel (not in dbSNP 135) of variants per exome for SNPs + indels, SNPs, then indels; and transition-to-transversion (Ti:TV) ratio for all and novel SNPs. Each result is shown for the complete exome, autosomal sites (chr1 to chr22), X chromosome in females, X chromosomes in males, and Y chromosomes in males. No high-confidence variants with >90% call ability were observed on the Y chromosome. “*” indicates Ti:Tv values not calculated due to division by zero.
From top-to-bottom is shown the number and % novel of variants observed in the Qatari population, including total variants (SNPs + indels), SNPs, then indels. Bottom row is the Ti:Tv ratio for all and novel SNPs. Each result is shown for the complete exome, autosomal sites (chr1 to chr22), X chromosome in females, X chromosomes in males, and Y chromosomes in males. “*” indicates Ti:Tv values not calculated due to division by zero. Variants in the Y chromosome are not listed due to low call ability.
After filtering of low-quality sites, the proportion of novel variants was counted at both the individual and population levels, where “novel” was defined as a variant not present in dbSNP 135. At the population level, a total of 132,303 variants were observed, 23% of which were novel (Table 1). This call set included 131,036 SNPs (23% novel) and 1,267 indels (53% novel). On average 17,487 variants were observed in each exome, including 17,399 SNPs and 88 indels. The proportion of novel variants was 2% for SNPs and 15% for indels. The SNP transition-to-transversion ratio (Ti:Tv) was 3.04 overall and 2.57 in novel SNPs. Stratified by gender, more novel SNPs and indels were observed in the X chromosomes of females (mean 113 SNPs and 0.75 indels discovered per exome) compared to the X chromosome of males (57 SNPs and 0.06 indels), a result consistent with the 2:1 ratio of these chromosomes, such that more novel SNPs are expected to be present in a larger sample of chromosomes. SNPs were observed in individual Y chromosomes; however the call rate was below 90% across all 32 male Qatari Y chromosomes and they were excluded on this basis, as described in Methods.
In a preliminary study of the exome sequences of 7 Qatari (3 Q1, 2 Q2, 2 Q3), we identified 38,427 autosomal SNPs in the Qatari population (Rodriguez-Flores et al., 2012). The majority of these variants (20,208 or 53%) were also observed in the 100 Qatari exomes. Because these are independent samples of the Qatari population, it provides an indication of the amount of rare genetic variation yet to be sampled in Qatar.
Coding Variant Allele Frequency
To verify the overall quality of the call set, the site frequency spectrum of coding variants among Qatari exomes was analyzed. The variants identified in 100 Qatari exomes were assigned to genes and functionally classified using SNPEFF (Cingolani et al., 2012). A total of 95,840 coding SNPs were observed in the ENSEMBL (Flicek et al., 2012) database of genes and transcripts (build 65; Table 2). Of these, 94% were nonsynonymous (49.4%) or synonymous (44.6%), and the remaining 6.0% included loss of function variants (splice donor, frameshift, splice acceptor, start loss, stop gained, start gained). The population frequency distribution of these 95,840 high-confidence coding variants with known function on 22 autosomes and the X chromosome was calculated for the Qatari population, accounting for gender at sites on the X chromosome (Supp. Figure S1). Nearly half (46%) were “personal variants,” single alleles observed in one exome (minor allele frequency 0.005 or 1 in 200 alleles), 36% were rare variants (minor allele frequency 0.005 to 0.1), 16.5% were common variants (minor allele frequency of 0.10 to 0.50). The variant (non-reference) allele was the major allele in Qataris for 5.8% of all variants; 0.5% of the Qatari variants had a non-reference frequency of 1.0 (fixed for the alternate allele).
Table 2.
Functional Classification of Variants in 100 Qatari Exomes1
| Class | Total | OMIM + HGMD | ||
|---|---|---|---|---|
|
| ||||
| Number | % | Total | % | |
| Nonsynonymous coding | 47,352 | 49.41 | 228 | 90.83 |
| Synonymous coding | 42,786 | 44.64 | 12 | 4.78 |
| Utr 3 prime | 2,752 | 2.87 | 0 | 0.00 |
| Utr 5 prime | 1,253 | 1.31 | 1 | 0.40 |
| Nonsense | 665 | 0.69 | 9 | 3.59 |
| Start gained | 260 | 0.27 | 0 | 0.00 |
| Splice site donor | 210 | 0.22 | 1 | 0.40 |
| Frameshift | 163 | 0.17 | 0 | 0.00 |
| Splice site acceptor | 163 | 0.17 | 0 | 0.00 |
| Start lost | 91 | 0.09 | 0 | 0.00 |
| Stop lost | 47 | 0.05 | 0 | 0.00 |
| Synonymous stop | 43 | 0.04 | 0 | 0.00 |
| Codon insertion | 36 | 0.04 | 0 | 0.00 |
| Codon change plus codon insertion | 16 | 0.02 | 0 | 0.00 |
| Nonsynonymous start | 3 | 0.00 | 0 | 0.00 |
| Codon change plus codon deletion | 0 | 0.00 | 0 | 0.00 |
| Codon deletion | 0 | 0.00 | 0 | 0.00 |
|
|
||||
| Total | 95,840 | 100 | 251 | 100.00 |
To identify variants linked to Mendelian disorders, the variants identified in 100 Qatari exomes were assigned to genes and functionally classified. Shown is a summary of 95,840 coding variants observed in the ENSEMBL (Flicek et al., 2012) database of genes and transcripts (build 65). For each variant, in cases where multiple transcripts are present, the most severe variant was selected, using a severity classification scheme established by SNPEFF (Cingolani et al., 2012). Of these coding variants, shown is the number and percentage in the OMIM (OMIM, 2012) or HGMD (Stenson et al., 2009) database of variants linked to genetic disorders with a known molecular basis.
Characterization of the Q1, Q2 and Q3 Qatari Exomes
Variants that differentiate the Q1, Q2 and Q3 Qatari sub-populations were identified by comparing the allele frequency for 95,840 coding variants using two methods, Fst and allele frequency difference comparisons of Q1 vs Q2, Q1 vs Q3, and Q2 vs Q3 (see Supp. Figure S2).
In the context that the use of “major allele” reference genomes are more effective at identification of population-specific variants (Dewey et al., 2011), future studies of Qatari exomes can benefit from a definition of a reference Qatari exome where the major allele in Q1, Q2 and Q3 is used in-lieu of the standard reference allele. For this purpose, the Qatari exomes were grouped into Q1, Q2 and Q3 populations (Supp. Figure S3), the major allele was determined for each population and major allele reference exomes were generated for Q1, Q2 and Q3 in FASTA (Pearson and Lipman, 1988) format. In each population-specific exome, the major allele in the Qatari subpopulation was substituted for the reference allele. This included a total of 14,192 sites, with 11,043 major allele sites in Q1, 11,033 major allele sites in Q2 and 11,183 major allele sites in Q3. The overlap among Q1, Q2 and Q3 (Figure 1B) represented 58% of the total major allele sites where the alternate allele was the major allele in at least one population (8,212 of 14,192).
Variants Linked to Mendelian Disorders
A total of 11,288 coding variants with potential effect on protein function were identified in 3,293 OMIM or HGMD genes, including 11,000 nonsynonymous, 139 missense, 46 splice site donor, 42 splice site acceptor, 22 frameshift, 17 start lost, 9 codon insertion, 9 stop lost, and 4 codon change plus codon insertion (Supp. Table S2). Of these, a total of 251 coding variants in the Qatari exomes were previously linked to a disease phenotype in the OMIM and HGMD databases and were assigned to a dbSNP rsID. Of these, 91% were nonsynonymous, 5.3% were synonymous, 3.1% were nonsense, and 0.3% were splice site donor variants (Table 2). Many of the variants discovered were for complex disorders or were at frequencies too high to be consistent with a deleterious/penetrant disorder as identified in OMIM. However, when limiting this set by manual curation of the literature to those with strongest available evidence for being true Mendelian disease-causative variants (see Methods), we identified a list of 37 variants in 33 genes, representing 36 disorders, that had genotype frequencies consistent with deleterious Mendelian effects (Table 3, Supp. Table S3). All of these were present in OMIM; an additional five variants present in HGMD and not in OMIM were filtered out using these criteria (BMP4, MIM# 112262, c.1070G>A, p.R287H, modifier mutation; TINF2, MIM# 604319, c.1076C>A, p.S245Y, observed in controls; PHKB, MIM# 172490, c.607G>T, p.M185I, insufficient evidence to determine causality; AGTR2, MIM# 300034, rs12917810, SNPEFF and HGMD disagree on coding function; GNPTG, MIM# 607838, rs193302860, SNPEFF and HGMD disagree on coding function). A full list of variants excluded and the reason for exclusion is in Supp. Table S4. On the average, we observed 2 curated OMIM variants per person; the highest number of curated OMIM variants was 5 for one individual.
Table 3.
OMIM Variants Linked to Mendelian Disorders Observed in the 100 Qataris1
| OMIM Information2 | Disease allele3 | Qatari genotype counts and disease allele frequency (d = disease, wt = wild-type)4 |
1000 Genomes genotype counts and disease allele frequency (d = disease, wt = wild-type)5 |
||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||||||||||
| Phenotype | Gene | Mutation | Inheritance mode (R = recessive, D = dominant, X = X-linked |
DbSNP | Ref>alt | Disease allele | Q d/d | Q d/wt | Q wt/wt | Q d | Q1 d | Q2 d | Q3 d | Population specificity of variant |
1000G d/d | 1000G d/wt | 1000G wt/wt | MAX d 1000G | MAX Pop | ||||
|
|
|
|
|||||||||||||||||||||
| Category | Name | MIM | Symbol | MIM | Coding base change |
Amino acid change |
|||||||||||||||||
| Auditory | Deafness autosomal recessive 1A | 220290 | GJB2 | 121011 | c.286G>A | p.Trp24Ter | R | rs104894396 | C>T | Alt | 0 | 1 | 99 | 0.005 | 0.000 | 0.000 | 0.019 | Q3 | 0 | 0 | 0 | 0.000 | |
| Hematologic | Thrombophilia due to factor V Leiden | 188055 | F5 | 612309 | c.1601G>A | p.Arg506Gln | D | rs6025 | T>C | Ref | 0 | 2 | 98 | 0.010 | 0.000 | 0.026 | 0.000 | Q2 | 1 | 8 | 1083 | 0.029 | CEU |
| Hemoglobin S sickle cell anemia | 603903 | HBB | 141900 | c.70A>T | p.Glu6Val | R | rs77121243 | T>A | Alt | 0 | 2 | 98 | 0.010 | 0.000 | 0.013 | 0.019 | Q2, Q3 | 1 | 47 | 1044 | 0.098 | LWK | |
| Hemoglobin E beta-plus-thalassemia | 613985 | c.129G>A | p.Glu26Lys | R | rs33950507 | C>T | Alt | 0 | 1 | 99 | 0.005 | 0.000 | 0.000 | 0.019 | Q3 | 0 | 0 | 0 | 0.000 | ||||
| Hemoglobin D (Punjab; Chicago; Los Angeles; North Carolina; Portugal; Oak ridge) | * | c.414G>C | p.Glu121Gln | R | rs33946267 | C>G | Alt | 0 | 2 | 98 | 0.010 | 0.000 | 0.013 | 0.019 | Q2, Q3 | 0 | 0 | 0 | 0.000 | ||||
| Thrombocythemia 2 | 601977 | MPL | 159530 | c.162G>T | p.Lys39Asn | R | rs17292650 | G>T | Alt | 0 | 2 | 98 | 0.010 | 0.000 | 0.000 | 0.038 | Q3 | 1 | 23 | 1068 | 0.074 | ASW | |
| Familial Mediterranean fever | 249100 | MEFV | 608107 | c.2270G>T | p.Ala744Ser | D | rs61732874 | C>A | Alt | 0 | 1 | 99 | 0.005 | 0.000 | 0.000 | 0.019 | Q3 | 0 | 3 | 1089 | 0.009 | PUR | |
| c.2120A>G | p.Met694Val | rs61752717 | T>C | Alt | 0 | 1 | 99 | 0.005 | 0.000 | 0.013 | 0.000 | Q2 | 0 | 0 | 0 | 0.000 | |||||||
| Bone | Ellis-van Creveld syndrome | 225500 | EVC | 604831 | c.1512G>A | p.Arg443Gln | D | rs35953626 | G>A | Alt | 0 | 7 | 89 | 0.036 | 0.000 | 0.027 | 0.096 | Q2, Q3 | 10 | 100 | 982 | 0.232 | LWK |
| Holoprosencephaly 4 | 142946 | TGIF | 602630 | c.636A>T | p.Gln107Leu | D | rs28939693 | A>T | Alt | 0 | 4 | 96 | 0.020 | 0.056 | 0.000 | 0.000 | Q1 | 0 | 0 | 0 | 0.000 | ||
| Odontoonychodermal dysplasia | 257980 | WNT10A | 606268 | c.1145T>A | p.Phe228Ile | D | rs121908120 | T>A | Alt | 0 | 1 | 90 | 0.005 | 0.016 | 0.000 | 0.000 | Q1 | 5 | 16 | 1071 | 0.036 | PUR | |
| Eye | Stargardt disease 1 | 248200 | ABCA4 | 601691 | c.2895G>A | p.Val931Met | R | rs58331765 | C>T | Alt | 0 | 1 | 98 | 0.005 | 0.000 | 0.000 | 0.019 | Q3 | 0 | 11 | 1081 | 0.052 | LWK |
| Retinitis pigmentosa 26 | 608380 | CERKL | 608381 | c.870C>T | p.Arg257Ter | R | rs121909398 | G>A | Alt | 0 | 1 | 99 | 0.005 | 0.000 | 0.013 | 0.000 | Q2 | 0 | 0 | 0 | 0.000 | ||
| Glaucoma 3, primary congenital A | 231300 | CYP1B1 | 601771 | c.1506G>A | p.Arg368His | R | rs79204362 | C>T | Alt | 0 | 3 | 97 | 0.015 | 0.028 | 0.013 | 0.000 | Q1, Q2 | 1 | 1 | 1090 | 0.006 | CEU | |
| Retinitis pigmentosa 1 | 180100 | RP1 | 603937 | c.1266C>T | p.Thr373Ile | R | rs77775126 | C>T | Alt | 0 | 4 | 96 | 0.020 | 0.000 | 0.053 | 0.000 | Q2 | 2 | 15 | 1075 | 0.041 | TSI | |
| Cardiovascular | Long QT syndrome 9 | 611818 | CAV3 | 601253 | c.310C>A | p.Thr78Met | D | rs72546668 | C>T | Alt | 0 | 2 | 88 | 0.011 | 0.016 | 0.015 | 0.000 | Q1, Q2 | 0 | 4 | 1088 | 0.015 | TSI |
| Tetralogy of Fallot | 187500 | NKX2-5 | 600584 | c.302C>T | p.Arg25Cys | D | rs28936670 | G>A | Alt | 0 | 4 | 90 | 0.021 | 0.014 | 0.000 | 0.060 | Q1, Q3 | 1 | 9 | 1082 | 0.009 | PUR | |
| Arterial tortuosity syndrome | 208050 | SLC2A10 | 606145 | c.340C>G | p.Ser81Arg | R | rs80358230 | C>G | Alt | 0 | 3 | 97 | 0.015 | 0.042 | 0.000 | 0.000 | Q1 | 0 | 0 | 0 | 0.000 | ||
| Inflammatory | Granulomatous disease chronic autosomal recessive cytochrome b-positive type II | 233710 | NCF2 | 608515 | c.1458C>T | p.Arg395Trp | R | rs13306575 | G>A | Alt | 0 | 1 | 99 | 0.005 | 0.000 | 0.000 | 0.019 | Q3 | 1 | 34 | 1057 | 0.068 | MXL |
| Familial cold autoinflammatory syndrome 2 | 611762 | NLRP12 | 609648 | c.1070C>T | p.Arg284Ter | D | rs104895564 | G>A | Alt | 0 | 1 | 99 | 0.005 | 0.000 | 0.000 | 0.019 | Q3 | 0 | 14 | 1078 | 0.072 | LWK | |
| Familial cold autoinflammatory syndrome 1 | 120100 | NLRP3 | 606416 | c.1344G>A | p.Val198Met | D | rs121908147 | G>A | Alt | 0 | 1 | 99 | 0.005 | 0.000 | 0.013 | 0.000 | Q2 | 1 | 10 | 1081 | 0.022 | FIN | |
| Immunity | Masp2 deficiency | 613791 | MASP2 | 605102 | c.380A>G | p.Asp105Gly | R | rs72550870 | T>C | Alt | 0 | 1 | 99 | 0.005 | 0.000 | 0.013 | 0.000 | Q2 | 12 | 23 | 1057 | 0.076 | CEU |
| Kidney | Cystinuria | 220100 | SLC7A9 | 604144 | c.661G>A | p.Ala182Thr | D | rs79389353 | C>T | Alt | 0 | 2 | 97 | 0.010 | 0.000 | 0.027 | 0.000 | Q2 | 1 | 3 | 1088 | 0.010 | TSI |
| Liver | North American Indian childhood cirrhosis | 604901 | CIRH1A | 607456 | c.870C>T | p.Arg565Trp | R | rs119465999 | C>T | Alt | 0 | 1 | 99 | 0.005 | 0.000 | 0.013 | 0.000 | Q2 | 0 | 5 | 1087 | 0.025 | CLM |
| Metabolic | Cystinosis atypical nephropathic | 219800 | CTNS | 606272 | c.594G>A | p.Val42Ile | R | rs35086888 | G>A | Alt | 0 | 2 | 98 | 0.010 | 0.000 | 0.013 | 0.019 | Q2, Q3 | 0 | 35 | 1057 | 0.067 | LWK |
| Maturity-onset diabetes of the young type 7 | 610508 | KLF11 | 603301 | c.821C>T | p.Thr220Met | D | rs34336420 | C>T | Alt | 0 | 2 | 98 | 0.010 | 0.000 | 0.013 | 0.019 | Q2, Q3 | 0 | 17 | 1075 | 0.046 | LWK | |
| Combined hyper-lipidemia familial | 144250 | LPL | 609708 | c.476G>A | p.Asp9Asn | D | rs1801177 | G>A | Alt | 0 | 5 | 94 | 0.025 | 0.014 | 0.013 | 0.058 | Q1, Q2, Q3 | 2 | 29 | 1061 | 0.041 | LWK | |
| Protoporphyria erythropoietic | 177000 | FECH | 612386 | c.918G>A | p.Met267Ile | R | rs118204037 | C>T | Alt | 0 | 1 | 99 | 0.005 | 0.014 | 0.000 | 0.000 | Q1 | 0 | 0 | 0 | 0.000 | ||
| c.280G>T | p.Gly55Cys | rs3848519 | C>A | Alt | 0 | 3 | 97 | 0.015 | 0.000 | 0.026 | 0.019 | Q2, Q3 | 2 | 32 | 1058 | 0.062 | GBR | ||||||
| Albinism oculocutaneous type IB | 203100 | TYR | 606933 | c.1299C>T | p.Pro406Leu | R | rs104894313 | C>T | Alt | 0 | 1 | 99 | 0.005 | 0.000 | 0.013 | 0.000 | Q2 | 0 | 0 | 0 | 0.000 | ||
| Beta-ureidopropionase deficiency | 613161 | UPB1 | 606673 | c.375C>A | p.Ala85Glu | R | rs34035085 | C>A | Alt | 0 | 1 | 99 | 0.005 | 0.000 | 0.000 | 0.019 | Q3 | 0 | 6 | 1086 | 0.015 | LWK | |
| Muscular | Miyoshi myopathy, muscular dystrophy limb-girdle type 2B | 254130 | DYSF | 603009 | c.4268A>G | p.Ile1298Val | R | rs121908954 | A>G | Alt | 0 | 1 | 95 | 0.005 | 0.000 | 0.014 | 0.000 | Q2 | 1 | 4 | 1087 | 0.009 | PUR |
| Muscular dystrophy-dystroglycanopathy (limb-girdle) type C3 | 613157 | POMGNT1 | 606822 | c.1666G>A | p.Asp556Asn | R | rs74374973 | C>T | Alt | 0 | 1 | 99 | 0.005 | 0.014 | 0.000 | 0.000 | Q1 | 3 | 13 | 1076 | 0.027 | FIN | |
| Neurologic | Mental retardation X-linked 88 | 248200 | AGTR2 | 300034 | c.1178G>A | (p.Arg324Gln | X | rs35474657 | G>A | Alt | 0 | 1 | 99 | 0.006 | 0.000 | 0.000 | 0.024 | Q3 | 0 | 3 | 564 | 0.014 | ASW |
| Aicardi-Goutieres syndrome 3 | 610329 | RNASEH2C | 610330 | c.385C>T | p.Arg69Trp | R | rs78635798 | G>A | Alt | 0 | 1 | 99 | 0.005 | 0.000 | 0.013 | 0.000 | Q2 | 0 | 0 | 0 | 0.000 | ||
| Reproductive | Premature ovarian failure 4 | 300510 | BMP15 | 300247 | c.587G>A | p.Ala180Thr | X | rs104894767 | G>A | Alt | 0 | 1 | 99 | 0.005 | 0.000 | 0.015 | 0.000 | Q2 | 0 | 2 | 565 | 0.010 | Many |
| Premature ovarian failure 2B | 300604 | POF1B | 300603 | c.1132G>A | p.Arg329Gln | X | rs75398746 | C>T | Alt | 0 | 1 | 99 | 0.005 | 0.017 | 0.000 | 0.000 | Q1 | 0 | 4 | 563 | 0.048 | CLM | |
Variants observed in 100 Qatari exomes were compared to the OMIM and HGMD database of allelic variants linked to disorders with a known molecular basis. The list was filtered to include only variants with ≥90% Qataris genotyped with confidence, and with variants linked to disorders where listed phenotype is a disorder, the disease allele can be determined, and the effect of the variant has literature evidence of autosomal dominant, recessive or X-linked inheritance. For a complete description of the exclusion critera and variant-by-variant reason for exclusion, see Supp. Table S4. Out of 251 variants, filtering resulted in the 37 variants in 33 genes listed. None of the HGMD variants passed the applied strict filtering. The table is sorted by category of the disorder.
Shown is the OMIM information (category, phenotype with MIM number, gene with MIM number, mutation in wildtype>disease format with coding base change and amino acid change, inheritance mode of the disorder). The gene is bolded and underlined if the Qatari allele frequency is higher than 1% above the maximum frequency observed in the 1000 Genomes; bolded if the Qatari allele frequency is not higher than 1% above the maximum frequency observed in the 1000 Genomes, but the Q1, Q2, or Q3 allele frequency is higher than 1% above the maximum frequency observed in 1000 Genomes; in italics if the variant is observed in only one Qatari population cluster and bolded and italic for combinations; for example RP1 c.1226C>T p.Thr373Ile observed only in Q2 with 0.053 higher than maximum 0.041 in TSI but not higher when considering whole Qatari population frequency 0.020. The inheritance mode was determined based on the literature and OMIM synopsis: if the variant is on the X chromosome it is marked as “X-linked”, if the variant is observed in cases only in the homozygous state it is marked as “auto-somal recessive”, and if the variant is observed in cases in the heterozygous state it is marked as “autosomal dominant”.
The alternate allele is not always the disease allele. In order to determine the disease allele, the variant function (such as F5 c.1601G>A p.Arg506Gln) was compared “ref>alt” vs “wildtype>disease”. For cases where the reference allele was the disease allele (e.g., F5 Leiden c.1601G>A p.Arg506Gln), the disease allele homozygous genotype count is for the reference allele. The first column is the dbSNP ID, which can be linked to multiple alternate alleles. The second column indicates the reference and alternate alleles in “ref>alt” format. The third column indicates which allele is the disease allele. Genotype counts and allele frequencies are based on the disease/wildtype orientation.
Disease allele frequency was calculated for the QE100 and for each Qatari population cluster (Q1, Q2, Q3). If the variant was observed in only one population cluster, the population name was noted in the column under “population specificity of variant”.
Shown is the genotype frequency for the total of 1,092 individuals from 14 populations (FIN, GBR, CEUR, IBS, TSI, JPT, CHB, CHS, MXL, CLM, PUR, ASW, LWK, YRI) in the 1000 Genomes Project Phase 1 V3 release of genotypes, followed by the maximum disease allele frequency and the population with such frequency. If the variant was not present in 1000G the maximum is 0.0 and the population is not listed “-“. No OMIM variants were observed with Qatari allele frequency lower than all 14 continents. Max = maximum; Pop = population. If multiple populations have the maximum frequency the population is marked as “Many”.
Of the 37 potential disease-causative variants, the most common disorders were hematologic disorders (8 variants, 2 involving 1 gene, 3 involving another gene, and 3 each in a different gene), metabolic disorders (5 variants, 2 in 1 gene, and 3 each in a different gene), eye disorders (4 variants, each in a different gene), inflammatory disorders (3 variants, each in a different gene), cardiovascular (3 variants, each in a different gene) and neurologic (3 variants, each in a different gene; Table 3 and Supp. Table S3). The disorders/genes with more than 1 variant included hemoglobin (HBB, MIM# 141900, 3 variants; hemoglobin S sickle cell anemia, MIM# 603903; hemoglobin D (no MIM number); and hemoglobin E beta-plus thalassemia, MIM# 613985), familial Mediterranean fever (MEFV, MIM# 608107, 2 variants; MIM# 249100) and erythropoietic porphyria (MIM# 177000, FECH2, 2 variants; MIM# 612386).
The majority (56%) of the disorders were recessive, with the remainder dominant (37%) or X-linked (7%). After carefully reviewing the literature for the 13 dominant disorders, we classified the 13 variant-disease links into 3 categories, “mild phenotype, difficult to detect”, “physically obvious phenotype”, and “serious, responsible for deaths”. We classified 9 as “mild phenotype, difficult to detect” (F5, MIM# 612309, c1601G>A, p.Arg506Gln; MEFV, MIM# 608107, c.2270G>T, p.Ala744Ser; MEFV, MIM# 608107, c.2120A>G, p.Met694Val; NKX2-5, MIM# 600584, c.302C>T, p.Arg25Cys; NLRP12, MIM# 609648, c.1070C>T, p.Arg284Ter; NLRP3, MIM# 606416, c.1344G>A, p.Val198Met; SLC7A9, MIM# 604144, c.661G>A, p.Ala182Thr; KLF11, MIM# 603301, c.821C>T, p.Thr220Met; LPL, MIM# 609708, c.476G>A, p.Asp9Asn), 3 as “physically obvious phenotype” (EVC, MIM# 225500, c.1512G>A, p.Arg443Gln; TGIF, MIM# 602630, c.636A>T, p.Gln107Leu; WNT10A, MIM# 606268, c.1145T>A, p.Phe228Ile), and one as “serious, responsible for deaths” (CAV3, MIM# 601253, c.310C>A, p.Thr78Met). For the mild mutations discovered, we believe that our pre-screening protocols and that our medical questionnaires are not detailed enough to provide a direct confirmation as to whether these subjects do have the disease. For the 3 physically obvious and 1 severe mutations, our results call into question the penetrance of these variants in the Qatari population.
For the SNP disease variants in the panel of genetic tests currently conducted in the Laboratory Medicine and Pathology, Hamad Medical Corporation, Doha, Qatar, which is used to screen Qatari couples before marriage, only 4 of the variants identified in the present study are represented, including a GJB2 (connexin 26; MIM# 121011) variant linked to deafness (MIM# 220290), SLC2A10 (MIM# 606145) variants linked to arterial tortuosity syndrome (MIM# 208050), HBB (MIM# 141900) variants linked to sickle cell disease (MIM# 603903) and beta-thalassemia (MIM# 613985), and F5 (MIM# 612309) variants linked to thrombophilia (MIM# 188055) (Table 3 and Supp. Table S3). Of interest, our survey did not detect 2 disorders screened for in Qataris, including homocystinuria (MIM# 236200) due to CBS (MIM# 613381) c.1006C>T, p.Arg336Cys and cystic fibrosis (MIM# 219700) due to CFTR (MIM# 602421) c.3700A>G, p. Ile123Val. Other common variants in Qataris such as deletions of SMN1 (MIM# 600354) or HBA2 (MIM# 141850) would not be detected by exome sequencing.
Upon query of the Center for Arab Study Database, we found that only 13 of these variants have been previously identified in this database; only one of these, arterial tortuosity syndrome (MIM# 208050), has been previously identified in a Qatari family and reported in a Pubmed-indexed academic journal. These variants include autosomal recessive deafness (MIM# 220290) (GJB2; MIM# 121011); familial Mediterranean fever (MIM# 249100) (MEFV; MIM# 608107); Ellis-van Creveld Syndrome (MIM# 225500) (EVC; MIM# 604831); holoprosencephaly (MIM# 142946) (TGIF; MIM# 602630); Stargardt disease (MIM# 248200) (ABCA4; MIM# 601691); primary congenital glaucoma (MIM# 231300) (CYP1B1; MIM# 601771); tetralogy of Fallot (MIM# 187500) (NKX2-5; MIM# 600584); autosomal recessive chronic granulomatous disease (MIM# 233710), (NCF2; MIM# 608515); cystinuria (MIM# 220100) (SLC7A9; MIM# 604144); Myoshi myopathy (MIM# 254130) (DYSF; MIM# 603009). Only three of the disorders have been previously observed in Qatar, including arterial tortuosity syndrome (MIM# 208050) (SLC2A10; MIM# 606145); sickle cell anemia (MIM# 603903) (HBB; MIM# 141900); and oculocutaneous albinism (MIM# 606952) (TYR; MIM# 606933), and the specific causative variant that our study identified was observed previously only for SLC2A10.
Unique and High Frequency Qatari Mendelian Disorders Compared to 1000 Genomes
Of the alleles observed for the 36 disorders, 10 were not present in the 1000 Genomes populations (Table 3). Of these 10, 9 were present in only one subpopulation. These included 2 variants specific to the Q1 population, including TGIF (MIM# 602630) c.636A>T, p.Gln107Leu linked to holoprosencephaly (MIM# 142946) (Aguilella et al., 2003) and SLC2A10 (MIM# 606145) c.340C>G, p.Ser81Arg linked to the arterial tortuosity syndrome (MIM# 208050) (Coucke et al., 2006; Faiyaz-Ul-Haque et al., 2008). SLC2A10 (MIM# 606145) c.340C>G, p.Ser81Arg was not observed in the 13,000 European Americans and African Americans sampled by the NHLBI Exome Project 9 (http://evs.gs.washington.edu/EVS/) with average depth 72x for other variants in the SLC2A10 gene. The frequency of TGIF (MIM# 602630) c.636A>T, p.Gln107Leu was 0.0002 in European Americans and 0.0002 in African Americans from the NHLBI Exome Project.
For the 27 variants that were present in at least one of the 1000 Genomes populations, we compared the frequencies of these variants to the entire 1000 Genomes sample and to 1000 Genomes populations to benchmark which of these variants are at unusually high frequency in Qatar. After a multiple test correction, we found that only one variant CYP1B1 (MIM# 601771) linked to primary congenital glaucoma (MIM# 231300) (Bejjani et al., 2000; Vincent et al., 2002; Vasiliou and Gonzalez, 2008) was significant compared to the entire 1000 Genomes sample (p<0.00004), while SLC7A9 (MIM# 604144) linked to cystinuria (MIM# 220100) (Feliubadalo et al., 1999) was close to significant (p<0.0061) after correcting for multiple tests. When comparing the subpopulation frequencies of these two variants (Q1 and Q2 for CYP1B1 and Q2 for SLC7A9) to each of the 1000 Genomes populations segregating for these variants, all comparisons produced relatively low p values (greatest p<0.075).
Assessment of Disease-related SNP Frequency
In order to validate the frequency and population specificity for the variants, 4 variants with single-population specificity and specific frequency higher than all 1000 Genomes populations (TGIF, MIM# 602630; RP1, MIM# 603973; SLC2A10, MIM# 606145; and SLC7A9, MIM# 604144) were assessed using TaqMan assays in 386 Qataris (Table 4). For all of the variants tested, the variant was not observed in Q3, consistent with the exome study results of population specificity of the variants in Q1 and Q2. For 2 of the 4 variants, (SLC2A10 c.340C>G, p.Ser81Arg and RP1 c.1266C>T, p.Thr373Ile), the trend of population specificity was conserved, such that if a variant was only observed in one subpopulation in the exome sample, the same group had the highest allele frequency in the assessed group. It is likely that the observation of Q1 specific variants in Q2 (and vice versa) is the result of admixture across Qatari populations (Omberg et al., 2012).
Table 4.
Assessment of Frequency of OMIM Variants in the Qatari Population Linked to Mendelian Disorders1
| Gene | Variant | Disorder | dbSNP | Q1 | Q2 | Q3 | |||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Exome | TaqMan | Exome | TaqMan | Exome | TaqMan | ||||
|
| |||||||||
| cohort (n=36) | cohort (n=217) | cohort (n=38) | cohort (n=154) | cohort (n=26) | cohort (n=15) | ||||
| RP1 | p.Thr373Ile | Retinitis pigmentosa 1 | rs77775126 | 0.000 | 0.014 | 0.053 | 0.029 | 0.000 | 0.000 |
| SLC2A10 | p.Ala182Thr | Arterial tortuosity syndrome | rs80358230 | 0.042 | 0.014 | 0.000 | 0.003 | 0.000 | 0.000 |
| SLC7A9 | p.Ala182Thr | Cystinuria | rs79389353 | 0.000 | 0.000 | 0.027 | 0.000 | 0.000 | 0.000 |
| TGIF | p.Gln107Leu | Holoprosencephaly 4 | rs28939693 | 0.056 | 0.002 | 0.000 | 0.016 | 0.000 | 0.000 |
Variants in Table 3 were selected for allele frequency assessment by TaqMan in an additional n=386 Qataris, including Q1 (n=217), Q2 (n=154) and Q3 (n=15). Shown is the gene, variant, disorder dbSNP rsID, and frequency in Q1, Q2 and Q3 Qatari. For each subpopulation, disease allele frequency is shown for two cohorts, on the left is the frequency in the exome sequencing cohort, and on the right is the allele frequency in the TaqMan genotyping cohort. The number of genomes sampled in each cohort is shown in parenthesis.
Discussion
The value of deep coverage exome sequencing has been demonstrated repeatedly for studies where the target is identifying the allele responsible for a disease of relatively simple inheritance and where the sampling design includes related individuals from families with high incidences of the disease. The present study highlights the value of the reverse approach: exome sequencing of a random sample of individuals for a population, without regard to specific phenotype, to identify the prevalence of variants previously linked to Mendelian disorders. Overall, with a median depth of 67x Illumina exome sequencing of a sample of 100 Qataris providing an overall sampling of SNP and indel exome variation in this population, we were able to identify 37 variants in 33 genes representing 36 known Mendelian disorders where the causal SNP has been reported, has a recessive, dominant or X-linked inheritance pattern, and where there are alleles segregating in the Qatari population.
With respect to available health informatics resources local to Qatar, this study adds a considerable amount of information. For example, only 13 of these variants have been identified in any regional Arabian populations as reported by the Center for Arab Study Database. This indicates this database is incomplete; a situation that we suspect will be mirrored in other local databases. As another example, the current panel of genetic tests conducted on a national basis in Qatar by the Laboratory Medicine and Pathology, Hamad Medical Corporation, Doha, Qatar, which is used to screen Qatari couples before marriage, includes only 4 of the variants identified in the present study: GJB2 (connexin 26; MIM# 121011) variant linked to deafness (MIM# 220290), SLC2A10 (MIM# 606145) variant linked to arterial tortuosity syndrome (MIM# 208050), HBB (MIM# 141900) variant linked to sickle cell disease (MIM# 603903) and beta-thalassemia (MIM# 613985), and F5 (MIM# 612309) variants linked to thrombophilia (MIM# 188055). Given many of the additional variants we identified are relatively severe recessive disorders, including POMGNT1 (MIM# 606822) c.1666G>A, p.Asp556Asn linked to muscular dystrophy-dystroglycanopathy (limb-girdle) type C,3 (MIM# 613157) and RP1 (MIM# 603937) c.1266C>T, p.Thr373Ile linked to retinitis pigmentosa (MIM# 180100) (Khaliq et al., 2005), this study provides additional candidates for this testing panel, after further characterization of disease causation and high penetrance in Qatar. In general, we suspect that direct exome studies conducted in understudied populations will similarly reveal the incompleteness of local genetic testing. Such studies are a cost-effective strategy for identifying candidate variants that could have an impact on population health.
Another more global benchmark of the value of exome studies of this type is provided by the analysis of representation and frequency of the 37 candidate disease-causative variants we identified in the 1000 Genomes populations. For example, we found 10 of our variants were not represented at all in the 1000 Genomes populations, where only arterial tortuosity syndrome (MIM# 208050), Hemoglobin S (MIM# 603903), and oculocutaneous albinism type I (MIM# 203100) had been previously known to be in Qatar and where the bulk of these were specific to the Q1-Bedouin and Q2-Persian subpopulations as expected, given that these are less closely related to 1000 Genome populations as compared to the Q3-African subpopulation. This study therefore added a considerable number of discoveries concerning what Mendelian variants are comparatively unique to the Qatari population.
The current study highlights the importance of careful consideration of both medical genetic and population genetic information when discovering Mendelian disease alleles through random exome sequencing. As a starting point, we used the OMIM and HGMD databases to identify potentially deleterious disease alleles. However, since we were focused on alleles that have a high likelihood of having a disease impact in the population (i.e., where the causal mutation is reported, the result has been confirmed in more than one family and has a clear Mendelian inheritance pattern), it was necessary to contend with the issue that OMIM and HGMD include a large number of entries that are not very useful for exome-based discovery. We therefore excluded a large number of records in our final list, including alleles reported for complex traits, where the causal mutation has been tagged rather than known, the disease allele requires interaction with a second allele on the same haplotype, and cases where the mechanism of inheritance was unclear. We found a need for a review of the primary literature to get an accurate assessment of each of these criteria and to identify other issues, such as cases where the reference allele is the disease allele, multiple alternate alleles are linked to the same dbSNP rsID, or where databases disagree on the residue number. While there have been some recent attempts to provide curated sub-lists for OMIM (Liao and Zhang, 2008; Bell et al., 2011), overall, studies of the type implemented here will continue to need careful medical informatic analysis, including surveying the primary literature for reported disease alleles.
After an extensive and thorough filtering process, over 80% of the OMIM/HGMD variants previously linked to a Mendelian disorder were filtered out based on disease allele prevalence and literature support. Among the remaining variants, additional false positives may remain; however, without further characterization of the variants, it is difficult to distinguish between low penetrance and false positive. For the 3 physically obvious and 1 severe disorders where the inheritance mode is autosomal dominant, the false positive rate could be as high as 100%, while for recessive variants identified in heterozygotes, the false positive rate is unknown, although after our extensive filtering process, we expect it to be nearer the other end of the spectrum, i.e., closer to 0%.
The long-term objective of this study is to provide a foundation for improving and expanding the set of genetic tests used for premarital and prenatal screening in Qatar. Among the variants discovered, there are reassuring cases where the variant and disease was previously observed in Qatar, such as SLC2A10 (MIM# 606145) c.340C>G, p.Ser81Arg and arterial tortuosity syndrome (MIM# 208050); HBB (MIM# 141900) c.129G>A, p.Glu26Lys and Hemoglobin E beta-plus-thalassemia (MIM# 613985); GJB2 (MIM# 121011) c.286G>A, p.Trp24Ter and auto-somal recessive deafness (MIM# 220290). For SLC2A10, our study serves as a confirmation that the disease allele is segregating in the population despite a national screening program. For HBB and GJB2, the Hamad list of genetic tests mentions these genes but does not specify these variants, hence we have evidence that could potentially narrow the focus of screening in these genes. It is expected that true causative variants for Mendelian disorders remain at minor allele frequency below 1% (Pritchard and Cox, 2002), hence some of the more interesting variants are those not observed in any of the 1000 Genomes Project populations and are rare in the Qatari population (1 in 200 alleles). This is the case for the three variants mentioned above (SLC2A10, HBB, GJB2). Three other variants that meet this criteria (never observed in 1000G, <1% disease allele frequency in Qatar) include MEFV (MIM# 608107) c.2120A>G, p.Met694Val, CERKL (MIM# 608381) c.870C>T, p.Arg257Ter, and RNASEH2C (MIM# 610330) c.385C>T, p.Arg69Trp. MEFV c.2120A>G, p.Met694Val is a well-studied variant that causes familial Mediterranean fever (MIM# 249100) in up to 80% of affected members of various origins (Jewish, Armenian, Turkish, Arabian) (French FMF Consortium, 1997). Given the prior observations in the region, it would not be surprising that the allele explains a number of familial Mediterranean fever cases in Qatar as well. The CERKL c.870C>T, p.Arg257Ter variant was previously observed in Spanish families with retinitis pigmentosa (Tuson et al., 2004) (MIM# 608380), and it would be interesting to confirm its existence in Qatar and causality. RNASEH2C c.385C>T, p.Arg69Trp was previously observed in Pakistani families with Aicardi-Goutieres syndrome (Crow et al., 2006) (MIM# 610329), with evidence of a founder effect. All three of these variants were observed in the Q2 subpopulation, where genetic origin is the least well understood of the three Qatari sub-populations.
Consideration of subpopulation genetic analyses of the Qatari population is an important aspect of this study. Previous studies from our group identified three genetic subpopulations within the Qatari population that are of Bedouin (Q1), Persian-South Asian (Q2), and African (Q3) ancestry (Hunter-Zinck et al., 2010; Omberg et al., 2012). We found that several of the Mendelian alleles were either exclusively or mostly present in one of these three subpopulations, such that ignoring population structure would have resulted in lower frequency estimates for these disorders. The Qatari population history suggests a low degree of intermarriage between the genetic subpopulations, such that ignoring this population structure would lead to an inaccurate picture of the potential impact of these disorders, e.g., how often recessive homozygotes are expected to occur in each subpopulation. Additional sampling and sequencing of non-Qatari Arab populations will be of value for future studies, with a long-term objective of discovering country-specific, population-specific and tribe-specific genetic variations linked to Mendelian disorders. The unique population structure of the Qatari and neighboring countries lends itself to such a study of genetic variation within population isolates.
Finally, we note that it is not unexpected to discover such a set of Mendelian diseases using a random exome sequencing approach in a population that has not been sampled extensively and is not closely related to previously sampled populations. What we demonstrate is that a relatively small sample of exomes can be used to discover Mendelian disorders of interest to populations where there is relatively little genetic information. Studies of this type are therefore a cost-effective approach for discovering Mendelian disease variants that are relevant for population-wide medical genetics.
Supplementary Material
Acknowledgments
Grant sponsor: These studies were supported, in part, by the Qatar Foundation and Weill Cornell Medical College in Qatar, and by the National Science Foundation (#0922432). JLRF is supported, in part, by NIH T32 HL09428.
We thank the Taino Genome Project and the 1000 Genomes Project Exome Working Group for helpful advice; M Staudt, Y Strulovici-Barel for help with the study; DN McCarthy and N Mohamed for help in preparing this manuscript; and Dr. Mohammad Fathy Saoud, President of Qatar Foundation, and Her Highness Sheikha Moza Bint Nasser, Chair of Qatar Foundation, for their continued encouragement and support. These studies were supported, in part, by the Qatar Foundation and Weill Cornell Medical College in Qatar, and by the National Science Foundation (#0922432). JLRF is supported, in part, by NIH T32 HL09428.
Footnotes
Conflict of interest: The authors have no conflict to declare
Supporting Information for this preprint is available from the Human Mutation editorial office upon request (humu@wiley.com)
References
- 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilella C, Dubourg C, ttia-Sobol J, Vigneron J, Blayau M, Pasquier L, Lazaro L, Odent S, David V. Molecular screening of the TGIF gene in holoprosencephaly: identification of two novel mutations. Hum Genet. 2003;112:131–134. doi: 10.1007/s00439-002-0862-8. [DOI] [PubMed] [Google Scholar]
- Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002;12:1805–1814. doi: 10.1101/gr.631202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejjani BA, Stockton DW, Lewis RA, Tomey KF, Dueker DK, Jabak M, Astle WF, Lupski JR. Multiple CYP1B1 mutations and incomplete penetrance in an inbred population segregating primary congenital glaucoma suggest frequent de novo events and a dominant modifier locus. Hum Mol Genet. 2000;9:367–374. doi: 10.1093/hmg/9.3.367. [DOI] [PubMed] [Google Scholar]
- Bell CJ, Dinwiddie DL, Miller NA, Hateley SL, Ganusova EE, Mudge J, Langley RJ, Zhang L, Lee CC, Schilkey FD, Sheth V, Woodward JE, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med. 2011;3:65ra4. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann HM, de TC, Cazes L, Legrand MF, Morel V, Piouffre L, Bodmer J, Bodmer WF, Bonne-Tamir B, Cambon-Thomsen A, Chen Z, Chu J, et al. A human genome diversity cell line panel. Science. 2002;296:261–262. doi: 10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang lL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w (1118); iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coucke PJ, Willaert A, Wessels MW, Callewaert B, Zoppi N, De BJ, Fox JE, Mancini GM, Kambouris M, Gardella R, Facchetti F, Willems PJ, et al. Mutations in the facilitative glucose transporter GLUT10 alter angiogenesis and cause arterial tortuosity syndrome. Nat Genet. 2006;38:452–457. doi: 10.1038/ng1764. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, Baumann C, Baxter P, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38:910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del AG, Rivas MA, Hanna M, McKenna A, Fennell TJ, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey FE, Chen R, Cordero SP, Ormond KE, Caleshu C, Karczewski KJ, Whirl-Carrillo M, Wheeler MT, Dudley JT, Byrnes JK, Cornejo OE, Knowles JW, et al. Phased whole-genome genetic risk in a family quartet using a major allele reference sequence. PLoS Genet. 2011;7:e1002280. doi: 10.1371/journal.pgen.1002280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faiyaz-Ul-Haque M, Zaidi SH, Wahab AA, Eltohami A, Al-Mureikhi MS, Al-Thani G, Peltekova VD, Tsui LC, Teebi AS. Identification of a p.Ser81Arg encoding mutation in SLC2A10 gene of arterial tortuosity syndrome patients from 10 Qatari families. Clin Genet. 2008;74:189–193. doi: 10.1111/j.1399-0004.2008.01049.x. [DOI] [PubMed] [Google Scholar]
- Feliubadalo L, Font M, Purroy J, Rousaud F, Estivill X, Nunes V, Golomb E, Centola M, Aksentijevich I, Kreiss Y, Goldman B, Pras M, et al. Non-type I cystinuria caused by mutations in SLC7A9, encoding a subunit (bo,+AT) of rBAT. Nat Genet. 1999;23:52–57. doi: 10.1038/12652. [DOI] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, Gil L, Gordon L, et al. Ensembl 2012. Nucleic Acids Res. 2012;40:D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet. 1997;17:25–31. doi: 10.1038/ng0997-25. [DOI] [PubMed] [Google Scholar]
- Holsinger KE, Weir BS. Genetics in geographically structured populations: defining, estimating and interpreting F(ST) Nat Rev Genet. 2009;10:639–650. doi: 10.1038/nrg2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter-Zinck H, Musharoff S, Salit J, Al-Ali KA, Chouchane L, Gohar A, Matthews R, Butler MW, Fuller J, Hackett NR, Crystal RG, Clark AG. Population genetic structure of the people of Qatar. Am J Hum Genet. 2010;87:17–25. doi: 10.1016/j.ajhg.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq S, Abid A, Ismail M, Hameed A, Mohyuddin A, Lall P, Aziz A, Anwar K, Mehdi SQ. Novel association of RP1 gene mutations with autosomal recessive retinitis pigmentosa. J Med Genet. 2005;42:436–438. doi: 10.1136/jmg.2004.024281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao BY, Zhang J. Null mutations in human and mouse orthologs frequently result in different phenotypes. Proc Natl Acad Sci U S A. 2008;105:6987–6992. doi: 10.1073/pnas.0800387105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omberg L, Salit J, Hackett N, Fuller J, Matthew R, Chouchane L, Rodriguez-Flores JL, Bustamante C, Crystal RG, Mezey JG. Inferring genome-wide patterns of admixture in Qataris using fifty-five ancestral populations. BMC Genet. 2012;13:49. doi: 10.1186/1471-2156-13-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheimer S. Out-of-Africa, the peopling of continents and islands: tracing uniparental gene trees across the map. Philos Trans R Soc Lond B Biol Sci. 2012;367:770–784. doi: 10.1098/rstb.2011.0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci U S A. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Cox NJ. The allelic architecture of human disease genes: common disease-common variant…or not? Hum Mol Genet. 2002;11:2417–2423. doi: 10.1093/hmg/11.20.2417. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qatar Statistics Authority. Results of the 2010 Census of Population, Housing and Establishments 2010 [Google Scholar]
- Rodriguez-Flores JL, Fuller J, Hackett NR, Salit J, Malek JA, Al-Dous E, Chouchane L, Zirie M, Jayoussi A, Mahmoud MA, Crystal RG, Mezey JG. Exome sequencing of only seven qata-ris identifies potentially deleterious variants in the qatari population. PLoS One. 2012;7:e47614. doi: 10.1371/journal.pone.0047614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandridge AL, Takeddin J, Al-Kaabi E, Frances Y. Consanguinity in Qatar: knowledge, attitude and practice in a population born between 1946 and 1991. J Biosoc Sci. 2010;42:59–82. doi: 10.1017/S002193200999023X. [DOI] [PubMed] [Google Scholar]
- Schaffner SF. The X chromosome in population genetics. Nat Rev Genet. 2004;5:43–51. doi: 10.1038/nrg1247. [DOI] [PubMed] [Google Scholar]
- Tadmouri GO. Genetic Disorders in the Arab World: Qatar. Duabi, UAE: Centre for Arab Genomic Studies; 2012. [Google Scholar]
- Tuson M, Marfany G, Gonzalez-Duarte R. Mutation of CERKL, a novel human ceramide kinase gene, causes autosomal recessive retinitis pigmentosa (RP26) Am J Hum Genet. 2004;74:128–138. doi: 10.1086/381055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiliou V, Gonzalez FJ. Role of CYP1B1 in glaucoma. Annu Rev Pharmacol Toxicol. 2008;48:333–358. doi: 10.1146/annurev.pharmtox.48.061807.154729. [DOI] [PubMed] [Google Scholar]
- Vincent AL, Billingsley G, Buys Y, Levin AV, Priston M, Trope G, Williams-Lyn D, Heon E. Digenic inheritance of early-onset glaucoma: CYP1B1, a potential modifier gene. Am J Hum Genet. 2002;70:448–460. doi: 10.1086/338709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.