

Abstract
This investigation aimed to develop nimesulide (NMS)-loaded poly(lactic-co-glycolic acid) (PLGA)-based nanoparticulate formulations as a biodegradable polymeric drug carrier to treat rheumatoid arthritis. Polymeric nanoparticles (NPs) were prepared with two different nonionic surfactants, vitamin E d-α-tocopheryl polyethylene glycol 1000 succinate (vitamin E TPGS) and poly(vinyl alcohol) (PVA), using an ultrasonication solvent evaporation technique. Nine batches were formulated for each surfactant using a 32 factorial design for optimal concentration of the emulsifying agents, 0.03–0.09% for vitamin E TPGS and 2–4% for PVA. The surfactant percentage and the drug/polymer ratio (1:10, 1:15, 1:20) of the NMS-loaded NPs were investigated based on four responses: encapsulation efficiency, particle size, the polydispersity index, and the surface charge. The response surface plots and linearity curves indicated a relationship between the experiment’s responses and a set of independent variables. The NPs produced with both surfactants exhibited a negative surface charge, and scanning electron micrographs revealed that all of the NPs were spherical in shape. A narrower size distribution and higher drug loadings were achieved in PVA-emulsified PLGA NPs than in the vitamin E TPGS emulsified. Decreasing amounts of both nonionic surfactants resulted in a reduction in the emulsion’s viscosity, which led to a decrease in the particle size of NPs. According to the ANOVA results obtained in this present research, vitamin E TPGS exhibited the best correlation between the independent variables, namely drug/polymer ratio and the surfactant percentage, and the dependent variables (encapsulation efficiency R2 = 0.9603, particle size R2 = 0.9965, size distribution R2 = 0.9899, and surface charge R2 = 0.8969) compared with PVA.
KEY WORDS: ANOVA, factorial design, nanoparticles, nimesulide, PLGA, PVA, vitamin E TPGS
INTRODUCTION
Arthritis refers to various medical conditions associated with joint disorders of the primary structures, such as the synovial membranes, bones, and cartilage. Among the different types of arthritis, such as osteoarthritis (OA), rheumatoid arthritis (RA), ankylosing spondylitis, septic arthritis, and juvenile idiopathic arthritis, OA and RA are the most common worldwide, affecting 40 million people in the USA in 1995, with expectations that this number will reach 59.4 million people by 2020 (1,2). OA is a disease of the cartilage characterized by progressive deterioration as the structure loses the natural ability to repair itself. It can be difficult to make a distinction between the histological structure of OA and RA (3). In the cartilage, there is sclerosis of the bones, and overgrowth is secondary to this manifestation. Generally, degeneration of the joints is a common inflammatory arthritic disease among the elderly that continually progresses, leading to chronic pain and a reduced quality of life (4). Its chronic nature poses significant challenges to the discovery and delivery of drugs that will retard the degeneration of joint tissues.
The administration of nonsteroidal anti-inflammatory drugs (NSAIDs), such as flurbiprofen, diclofenac, indomethacin, ibuprofen, dexibuprofen, aspirin, ketoprofen, piroxicam, and, in particular, nimesulide, mainly by the oral, parenteral, or topical routes, are the common treatments for arthritic diseases (1,5). There is a body of evidence to support NSAIDs as a more effective treatment for these diseases when compared to common, long-acting analgesics. However, because most NSAIDs used in the treatment of arthritis have short biological half-lives, repeat administration three to four times a day is required. These frequent dosages and the route of administration, whether oral or otherwise, are further impacted by numerous side effects, such as gastric irritation, dyspepsia, peptic ulceration, gastric bleeding, esophagitis, constipation, sodium and water retention, chronic renal failure, and intestinal nephritis (1,3,6,7). Because such inflammatory arthritic diseases are localized, intra-articular delivery may be a suitable alternative by compartmentally targeting drugs into arthritic joints, and are widely used by patients suffering inflammatory arthritic diseases (6–10). The most important advantage of intra-articular treatment of arthritis is targeting the drug to the specific inflammatory site and minimizing its systemic toxic effects (2). However, current preparations of intra-articular drug therapies often require frequent injections that rapidly degrade and clear injected pharmacologic agents, impacting on finances and the patient’s quality of life, as well as increasing the risk of complications. In addition to these effects, after delivering the drugs via this intra-articular method, the agents can also leave the articular cavity shortly after administration. Hence, to ensure sustained release of anti-inflammatory drugs, different drug delivery systems have been developed (2,11).
Nano- and/or microparticulate drug delivery is one of the most widely used strategies developed for this approach (5,10,12–14). The novel nanoparticulate drug delivery technology reduces the dosing requirements and eliminates gastrointestinal irritation, thus ultimately improving the patient’s compliance in the treatment of arthritis. The goal of the nanoparticulate system is to promptly deliver a therapeutic amount of the drug to the target site and maintain the desired concentration to deliver the drug at a rate determined by the body’s needs over an entire period of treatment (7,15,16). These systems could also reduce the clearance of the drug and improve and prolong its retention within the joint cavity (2,17). With the aim of intra-articular administration of nanoparticles (NPs), polymeric materials, such as gelatin, chitosan, poly(lactic-co-glycolic acid) (PLGA), and poly(lactic acid) (PLA), have gained widespread attention due to their biodegradability and biocompatibility (4). One of the most successful is PLGA because of its hydrolysis, which leads to metabolite monomers, lactic acid, and glycolic acid. Another important advantage of biocompatible nanosized particles over nonbiocompatible particles in intra-articular administration is not inducing the undesired “crystal-induced pain” effect. PLGA nanocarriers may be adapted for the development of arthritic lesion delivery systems for anti-inflammatory drugs due to their biodegradability, biocompatibility, and nanosize (11,18). After administering PLGA micro- and nanoparticles directly into the joint cavity for an intra-articular delivery system on the phagocytosis of rat synovium, Horisawa et al. (8) investigated the size dependency and showed that PLGA NPs, with an average diameter of 265 nm, would be more suitable for delivery to inflamed synovial tissue than microspheres, with an average diameter of 26.5 μm, due to their penetrability. Their study showed that PLGA NPs directly injected into the joint cavity were selectively phagocytosed by macrophages and disseminated into underlying tissue, whereas the PLGA microparticles remained in the synovium (8).
4-Nitro-2-phenoxymethanesulfonanilide, nimesulide (NMS), is a second-generation NSAI agent widely used in the long-term treatment of rheumatoid arthritis to alleviate pain and inflammation. Nonsteroidal anti-inflammatory drugs (NSAIDs) are generally effective by inhibiting cyclooxgenase (COX)-2, the main isozyme associated with inflammation. However, the simultaneous inhibition of COX-1 disrupts gastric and renal functioning. Compared to drugs of the same class, NMS is 5 to 16 times more selective for the inhibition of COX-2 and has been employed to treat major pain (19). Several studies have been conducted in recent years on the preparation and investigation of PLA microspheres (20) for parenteral administration and the oral delivery of NMS by ethylcellulose and methylcellulose micro-and nanoparticles (21). However, no research was found investigating the design and the characterization of NMS-loaded biodegradable polymeric NPs as possible treatment for inflammatory arthritic diseases. Therefore, for this study, using an emulsion solvent evaporation technique, NMS-loaded PLGA-based biodegradable NPs were prepared. Two different nonionic surfactant emulsifiers were selected as stabilizers in the formulations: poly(vinyl alcohol) (PVA), the most commonly used to stabilize emulsions caused by the formation of relatively small-sized particles with uniform size distribution and which has excellent stabilizing properties on the NP emulsion (22), and vitamin E d-α-tocopheryl polyethylene glycol 1000 succinate (TPGS), which is a new water-soluble derivative of natural vitamin E and is formed by esterification of vitamin E succinate with PEG 1000. Used as an absorption enhancer, emulsifier, and solubilizer in pharmaceutical formulations, its biocompatibility, amphiphilicity, and low molecular weight makes vitamin E TPGS potentially suitable as an emulsifier for the encapsulation of both hydrophilic and hydrophobic drugs into nanosized particles (23,24). The focus of this study is to optimize and investigate the drug/polymer ratio, emulsifier type, and percentages used in the NP formulations on the physicochemical properties of the NMS-loaded PLGA NPs by using a 32 factorial design. Factorial experimental designs allow for the rapid and efficient screening of design variables with a limited number of planned experiments (25,26). The design of the experiments and the response surface methodology (RSM) approaches were implemented for optimal processing. The independent variables were the drug/polymer ratio (X1) and the emulsifier percentages (X2–X3). The dependent (response) variables investigated to characterize the particles were the encapsulation efficiency percentage (Q1–Y1), particle size (Q2–Y2), polydispersity index (Q3–Y3), and surface charge (Q4–Y4) of the PLGA NPs. This research also employed differential scanning calorimetry (DSC), field emission scanning electron microscopy (FESEM), and gas chromatography (GC) analysis.
MATERIALS AND METHODS
Materials
The materials used were NMS (Ulkar Chem. Co., Istanbul, Turkey), PLGA with a copolymer ratio of d,l-lactide to glycolide of 50:50 (MW 40–75 kDa), PVA (MW 30–70 kDa) (Sigma-Aldrich Chem. Co., Munich, Germany), vitamin E TPGS (Eastman Chem. Co., USA), dichloromethane (DCM) (Merck Chem. Co., Darmstadt, Germany), and purified water (Milli-Q, Millipore Corp., MA, USA). All other chemicals used were at least of reagent grade.
Preparation of NMS-Loaded PLGA NPs
Emulsification by a sonication-solvent evaporation method was used to prepare the PLGA NP formulations (24), by dissolving various amounts of polymer and drug in 4 mL of DCM. For the aqueous phase, PVA or vitamin E TPGS was dissolved in 60 mL Milli-Q water. The organic phase was emulsified into the aqueous phase for 5 min by probe sonication (Bandelin Sonopuls HD 2070, Bandelin Elec., Germany) at 80% output energy and pulse duty of 0.7 s per cycle. After emulsification, the resulting dispersion was magnetically stirred overnight to evaporate the organic phase. By centrifuge, the NPs formed were recovered at 20,000 rpm for 15 min (Sorvall Superspeed Centrifuge SS-3, DJB Labcare Limited, Buckinghamshire, England) and washed with Milli-Q water to remove excess surfactant, and the suspension was lyophilized (Christ Gamma 2-16 LSC, Martin Christ Gef., Germany) for 60 h until a dry powder was obtained.
Characterizations of the PLGA NPs
To determine the encapsulation efficiency of the lyophilized NPs, a set amount of the lyophilized NMS-loaded PLGA NPs was dissolved in DCM using the direct method (22,24) and ultrasonicated for 10 min with 40% output energy and pulse duty of 0.7 s per cycle (Bandelin Sonopuls HD 2070, Bandelin Electronics, Germany). UV–visible spectrophotometry determined the entrapped NMS concentration (Shimadzu UV-1240, Japan) at a wavelength of 297 nm (n = 3). The analytical method used for the assay of NMS was validated with respect to precision (repeatability, reproducibility), accuracy, specificity, linearity, and range (Table I).
Table I.
Analytical Parameters for the Assay of NMS by UV Spectrophotometric Method
| Parameter | Result |
|---|---|
| Linearity range (μg mL−1) | 2.5–35 |
| Slope | 0.0257 |
| Intercept | −0.0128 |
| Determination coefficient (R 2) | 0.9999 |
| SE of slope | 0.0002 |
| SE of intercept | 0.0013 |
| Detection limit (μg mL−1) | 0.2628 |
| Quantification limit (μg mL−1) | 0.8761 |
| RSD of interday precision (%) | 0.1091 |
| RSD of intraday precision (%) | 0.0641 |
SE standard error, RSD relative standard deviation
A particle size analyzer was used to determine the particle size and polydispersity index (PDI) (Nano ZS, Malvern Inst., Malvern, Worcestershire, UK), based on the principle of dynamic light scattering. Following a 1/10 (v/v) dilution of the NPs in Milli-Q water, five size measurements were performed.
The zeta potential of NMS-loaded PLGA NPs was measured using laser Doppler anemometry (LDA) (Nano ZS, Malvern Inst., Malvern, Worcestershire, UK). Prior to the zeta potential measurements, the NPs were diluted with Milli-Q water at a ratio of 1/15 (v/v) and sonicated in an ultrasonic bath. For each NP formulation, five measurements were made and the averages of these replicates were calculated.
The shape of the NMS-loaded PLGA NPs was observed by use of an FEI Nova Nano SEM 430 FESEM (FEI, Eindhoven, Netherlands) at 10 kV. A specific amount of lyophilized NPs was fixed to double-sided carbon tape and placed on a metallic surface coated with a gold layer for 40 s.
One concern of pharmaceutical applications is the residual solvent contained in the developed formulations of NPs prepared by the emulsification procedure using organic solvents that must fulfill the requirements of the USP XXIII for volatile impurities (27,28). Using GC analysis, the presence of DCM remaining in PLGA NPs was determined. Chromatographic analysis by GC was carried out on an Agilent 6,890N Network GC System with flame-ionization detector (FID) (Agilent Technologies, Inc., Santa Clara, USA). The capillary column used was Supelco SP-2380 (60.0 m × 0.25 mm × 0.25 μm). DCM was extracted from PLGA NPs by using an n-Hexan, via partitioning without dissolving the polymer. A certain amount of PLGA NPs was treated with n-Hexan in a vial for a couple of hours with vortexing. After that procedure, the NPs were separated by centrifuge, and a supernatant was injected into a column of a gas chromatography device. Pure n-Hexan and DCM were also injected into the column to obtain the peaks.
The DSC study was performed to characterize the physical state of NMS in PLGA NPs. Thermograms were obtained using a DSC (DSC 60 with a software of TA/60 WS, Shimadzu, Japan). About 5 mg of the NP sample was weighted, crimped into an aluminum pan, and analyzed at a scanning temperature ranging from 25 to 300°C at a heating rate of 5°C/min. Baseline optimization was performed before each run.
Vitamin E TPGS and PVA-emulsified NMS-loaded PLGA NPs were also evaluated for the storage stability. For this purpose, lyophilized NPs were stored at 4°C during the third month period, and samples were investigated in terms of the encapsulation efficiency, mean particle size, PDI, and surface charge. Three replications per formulation were performed for all experiments. The results were statistically compared with each other using one-way analysis of variance (ANOVA).
Optimization by 32 Full Factorial Design with Center Point
PVA and vitamin E TPGS were evaluated as surfactants in the production of NMS-loaded PLGA NPs using a two-level full factorial design with a center point. Nine nanoparticulate formulations were prepared per the 32 factorial design to determine the effect of two independent variables, drug/polymer ratio (X1) and surfactant percentage (X2 coded for PVA and X3 coded for vitamin E TPGS), on the four responses: encapsulation efficiency percentage (Q1–Y1), mean particle size (Q2–Y2), polydispersity index (Q3–Y3), and surface charge (Q4–Y4) of the NMS-loaded PLGA NPs. Three replications per formulation were performed in order to assess the process and system reproducibility. Each factor was tested at three levels, with −1 designated the lower level, 0 the center point, and +1 the upper level (Table II and Fig. 1). At the center point, preparations were made in quintuple in order to estimate the experimental error. The regression equations of the fitted model for the responses were calculated using Eq. 1.
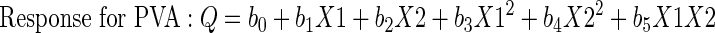 |
1 |
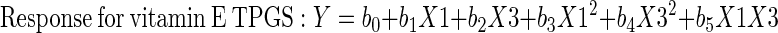 |
Table II.
32 Full Factorial Design for the Preparation of NMS-loaded PLGA NPs, Providing the Two Levels for Each Evaluated Factor
| Factors | Levels | ||
|---|---|---|---|
| −1 | 0 | +1 | |
| Drug/polymer ratio (X1) | 1:10 | 1:15 | 1:20 |
| PVA % (X2) | 2 | 4 | 6 |
| Vitamin E TPGS % (X3) | 0.03 | 0.06 | 0.09 |
PVA poly(vinyl alcohol), TPGS d-α-tocopheryl polyethylene glycol 1000 succinate
Fig. 1.
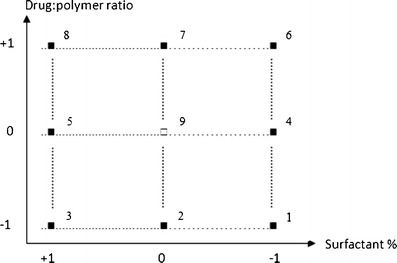
32 full factorial design points (black squares) and center point (white squares) of NMS-loaded NPs
The results from the factorial design were evaluated on the basis of a three-level factorial design by using Design Expert® 6.0.8 software (Stat-Easy Inc., Minneapolis, MN, USA). To evaluate the responses, a statistical model incorporating interactive and polynomial terms was utilized, where Q/Y is the independent variable, b0 is the arithmetic mean response, and b1, b2, b3, b4 and b5 are the estimated coefficients for the factor X1 and X2 (X3 for vitamin E TPGS instead of X2). The interaction term (X1X2) X3 for vitamin E TPGS [instead of X2] shows the response changes when two factors are simultaneously changed. To investigate nonlinearity, the polynomial terms [(X12 and X22) X32 for vitamin E TPGS instead of X22] are included. The results from the factorial design were statistically analyzed using ANOVA (29).
Linearity and Response Surface Plots
The RSM helps with understanding how changes in one variable can influence the physicochemical properties of prepared NPs. The RSM was employed to plot response behavior against factor levels. Linearity plots can reveal the relationship between the observed and the predicted responses. Graphs give similar information to the mathematical equations obtained from statistical analyses (26,30). In this research, four responses were evaluated, and each response was plotted in relation to the modified factor. Both the experimental design and the linearity and response surface plots were set using Design Expert 6.0.8 software.
RESULTS AND DISCUSSION
Analytical parameters for the determination of NMS by UV spectrophotometric method were given in Table I. The concentration range between 2.5 and 35 μg/mL with R2 = 0.9999 confirmed the linearity of the response. The low values of the standard error (SE) of slope and intercept, and greater than 0.999 determination coefficient established the precision of the proposed method. In addition, since the drug quantification was not interfered with any of the excipients, the specificity of the method was provided. Nine NMS-encapsulated PLGA NP formulations were successfully prepared by an emulsification-solvent evaporation method using vitamin E TPGS and PVA as surfactants. The desirable properties of NPs are high encapsulation efficiency, narrow size distribution, and suitable particle size. However, there are several factors that can affect the desired features of the NPs. From our preliminary experiments, the drug/polymer ratio, surfactant type, and the concentration value were found to be the most important parameters that affected the final properties of the prepared NPs (22,24). Two nonionic surfactants having a different nature and structure were selected and used to produce NPs. The selected variables were the drug/polymer ratio and the surfactant concentration, and entrapment efficiency, particle size, size distribution, and surface charge were the response parameters. A 32 factorial design was selected as it helps research the effect on response parameters by changing both variables simultaneously with a minimum number of experimental runs.
Evaluation of PVA in the Preparation of NPs
For PVA-emulsified PLGA NP production, drug concentration was kept constant at 10 mg per formulation, and PLGA concentration was varied from 100 to 150 to 200 mg to give a drug/polymer ratio of 1:10, 1:15, and 1:20. These three different ratios were evaluated at three different PVA percentages of 2, 4, and 6. In this way, nine formulations were prepared in accordance with a 32 factorial design. Table III displays the observed and predicted response values belonging to PVA-emulsified PLGA NPs. The predicted values were derived from the polynomial equations, and the observed values were determined from experimental results.
Table III.
Observed and Predicted Responses of Two Factors: Drug/Polymer Ratio (X1) and PVA Percentage Used (X2)
| Responses | ||||||||
|---|---|---|---|---|---|---|---|---|
| Observed values | Predicted values | |||||||
| Q1a (%) | Q2a (nm) | Q3a | Q4a (mV) | Q1a (%) | Q2a (nm) | Q3a | Q4a(mV) | |
| Factorial points | ||||||||
| P1 | 89.54 ± 7.26 | 212.1 ± 2.01 | 0.101 ± 0.03 | −26.8 ± 0.06 | 87.05 | 209.3 | 0.120 | −26.9 |
| P2 | 86.24 ± 9.49 | 231.3 ± 3.34 | 0.141 ± 0.02 | −26.6 ± 2.48 | 93.63 | 231.8 | 0.120 | −25.3 |
| P3 | 84.94 ± 5.96 | 239.2 ± 2.22 | 0.226 ± 0.06 | −25.0 ± 3.13 | 80.04 | 241.5 | 0.240 | −26.2 |
| P4 | 91.09 ± 4.69 | 211.9 ± 1.97 | 0.182 ± 0.01 | −25.4 ± 2.72 | 93.94 | 219.7 | 0.140 | −25.0 |
| P5 | 72.86 ± 1.49 | 247.6 ± 2.78 | 0.223 ± 0.04 | −28.3 ± 1.63 | 80.51 | 245.1 | 0.220 | −25.9 |
| P6 | 100.0 ± 16.02 | 275.2 ± 1.77 | 0.177 ± 0.04 | −32.0 ± 1.88 | 99.65 | 270.2 | 0.200 | −32.2 |
| P7 | 96.7 ± 10.06 | 281.0 ± 1.29 | 0.199 ± 0.09 | −33.6 ± 1.27 | 99.81 | 285.8 | 0.170 | −32.2 |
| P8 | 82.55 ± 0.61 | 288.5 ± 2.36 | 0.235 ± 0.01 | −33.7 ± 0.40 | 79.80 | 288.7 | 0.230 | −34.9 |
| Center points | ||||||||
| P9 | 100.0 ± 2.79 | 240.4 ± 2.35 | 0.117 ± 0.04 | −24.1 ± 1.30 | 97.31 | 238.8 | 0.130 | −24.2 |
| P10 | 100.0 ± 0.45 | 238.1 ± 2.83 | 0.143 ± 0.08 | −23.3 ± 1.07 | 97.31 | 238.8 | 0.130 | −24.2 |
| P11 | 98.21 ± 1.11 | 239.4 ± 1.65 | 0.128 ± 0.07 | −22.3 ± 2.21 | 97.31 | 238.8 | 0.130 | −24.2 |
| P12 | 99.03 ± 2.43 | 240.5 ± 1.33 | 0.110 ± 0.06 | −24.5 ± 1.46 | 97.31 | 238.8 | 0.130 | −24.2 |
| P13 | 99.78 ± 1.78 | 240.9 ± 1.89 | 0.133 ± 0.03 | −24.0 ± 1.58 | 97.31 | 238.8 | 0.130 | −24.2 |
aThe values indicate mean ± standard deviation
To quantify the effect of independent variables on the response variables, it was necessary to construct a mathematical model which would help to predict the values of dependent variables within the limits of the design. Design Expert software was used to generate a mathematical model for each dependent variable and the subsequent statistical analysis. The results of the model coefficients estimated by the Design Expert software and the regression analysis of variance of the investigated model for all responses (Q1–Q4) belonging to this formulation group are summarized in Table IV. The quality of the model developed was evaluated based on the regression coefficient values.
Table IV.
Results of Model Coefficients and the Regression Analysis of Variance (ANOVA) of the Fitted Model for All Responses
| Responses | Model coefficients | Regression analysis of variance | ||||||
|---|---|---|---|---|---|---|---|---|
| Factor | Coefficients | P value | F | P value | R 2 | Adj R 2 | Adeq Precision | |
| Q1 | Intercept | 97.3052 | 5.17 | 0.0265 | 0.7869 | 0.6346 | 5.607 | |
| X1 | 3.0883 | 0.1930 | ||||||
| X2 | −6.7133 | 0.0166 | ||||||
| X12 | −0.5881 | 0.8577 | ||||||
| X22 | −10.0831 | 0.0153 | ||||||
| X1X2 | −3.2125 | 0.2609 | ||||||
| Q2 | Intercept | 238.7966 | 64.91 | <0.0001 | 0.9789 | 0.9638 | 26.110 | |
| X1 | 27.0167 | <0.0001 | ||||||
| X2 | 12.6833 | 0.0002 | ||||||
| X12 | 20.0121 | 0.0001 | ||||||
| X22 | −6.3879 | 0.0495 | ||||||
| X1X2 | −3.4500 | 0.1671 | ||||||
| Q3 | Intercept | 0.1347 | 7.35 | 0.0104 | 0.8400 | 0.7257 | 7.332 | |
| X1 | 0.0238 | 0.0491 | ||||||
| X2 | 0.0373 | 0.0074 | ||||||
| X12 | 0.0141 | 0.3722 | ||||||
| X22 | 0.0466 | 0.0161 | ||||||
| X1X2 | −0.0168 | 0.2148 | ||||||
| Q4 | Intercept | −24.1828 | 13.91 | 0.0016 | 0.9085 | 0.8432 | 10.232 | |
| X1 | −3.4833 | 0.0009 | ||||||
| X2 | −0.4667 | 0.4817 | ||||||
| X12 | −4.5603 | 0.0017 | ||||||
| X22 | −1.3103 | 0.1999 | ||||||
| X1X2 | −0.8750 | 0.2928 | ||||||
| Y1 | Intercept | 88.5969 | 33.86 | <0.0001 | 0.9603 | 0.9319 | 16.340 | |
| X1 | −14.3733 | 0.0006 | ||||||
| X3 | 8.8433 | 0.0081 | ||||||
| X12 | −30.2741 | <0.0001 | ||||||
| X32 | −11.4341 | 0.0149 | ||||||
| X1X3 | 0.8700 | 0.7776 | ||||||
| Y2 | Intercept | 238.4276 | 400.30 | <0.0001 | 0.9965 | 0.9940 | 68.307 | |
| X1 | 77.1000 | <0.0001 | ||||||
| X3 | 34.8000 | <0.0001 | ||||||
| X12 | 24.2035 | <0.0001 | ||||||
| X32 | 14.1035 | 0.0018 | ||||||
| X1X3 | 7.8000 | 0.0144 | ||||||
| Y3 | Intercept | 0.3704 | 137.33 | <0.0001 | 0.9899 | 0.9827 | 36.092 | |
| X1 | 0.02150 | 0.0008 | ||||||
| X3 | 0.0862 | <0.0001 | ||||||
| X12 | 0.0377 | 0.0003 | ||||||
| X32 | 0.0357 | 0.0004 | ||||||
| X1X3 | −0.0003 | 0.4761 | ||||||
| Y4 | Intercept | −29.1069 | 12.18 | 0.0024 | 0.8969 | 0.8233 | 11.116 | |
| X1 | −1.1500 | 0.0183 | ||||||
| X3 | −1.5500 | 0.0044 | ||||||
| X12 | −0.7259 | 0.2313 | ||||||
| X32 | −1.7259 | 0.0169 | ||||||
| X1X3 | 1.9250 | 0.0041 | ||||||
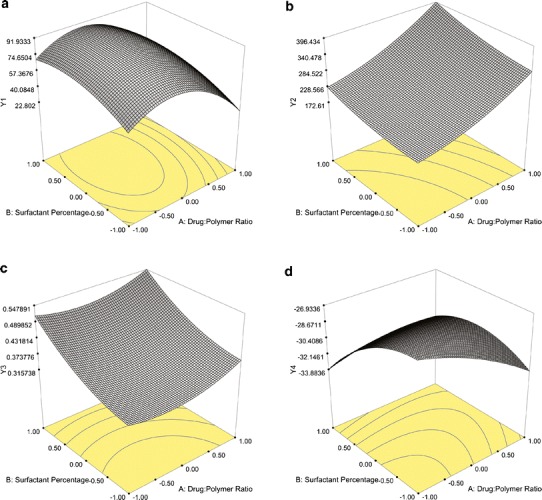
In this study, it was found that PVA-emulsified NPs were produced with high encapsulation efficiency (Q1), in the range of 72.86–100.0% (Table III). The regression coefficients which have a P value <0.05 are significant, while the terms which have a coefficient P value >0.05 are insignificant in the prediction response with 95% confidence (31). Within this perspective, the coefficient P values showed that the level of X2 had a significant effect on Q1 (P = 0.0166); however, X1 did not have a significant effect on Q1 (P = 0.1930) (Table IV). Moreover, there is no interactive effect between X1 and X2 (P = 0.2609). The surfactant percentage is a very important parameter, which affects the drug loading in the production of nanosized particles. As an excess amount of surfactant was used, drug incorporation could be reduced due to the interaction between the drug and the surfactant. In this study, we observed that the formulations containing 6% PVA have less drug loading than the other formulations, which produced 2% and 4% PVA. The positive sign in front of the coefficient of X1 indicated a positive effect of the drug/polymer ratio on Q1, whereas, the negative sign for the coefficient of X2 indicated that the surfactant percentage had a negative influence on encapsulation efficiency of NPs (Table IV). On the other hand, a model F value was assessed by the F statistics, which estimates the percentage of the variability in the outcome. The model F value of 5.17 was higher than the tabulated F value (Ftab = 4.46), indicating that the model was significant (P = 0.0265). The R2 value is a measure of total variability explained by the model. The R2 value for the model was found to be 0.7869, indicating that 78.69% variation was explained by the model (Table IV) (31,32). In addition, the RSM used in the experimental design studies was constructed to determine the optimal conditions for the drug/polymer ratio and PVA percentages used in the NMS-loaded PLGA NPs leading to the highest Q1. The surface plot (Fig. 2a) shows that Q1 of 100% could be achieved with the X1 range at 0.00 level (1:15) to 1.00 level (1:20) and X2 at two different levels, −1.00 (2%) to −0.50 (3%), as well as −0.50 (3.5%) to 0.00 (4%).
Fig. 2.
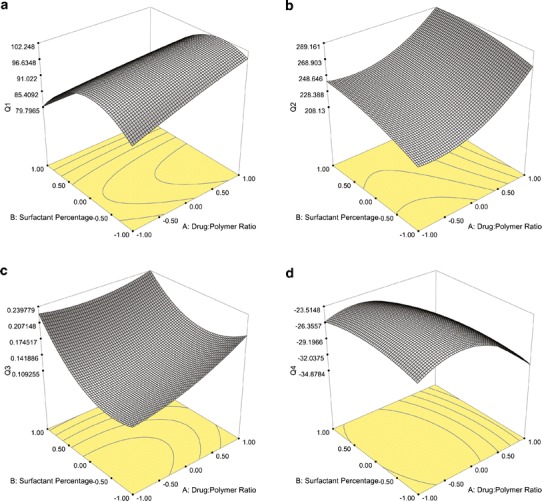
Surface response plots of PVA-emulsified PLGA NPs showing the influence of X1 and X2 on a Q1, b Q2, c Q3, and d Q4
For particle size response (Q2), the mean particle size of PVA-emulsified PLGA NPs ranged from 211.9 to 288.5 nm (Table III). The positive sign for the coefficient of X1 indicated that the drug/polymer ratio had a positive effect on the particle size of the NPs, and the positive sign for the coefficient of X2 indicated that Q2 increases when the surfactant concentration increases. The polymer concentration in the organic phase of the emulsion system led to increased viscosity, which resulted in an increase of Q2 due to the coalescence tendency of the inner phase droplets. As the polymer amount increased, generally, a more viscous internal phase occurred, causing larger droplets in the emulsion and thus larger particles (33,34). Similarly, as the PVA concentration increased, the size of the particles increased due to the increased viscosity of the aqueous phase, which, in turn, reduced the net shear stress available for droplet breakdown (35). The size data provided a good and significant fit (R2 = 0.9789, P < 0.0001) to the linear model (Table IV). Generally, Q2 was significantly affected by the level of both X1 (P < 0.0001) and X2 (P = 0.0002). While separately they have an effect on Q2, there is no interactive effect between X1 and X2 (P = 0.1671). The RSM plot of Q2 is shown in Fig. 2b. The RSM curve indicated that when the drug/polymer ratio and the surfactant percentages increased, the value of Q2 increased. The lowest Q2 was observed at the range of −1.00 level (1:10) to 0.00 level (1:15) for X1, and −1.00 level (2%) to −0.50 (3%) level for X2.
The other two responses investigated on PVA-emulsified PLGA NPs were PDI (Q3) and surface charge (Q4). PDI is a measure of dispersion homogeneity and generally expressed as smaller than 0.2 for narrow size distribution (22). It was observed that the factorial formulations that contain 6% PVA showed a wide size distribution (PDI > 0.2) based on the X1 levels investigated (Table III). As the PVA percentage decreased from 6% to 4% or 2%, Q3 values were found less than 0.2 indicating a uniform size distribution. The surfactant molecules must cover the organic/aqueous interfacial area of all the droplets for stabilization to be effective. Hence, a minimum number of surfactant molecules were required to achieve a small particle size and narrow size distribution. At very high concentrations of PVA, the particle size increased, and the size distribution of the formulation became wider due to the high viscosity of the aqueous phase. In the emulsion system, the total amount of PVA relative to PLGA was inadequate to stabilize the emulsion droplets and resulted in particle aggregation (35). For Q3, the P value (0.0104) was lower than 0.05, and the result implied that the model is significant. The positive sign for the coefficients of both of X1 and X2 indicated that both of the independent variables had a positive effect on the size distribution of the NPs (Table IV). Moreover, there is no interactive effect between X1 and X2 (P = 0.2148). Figure 2c shows the represented RSM curve of PVA-emulsified PLGA NPs illustrating the influence of X1 and X2 on Q3. The minimum PDI value can be achieved with a surfactant at a percentage range of 2% (−1.00 level) to 3% (−0.50 level) and with a drug/polymer ratio of 1:10 (−1.00 level) to 1:12.5 (−0.50 level).
Zeta potential is the potential at the hydrodynamic shear plane and provides information about particle stability in the dispersion (22,24). The zeta potential response (Q4) values ranged between −22.3 and −33.7 mV for the PVA-emulsified PLGA NPs. The negative surface charge determined from the zeta potential analysis may be due to ionized PVA carboxyl groups on the surface of the NPs (22). The ANOVA results of Q4 shown in Table IV indicate that X1 had a significant influence on Q4 (P = 0.0009). On the other hand, there is no significant interactive effect between X1 and X2 (P = 0.2928). As X1 increased to 1:20, Q4 increased significantly, while below the 1:20 drug/polymer ratio, Q4 was not found to be statistically significant. The P value for the Q4 response was 0.0016, a value less than 0.05 indicating that the model was significant. The R2 value was found to be 0.9085. As a result, a variation of 90.85% was explained by the model, and the result indicated that the model is a good fit. When the negative signs for the coefficients of both X1 and X2 were evaluated, it was understood that the zeta potential of the NPs was barely dependent on the independent variables (Table IV). It was concluded from the RSM plot (Fig. 2d) that the Q4 of −22.3 could be achieved at the 0.00 X1 level (1:15) and the 0.00 X2 level (4%) at the center point of the design with the PVA-emulsified formulations.
The term R2 adjusted (Adj R2) is used to compensate for the addition of variables to the model. As more independent variables are added to the regression model, R2 will increase. However, the adjusted R2 can increase or decrease depending on whether the additional variable adds or detracts to the explanatory power of the model. For this reason, Adj R2 is generally considered to be a more accurate goodness-of-fit measure than R2. Adj R2 will always be lower or equal to R2. In the present model, for all responses (Q1–Q4), the value of Adj R2 was found to be less than the R2 value.
The signal-to-noise ratio is measured by Adeq Precision, and a value greater than 4 is considered adequate. In the case of Q1, Q2, Q3, and Q4, the ratio values were 5.607, 26.110, 7.332, and 10.232, respectively, indicating an acceptable signal. Thus, the proposed model is suitable for navigation of the design space (Table IV).
Figure 3 shows the linear correlation plots of PVA-emulsified PLGA NPs between the observed and predicted values of Q1, Q2, Q3, and Q4. The plots from the predicted and actual responses belonging to Q2 indicate excellent fitting of the model (P < 0.001).
Fig. 3.
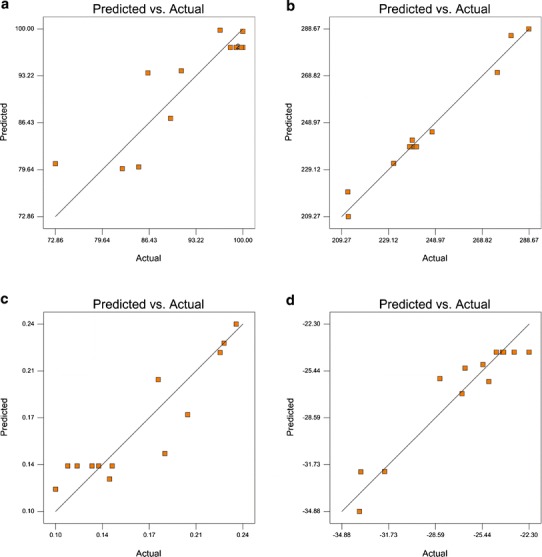
Linearity plots of PVA-emulsified PLGA NPs shown as observed versus predicted values a Q1, b Q2, c Q3, and d Q4
Evaluation of Vitamin E TPGS in the Preparation of NPs
The other nonionic surfactant evaluated for its effect in the preparation of PLGA-based NP formulations was vitamin E TPGS. In this factorial formulation group, the drug/polymer ratio used was the same as PVA-emulsified NP formulations where vitamin E TPGS percentages varied as 0.03, 0.06, and 0.09. Researchers frequently use vitamin E TPGS as an emulsifier in low concentrations ranging from 0.15% to 0.03% due to its potential to have an emulsification effect 67 times greater than PVA in PLGA NPs (36). Another nine formulations were prepared in accordance with the 32 factorial design with vitamin E TPGS. Table V shows the values of the observed and predicted responses belonging to this group.
Table V.
Observed and Predicted Responses of Two Factors: Drug/Polymer Ratio (X1) and Vitamin E TPGS Percentage Used (X3)
| Responses | ||||||||
|---|---|---|---|---|---|---|---|---|
| Observed values | Predicted values | |||||||
| Y1a (%) | Y2a (nm) | Y3a | Y4a (mV) | Y1a (%) | Y2a (nm) | Y3a | Y4a (mV) | |
| Factorial points | ||||||||
| V1 | 55.20 ± 1.28 | 168.0 ± 3.11 | 0.326 ± 0.10 | −27.6 ± 1.26 | 53.29 | 172.6 | 0.330 | −26.9 |
| V2 | 66.85 ± 1.39 | 188.6 ± 5.42 | 0.399 ± 0.09 | −28.6 ± 1.51 | 72.70 | 185.5 | 0.390 | −28.7 |
| V3 | 73.17 ± 1.86 | 228.2 ± 6.57 | 0.506 ± 0.07 | −33.3 ± 1.89 | 69.24 | 226.6 | 0.510 | −33.8 |
| V4 | 64.13 ± 2.16 | 226.5 ± 3.72 | 0.325 ± 0.11 | −27.8 ± 2.11 | 68.32 | 217.7 | 0.320 | −29.3 |
| V5 | 77.77 ± 1.64 | 283.7 ± 5.63 | 0.496 ± 0.13 | −33.4 ± 1.51 | 86.01 | 287.3 | 0.490 | −32.4 |
| V6 | 25.08 ± 4.49 | 307.1 ± 8.40 | 0.384 ± 0.05 | −33.9 ± 1.28 | 22.80 | 311.2 | 0.380 | −33.1 |
| V7 | 37.37 ± 2.43 | 341.8 ± 7.68 | 0.426 ± 0.04 | −30.6 ± 1.73 | 43.95 | 339.7 | 0.430 | −30.9 |
| V8 | 46.53 ± 3.03 | 398.5 ± 5.57 | 0.550 ± 0.12 | −31.9 ± 2.33 | 42.23 | 396.4 | 0.550 | −32.3 |
| Center points | ||||||||
| V9 | 91.86 ± 3.32 | 237.0 ± 5.21 | 0.370 ± 0.08 | −29.2 ± 2.43 | 88.60 | 238.4 | 0.370 | −29.1 |
| V10 | 90.85 ± 2.24 | 235.9 ± 3.85 | 0.355 ± 0.11 | −29.6 ± 1.58 | 88.60 | 238.4 | 0.370 | −29.1 |
| V11 | 88.67 ± 2.20 | 236.4 ± 4.66 | 0.367 ± 0.13 | −28.9 ± 1.67 | 88.60 | 238.4 | 0.370 | −29.1 |
| V12 | 91.26 ± 3.47 | 238.2 ± 5.73 | 0.373 ± 0.09 | −29.7 ± 1.84 | 88.60 | 238.4 | 0.370 | −29.1 |
| V13 | 92.77 ± 3.26 | 239.5 ± 4.69 | 0.378 ± 0.04 | −28.6 ± 2.01 | 88.60 | 238.4 | 0.370 | −29.1 |
aThe values indicate mean ± standard deviation
Table V summarizes that the encapsulation efficiency (Y1) of NPs varied widely from 25.08% to 92.77%. Vitamin E TPGS significantly decreased the encapsulation efficiency of NMS to as low as 25.08% compared to PVA. Zhang et al. reported that vitamin E TPGS was distributed on the surface of the NPs and exhibited a notable effect on the organic/aqueous interfacial area of the droplets in the NP dispersion; this interaction may have caused the reduction of the encapsulation of the drug into the polymeric matrix (36). As seen from the surface response plot of NPs (Fig. 4a), the highest Y1 value was observed at the NPs including a 1:15 drug/polymer ratio (0.00 level) and 0.06% vitamin E TPGS concentration (0.00 level) at the design’s center point of vitamin E TPGS-loaded PLGA NPs. When the response values were investigated, depending on the variation of X3 at each X1 variable, it was observed that the Y1 values increased with higher X3 levels. On the other hand, in RSM plot data (Fig. 4a), it was found that the effect of X1 on Y1 values is positive at low levels of X1 (1:10–1:15), while at higher levels of X1 (1:20), the Y1 values decrease. This is why the response surface presents a twisted view. When the PLGA concentration reached 200 mg (drug/polymer ratio 1:20), the entrapment efficiency of NMS was found to be very low (Table V), which is due to the adsorption of drug molecules on the NP surface. The hydrophobic molecule NMS was likely adsorbed more on the large surface of the NPs due to the overly large particle size, thereby leading to a lower amount of NMS entrapped in the NPs. When the statistical data of Y1 was evaluated based on the P values of the regression coefficients (Table IV), it was observed that the percentage of vitamin E TPGS had a significant influence on Y1 (P = 0.0081). The positive sign in front of the coefficient of the X3 value indicated that X3 had a positive effect on Y1. In contrast, the negative sign of the coefficient of X1 showed that the drug/polymer ratio had a negative influence on the encapsulation efficiency of NPs (P = 0.0006) (Table IV). In this formulation group, the model F value of 33.86 was higher than the tabulated F value (Ftab = 4.46), indicating model significance (P < 0.0001). The R2 statistics indicated that the model explains 96.03% of the variability in the encapsulation efficiency (Y1) (Table IV).
Fig. 4.
Surface response plots of vitamin E TPGS-emulsified PLGA NPs showing the influence of X1 and X3 on a Y1, b Y2, c Y3, and d Y4
When the particle size response (Y2) was examined, the mean particle size of vitamin E TPGS-emulsified PLGA NPs was found to range from 168.0 to 398.5 nm (Table V). The lowest Y2 was observed in both of the lowest levels of X1 (1:10) and X3 (0.03%) in batch V1. The positive sign for the coefficients of X1 and X3 indicated that both the drug/polymer ratio and the surfactant percentages used positively affected the particle size of the NPs. The ANOVA statistical results indicated that the model is a significant and good fit (R2 = 0.9965, P < 0.0001). The factors X1 and X3 were also found to significantly influence Y2 (P < 0.0001). The RSM plot of Y2 is shown in Fig. 4b. From the RSM plot, it was concluded that achieving a size of 168 nm was possible with X1 ranging from 1:10 (−1.00 level) to 1:12.5 (−0.50 level), and X3 ranging from 0.03% (−1.00 level) to 0.045% (−0.50 level). This plot illustrated that as both the drug/polymer ratio and the surfactant percentages increase, the value of the dependent variable, the particle size also increases. One possible reason could be the increase in viscosity of the polymeric solution in the organic phase due to the increase in the polymer amount, thereby posing difficulty in breaking down the emulsion droplets into smaller ones in contrast to the lower X1 levels. As the polymer amount increased, the organic phase showed greater viscosity, while during the emulsification process, the organic phase was barely dispersed in the aqueous phase, producing larger NPs. When the Y2 levels were investigated depending on the variation of surfactant concentration (X3) at each drug/polymer ratio (X1), it was found that as the X3 level increased, the mean particle size increased. This is understandable because the surfactant remains at the interface of the organic/aqueous phase, thus separating the phases from each other. As the vitamin E TPGS concentration was increased, the viscosity of the aqueous phase increased, and this reduced the net shear stress of the system that is available for droplet breakdown to smaller ones (37,38).
The response of PDI values (Y3) for vitamin E TPGS-emulsified NMS-loaded PLGA NPs ranged between 0.325 and 0.550 and inhibited a wide size distribution (PDI > 0.2) (Table V). As can be observed from the RSM plot provided in Fig. 4c, X1 and X3 have a positive effect on the Y3 levels. The increasing viscosity based on the polymer amount in the organic phase and the surfactant concentration in the aqueous phase of the emulsion led to an increase of the PDI value due to the insufficient dispersion of phases. The PDI data provided a significant and good fit (P < 0.0001, R2 = 0.9899) to the linear model. The positive sign for the coefficients of both of X1 and X3 indicated that both of the independent variables positively affected the PDI value of the vitamin E TPGS-emulsified NPs. When the coefficients of the model were estimated by ANOVA for Y3, it was observed that X1 and X3 had a statistically significant effect on Y3 (P = 0.0008 and P < 0.0001, respectively) (Table IV). The RSM curve showed that the minimum PDI value could be obtained with a drug/polymer ratio ranging from 1:10 (−1.00 level) to 1:15 (0.00 level) and with a percentage range of surfactant from 0.03% (−1.00 level) to 0.045% (−0.50 level) (Fig. 4c).
The zeta potential values (Y4) belonging to the vitamin E TPGS-emulsified NMS-loaded PLGA NPs in the factorial design study are summarized in Table V. The zeta potential (Y4) values ranged between −27.6 and −33.9 mV. NPs belonging to this formulation group exhibited negative surface charges because of the polar PEG1000 components of vitamin E TPGS, which extend outwards into the aqueous phase, while the lipophilic end is adsorbed into the matrix structure of the PLGA NP. The presence of PEG chains on the surface of the NPs gained the negative charges to the NPs. The ANOVA results in Table IV demonstrate the significant effect of X1 and X3 on Y4 (P = 0.0183 and 0.0044, respectively). The model F value was found as 12.18 (>Ftab = 4.46), and the result indicates that the model is significant. The negative sign for the coefficients of both X1 and X3 indicates that the both of the independent variables had a negative influence on the surface charge of the NPs. It was concluded from the RSM plot (Fig. 4d) that the minimum Y4 of the design was achievable with an X1 ranging from the −1.00 level (1:10) to the −0.50 level (1:15) and an X2 range from the −1.00 level (0.03%) to the −0.075 level (0.05%).
Since the values of R2 are relatively high for all of the dependent variables, 0.9603 for Y1, 0.9965 for Y2, 0.9899 for Y3, and 0.8969 for Y4, the polynomial equations form an excellent fit to the obtained data and are highly statistically valid. Similarly, the value of Adj R2 for the PVA-emulsified PLGA NPs and the Adj R2 value for all of the responses (Y1–Y4) of vitamin E TPGS-emulsified PLGA NPs were found to be less than the R2 value.
The observed Adeq Precision ratios given in Table IV were 16.340 for Y1, 68.307 for Y2, 36.092 for Y3, and 11.116 for Y4. All ratios were higher than 4, indicating an adequate signal and that the model can be used for navigation of the design space.
Figure 5 gives the linearity plots of vitamin E TPGS-emulsified PLGA NPs between observed and predicted values of Y1, Y2, Y3, and Y4. The plots obtained for the predicted and actual responses belonging to Y1, Y2, and Y3 indicate excellent fitting of the model (P < 0.001). The linearity graphics of data were supported with the ANOVA results in Table IV.
Fig. 5.
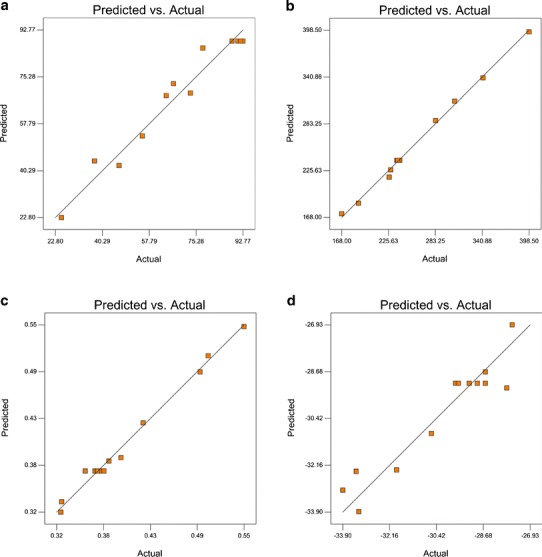
Linearity plots of vitamin E TPGS-emulsified PLGA NPs shown as observed versus predicted values a Y1, b Y2, c Y3, and d Y4
PLGA formulations produced at the center points of the factorial design were selected as the optimum formulations due to their high encapsulation efficiency in this research. Therefore, SEM, GC, and DSC studies were performed only on the optimum PLGA formulations. The shape of the NPs is shown in Fig. 6. As shown in the micrographs, all NPs had a spherical shape. Vitamin E TPGS-emulsified PLGA NPs had less of a spherical shape and showed a wider size distribution than the PVA-emulsified nanosized particles. The shape differences between the NP formulations prepared from two different nonionic surfactants occurred due to the difference in the hydrophilic–lipophilic balance (HLB) values of the agents. Although PVA and vitamin E TPGS are both nonionic surfactants, PVA has a HLB value of 18 while that of vitamin E TPGS is 13.2 (36,39). Therefore, PVA solutions can stabilize smaller and more uniform droplets than a vitamin E TPGS solution during the production process. Hence, the homogeneous droplets in the emulsion solidified in the PVA-emulsified PLGA NPs were more spherical in shape.
Fig. 6.

SEM micrographs of a P1-, b P9-, c V1-, and dV9-NP formulations
In the PLGA NP production procedure, DCM was used as an organic solvent to dissolve polymeric material. Because of PLGA’s insolubility in water, it is often dissolved in DCM before mixing with an aqueous solution containing various amounts of emulsifier. DCM is a controlled organic solvent, which can cause severe effects after oral administration, inhalation, or skin contact, and significantly increases the incidence of several types of tumors, such as lung, breast, and liver tumors (40). Because of these reasons, it is important to determine the residual DCM content in the developed formulations. GC chromatograms of pure DCM, n-hexane, and the NP formulations coded P9 and V9 are presented in Fig. 7. The retention times of pure DCM and n-hexane were 6.77 and 4.52 min, respectively (Fig. 7a, b). When the GC chromatograms of the PLGA NP formulations were examined (Fig. 7c, d), it was shown that the retention times were 4.52 min for both formulations, which was attributed to the presence of solely n-hexane. There was no peak signal obtained that proved the existence of DCM in the NP formulations.
Fig. 7.
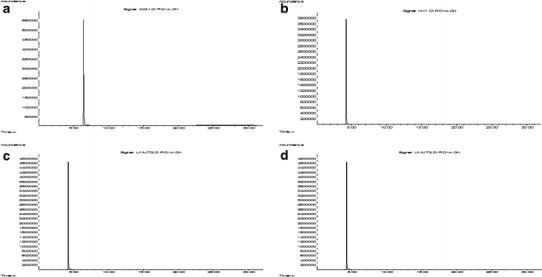
GC chromatograms of a pure DCM, b pure n-hexane, and c P9- and d V9-NP formulations
Different qualitative analytical methods have been developed that provide qualitative information regarding the physicality of materials. DSC is a thermal analyses method widely used in the pharmaceutical sciences and provides information about physical characteristics, such as the crystalline or amorphous state of raw materials, and the active agents used in the development of pharmaceutical formulations (41). Figure 8 shows the DSC curves of the pure NMS, raw PLGA, PVA, and vitamin E TPGS, and also the factorial NP formulations that were prepared at the center point of the design with both surfactants, respectively. The DSC curve of pure NMS sharply peaked endothermically at 148.49°C, as evidence of the absence of crystallinity and corresponding to its melting point (Fig. 8a). This result agrees with previous research by Nalluri et al. (42). The thermal transition of the raw polymer, PLGA, seen at 41.90°C, was attributed to the glass transition temperature (Tg) of the polymeric material (Fig. 8b); no melting point was observed at the curve because of the amorphous nature of PLGA, as Mu and Feng (37) also established. When the DSC curves of the nonionic surfactants, PVA and vitamin E TPGS, were examined, PVA had a broad melting endotherm at 184.55°C, while vitamin E TPGS showed a sharp melting endotherm at 40.38°C, which were attributed to the melting temperatures (Tm) of the surfactants, respectively (Fig. 8c, d). Similar findings have been reported by other researchers for both surfactants (37,43). The DSC curves of the NMS-loaded factorial PLGA NPs which were prepared with both surfactants (Fig. 8e, f) showed that the NMS endothermic peak at 148.49°C (Fig. 8a) disappeared. It can be concluded that the encapsulated NMS in the NPs was in an amorphous state in the polymeric matrix after the production process. Other authors observed similar findings using hydrophobic drugs encapsulated in the PLGA matrix (37,43). The DSC curve of the P9-NP formulation showed that the Tg of PLGA was observed at 46.24°C, and the Tm of PVA was found at 181.30°C (Fig. 8e). Examination of the DSC curve of the V9-coded NP formulation (Fig. 8f) revealed two melting endothermic peaks at 33.88 and 47.63°C, corresponding to the Tm of the surfactant and the Tg of the polymer, respectively. The increase in the Tg value of PLGA that was observed in both NP formulations prepared with different surfactants is presumed to be caused by the solubility of NMS in the polymeric structure. This finding is similar to the change in the Tg value of polymeric material, Eudragit RS 100, reported in our previous study and which agrees with our results (41). From the DSC curve of the P9-NP formulation (Fig. 8e), it was determined that the production procedure did not influence the Tm of PVA. In contrast, in the DSC curve of the V9-coded NP formulation (Fig. 8f), the Tm of vitamin E TPGS was found to decrease. During the solvent evaporation process and the dissolution of PLGA, the slower evaporation of the DCM and the longer mixing of the dispersion were probably responsible for the Tm of the surfactant decreasing from 40.38 to 33.88°C. This situation also causes the broadening of the Tm peak of vitamin E TPGS and is similar to a result mentioned before (41).
Fig. 8.
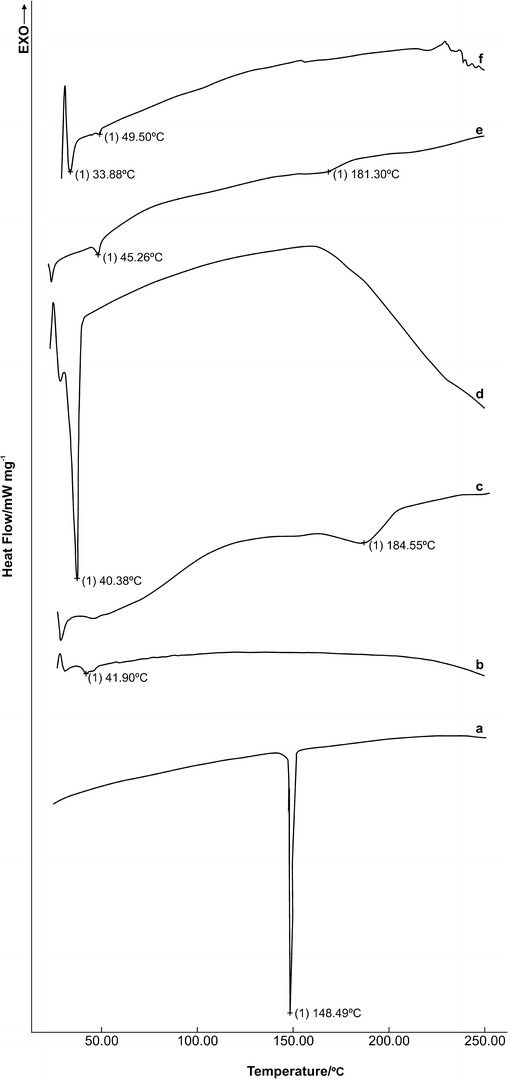
DSC curves of a NMS, b PLGA, c PVA, d vitamin E TPGS, and e P9- and f V9-NP formulations
The storage stability of the prepared PLGA NPs is a very important factor that affected the physicochemical properties of the nanosized particles such as drug content, mean particle size, size distribution, and surface charge. Table VI shows the storage stability results of the NPs. All of the investigated parameters did not show any significant changes during the third month storage period (P > 0.05).
Table VI.
Storage Stability Results of the PLGA NPs
| NP formulation coded P9 | NP formulation coded V9 | |
|---|---|---|
| Encapsulation efficiency (% ± SE) | 98.73 ± 1.32 | 89.63 ± 1.10 |
| Mean particle size (nm ± SE) | 241.2 ± 1.21 | 236.2 ± 3.37 |
| PDI ± SE | 0.137 ± 0.04 | 0.362 ± 0.007 |
| Surface charge (mV ± SE) | −22.1 ± 1.15 | −29.3 ± 1.17 |
SE standard error, PDI polydispersity index
CONCLUSION
This study demonstrated that the various physicochemical properties of polymeric NPs loaded with a hydrophobic drug could be manipulated by varying the contents of the drug, polymer, and surfactant according to the full factorial design studies and graphical analysis, which established the optimal formulation conditions with a reduced number of experiments. Both PVA and vitamin E TPGS-emulsified PLGA formulations prepared at the center points of the factorial study had the highest encapsulation efficiency, thus were chosen as the optimum formulations of the design. ANOVA results showed that independent and dependent variables seemed to have the best correlation in vitamin E TPGS-emulsified PLGA NPs compared to the PVA-emulsified formulations. However, PVA may be preferred to vitamin E TPGS for producing nanosized particles possessing the higher drug loading and narrow size distribution. The obtained data could be used to model the attributes of PVA- or vitamin E TPGS-emulsified PLGA NPs loaded with other similar hydrophobic drugs. However, further studies conducted with animal models are required to confirm the pharmacological activities.
References
- 1.Manjanna KM, Shivakumar B, Pramod Kumar TM. Microencapsulation: an acclaimed novel drug-delivery system for NSAIDs in arthritis. Crit Rev Ther Drug Carr Syst. 2010;27:509–45. doi: 10.1615/CritRevTherDrugCarrierSyst.v27.i6.20. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Z, Huang G. Micro- and nano-carrier mediated intra-articular drug delivery systems for the treatment of osteoarthritis. J Nanotechnol. 2012;2012:1–11. doi: 10.1155/2012/748909. [DOI] [Google Scholar]
- 3.Bianchi M, Ehrlich GE, Facchinetti F, Huskisson EC, Jenoure P, La Marca A, et al. Clinical applications of nimesulide in pain, arthritic conditions and fever. In: Rainsford KD, et al., editors. Nimesulide actions and uses. Basel: Birkhäuser; 2005. pp. 245–314. [Google Scholar]
- 4.Zhang Z, Bi XB, Huang G. Enhanced targeting efficiency of PLGA microspheres loaded with lornoxicam for intra-articular administration. Drug Deliv. 2011;18(7):536–44. doi: 10.3109/10717544.2011.596584. [DOI] [PubMed] [Google Scholar]
- 5.Schulze K, Koch A, Schöpf B, Petri A, Steitz B, Chastellain M, et al. Intrarticular application of superparamagnetic nanoparticles and their uptake by synovial membrane-an experimental study in sheep. J Magn and Magn Mater. 2005;293:419–32. doi: 10.1016/j.jmmm.2005.02.075. [DOI] [Google Scholar]
- 6.Gerwin N, Hops C, Lucke A. Intraraticular drug delivery in osteoarthritis. Adv Drug Deliv Rev. 2006;58:226–42. doi: 10.1016/j.addr.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 7.Liang LS, Jackson J, Min W, Risovic V, Wasan KM, Burt HM. Methotrexate loaded poly(L-lactic acid) microspheres for intra-articular delivery of methotrexate to joint. J Pharm Sci. 2004;93:943–56. doi: 10.1002/jps.20031. [DOI] [PubMed] [Google Scholar]
- 8.Horisawa E, Kubota K, Tuboi I, Sato K, Yamamoto H, Takeuchi H, et al. Size-dependency of D, L-lactide/glycolide copolymer particulates for intra-articular delivery system on phagocytosis in rat synovium. Pharm Res. 2002;19:132–139. doi: 10.1023/A:1014260513728. [DOI] [PubMed] [Google Scholar]
- 9.Ratcliffe JH, Hunneyball M, Smith A, Wilson CG, Davis SS. Preparation and evaluation of biodegradable polymeric systems for the intra-articular delivery of drugs. J Pharm Pharmacol. 1984;36:431–6. doi: 10.1111/j.2042-7158.1984.tb04419.x. [DOI] [PubMed] [Google Scholar]
- 10.Rothenfluh DA, Bermudez H, O’Neil CP, Hubbell JA. Bifunctional polymer nanoparticles for intra-articular targeting and retention in cartilage. Nat Mater. 2008;7:248–254. doi: 10.1038/nmat2116. [DOI] [PubMed] [Google Scholar]
- 11.Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161:505–22. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 12.Wang JX, Fan YB, Gao Y, Hu QH, Wang TC. TiO2 nanoparticles translocation and potential toxicological effect in rats after intraarticular injection. Biomater. 2009;30:4590–600. doi: 10.1016/j.biomaterials.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 13.Wang J, Gao Y, Hou Y, Zhao F, Pu F, Liu X, et al. Evaluation on cartilage morphology after intra-articular injection of titanium dioxide nanoparticles in rats. J Nanomater. 2012;452767:1–11. [Google Scholar]
- 14.Crowe LA, Tobalem F, Gramoun A, Delattre BMA, Grosdemange K, Salaklang J, et al. Improved dynamic response assessment for intra-articular injected iron oxide nanoparticles. Magn Reson Med. 2012;68:1544–52. doi: 10.1002/mrm.24166. [DOI] [PubMed] [Google Scholar]
- 15.Ding YDH, Shangli L, Ruofan M. The efficacy and safety of lornoxicam in treatment of rheumatoid arthritis and osteoarthritis. Chin J New Drugs. 2004;13:562–4. [Google Scholar]
- 16.Betre H, Liu W, Zalutsky MR, Chilkoti A, Kraus VB, Setton LA. A thermally responsive biopolymer for intra-articular drug delivery. J Control Release. 2006;115:175–82. doi: 10.1016/j.jconrel.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 17.Horisawa E, Hirota T, Kawazoe S, Yamada J, Yamamoto H, Takeuchi H, et al. Prolonged anti-inflammatory action of DL-lactide/glycolide copolymer nanospheres containing betamethasone sodium phosphate for an intra-articular delivery system in antigen-induced arthritic rabbit. Pharm Res. 2002;19:403–10. doi: 10.1023/A:1015123024113. [DOI] [PubMed] [Google Scholar]
- 18.Higaki M, Ishihara T, Izumo N, Takatsu M, Mizushima Y. Treatment of experimental arthritis with poly(D, L-lactic/glycolic acid) nanoparticles encapsulating betamethasone sodium phosphate. Ann Rheum Dis. 2005;64:1132–6. doi: 10.1136/ard.2004.030759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gohel M, Patel M, Amin A, Agrawal R, Dave R, Bariya N. Formulation design and optimization of mouth dissolve tablets of nimesulide using vacuum drying technique. AAPS Pharm Sci Tech. 2004;5(3). Article 36 [DOI] [PMC free article] [PubMed]
- 20.Freitas MN, Marchetti JM. Nimesulide PLA microspheres as a potential sustained release system for the treatment of inflammatory diseases. Int J Pharm. 2005;295:201–11. doi: 10.1016/j.ijpharm.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 21.Ravikumara NR, Madhusudhan B, Nagaraj TS, Hiremat SR, Raina G. Preparation and evaluation of nimesulide-loaded ethylcellulose and methylcellulose nanoparticles and microparticles for oral delivery. J Biomater Appl. 2009;24:47–64. doi: 10.1177/0885328209103406. [DOI] [PubMed] [Google Scholar]
- 22.Sengel Turk CT, Hascicek C, Dogan AL, Esendagli G, Guc D, Gönül N. Preparation and in vitro evaluation of meloxicam-loaded PLGA nanoparticles on HT-29 human colon adenocarcinoma cells. Drug Dev Ind Pharm. 2012;38:1107–16. doi: 10.3109/03639045.2011.641562. [DOI] [PubMed] [Google Scholar]
- 23.Dong Y, Zhang Z, Feng SS. D-α-Tocopheryl polyethylene glycol 1000 succinate (TPGS) modified poly(L_lactide) (PLLA) films for localized delivery of paclitaxel. Int J Pharm. 2008;350:166–71. doi: 10.1016/j.ijpharm.2007.08.043. [DOI] [PubMed] [Google Scholar]
- 24.Sengel CT, Hascicek C, Gönül N. Design of vitamin E d-a tocopheryl polyethylene Glycol 1000 succinate-emulsified poly (DL–lactide–co-glycolide) nanoparticles: influence of duration of ultrasonication energy. J Young Pharm. 2011;3:171–5. doi: 10.4103/0975-1483.83754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alayoubi A, Nazzal M, Sylvester PW, Nazzal S. “Vitamin E” fortified parenteral lipid emulsions: Plackett-Burman screening of primary process and composition parameters. Drug Dev Ind Pharm. 2013;39:363–73. doi: 10.3109/03639045.2012.682223. [DOI] [PubMed] [Google Scholar]
- 26.Gómez-Gaete C, Bustos GL, Godoy RR, Saez CK, Novoa GP, Fernández EM, et al. Successful factorial design for the optimization of methylprednisolone encapsulation in biodegradable nanoparticles. Drug Dev Ind Pharm. 2013;39:310–20. doi: 10.3109/03639045.2012.676049. [DOI] [PubMed] [Google Scholar]
- 27.United States Pharmacopoeia 23 – National Formulary 18, 1995
- 28.Nie H, Wang CH. Fabrication and characterization of PLGA/Hap composite scaffolds for delivery of BMP-2 plasmid DNA. J Control Release. 2007;120:111–21. doi: 10.1016/j.jconrel.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 29.El Gamal SS, Naggar VF, Allam AN. Optimization of acyclovir tablets based on gastroretention technology: factorial design analysis and physicochemical characteristics studies. Drug Dev Ind Pharm. 2011;37:855–67. doi: 10.3109/03639045.2010.546404. [DOI] [PubMed] [Google Scholar]
- 30.Blasi P, Giovagnoli S, Schoubben A, Puglia C, Bonina F, Rossi C, et al. Lipid nanoparticles for brain targeting I. formulation optimization. Int J Pharm. 2011;419:287–95. doi: 10.1016/j.ijpharm.2011.07.035. [DOI] [PubMed] [Google Scholar]
- 31.Yadav KS, Sawant KK. Modified nanoprecipitation method for preparation of cytarabine-loaded PLGA nanoparticles. AAPS PharmSciTech. 2010;11:1456–65. doi: 10.1208/s12249-010-9519-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yadav KS, Jacob S, Sachdeva G, Chunttani K, Mishra AK, Sawant KK. Long circulating PEGylated PLGA nanoparticles of cytarabine for targeting leukemia. J Microencapsul. 2011;28:729–42. doi: 10.3109/02652048.2011.615949. [DOI] [PubMed] [Google Scholar]
- 33.Sengel CT, Hascicek C, Gönül N. Development and in-vitro evaluation of modified release tablets including ethylcellulose microspheres loaded with diltiazem hydrochloride. J Microencapsul. 2006;23:135–52. doi: 10.1080/02652040500286474. [DOI] [PubMed] [Google Scholar]
- 34.Sengel-Turk CT, Hascicek C, Gönül N. Microspheres-based once-daily modified release matrix tablets for oral administration in angina pectoris. J Microencapsul. 2008;25:257–66. doi: 10.1080/02652040801967228. [DOI] [PubMed] [Google Scholar]
- 35.Budhian A, Siegel SJ, Winey KI. Haloperidol-loaded PLGA nanoparticles: systematic study of particle size and drug content. Int J Pharm. 2007;336:367–75. doi: 10.1016/j.ijpharm.2006.11.061. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z, Tan S, Feng SS. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomater. 2012;33:4889–906. doi: 10.1016/j.biomaterials.2012.03.046. [DOI] [PubMed] [Google Scholar]
- 37.Mu L, Feng SS. A novel controlled release formulation for the anticancer drug paclitaxel (Taxol®): PLGA nanoparticles containing vitamin E TPGS. J Control Release. 2003;86:33–48. doi: 10.1016/S0168-3659(02)00320-6. [DOI] [PubMed] [Google Scholar]
- 38.Vega E, Egea MA, Valls O, Espina M, García ML. Flurbiprofen loaded biodegradable nanoparticles for opthalmic administration. J Pharm Sci. 2006;95:2393–405. doi: 10.1002/jps.20685. [DOI] [PubMed] [Google Scholar]
- 39.Xu Q, Crossley A, Czernuszka J. Preparation and characterization of negatively charged poly(lactic-co-glycolic acid) microspheres. J Pharm Sci. 2009;98:2377–89. doi: 10.1002/jps.21612. [DOI] [PubMed] [Google Scholar]
- 40.Chen JL, Yeh MK, Chiang CH. Dichloromethane evaporative behaviour during the solidifying process of ovalbumin-loaded poly (DL lactic-co-glycolic acid) microparticles. J Food and Drug Anal. 2004;12:291–8. [Google Scholar]
- 41.Turk CT, Hascicek C, Gonul N. Evaluation of drug-polymer interaction in polymeric microspheres containing diltiazem hydrochloride. J Therm Anal Cal. 2009;95:865–9. doi: 10.1007/s10973-007-8605-7. [DOI] [Google Scholar]
- 42.Nalluri BN, Chowdary KPR, Murthy KVR, Hayman AR, Becket G. Physicochemical characteristics and dissolution properties of nimesulide cyclodextrin binary systems. AAPS PharmSciTech. 2003;4: Article 2. [DOI] [PMC free article] [PubMed]
- 43.Mccarron P, Donnelly RF, Marouf W. Celecoxib-loaded poly(D, L-lactide-co-glycolide) nanoparticles prepared using a novel and controllable combination of diffusion and emulsification steps as part of the salting-out procedure. J Microencapsul. 2006;23:480–98. doi: 10.1080/02652040600682390. [DOI] [PubMed] [Google Scholar]