

Abstract
Ursolic acid (UA), which is a natural pentacyclic triterpenoid, has the potential to be developed as an anticancer drug, whereas its poor aqueous solubility and dissolution rate limit its clinical application. The aim of the present study was to develop UA nanocrystals to enhance its aqueous dispersibility, dissolution rate and anticancer activity. Following the investigation on the effects of stabiliser, the ratio of organic phase to aqueous solution and drug concentration, the UA nanocrystals without stabiliser were successfully prepared by anti-solvent precipitation approach. The nanocrystals maintained similar crystallinity with particle size, polydispersion index and zeta potential values of 188.0 ± 4.4 nm, 0.154 ± 0.022, and −25.0 ± 5.9 mV, respectively. Compared with the raw material, the UA nanocrystals showed good aqueous dispensability and a higher dissolution rate, and they could be completely dissolved in 0.5% SDS solution within 120 min. Moreover, the suspension of UA nanocrystals was physically stable after storage at 4°C for 7 weeks. By inducing G2/M phase cell cycle arrest, the UA nanocrystals significantly induced stronger cell growth inhibition activity against MCF-7 cells compared with free drug in vitro, although the uptake of free UA was approximately twice higher than that of the UA nanocrystals. The UA nanocrystals may be used as a potential delivery formulation for intravenous injection with enhanced dissolution velocity and anticancer activity.
Key words: anticancer, dissolution, MCF-7, nanocrystals, ursolic acid
INTRODUCTION
Ursolic acid (UA), a natural pentacyclic triterpenoid, is widely found in plants such as Sambucus chinensis (Lindl.) and Eriobotrya japonica (Thunb.). It has been reported that UA possesses a variety of pharmacological activities, including antitumour, anti-inflammatory, antioxidant, anti-bacterial, antiviral, and hepatoprotective effect (1). UA also inhibits the growth of various cancer cell lines, such as endometrial cancer cells (2), breast cancer cells (3), T24 bladder cancer cells (4), ovarian cancer cells (5), pancreatic cancer cells (6), lung cancer cells (7), etc. A toxicity study showed that no mice died at 72 h after a single hypodermic injection of 3.5 g/kg UA, suggesting that UA was relatively slightly toxic (8). Although UA has the potential to be developed as an anticancer drug, its poor aqueous solubility, dispersibility and dissolution rate limit its clinical application, and there is no current commercial product of UA. In the last decade, several UA formulations with low drug loading have been developed. The UA phospholipid nanopowders were prepared with high ratio of excipients in the system, so the drug loading was just 12.8% (9). Similarly, using copolymers (mPEG-PCL) as carriers, UA-loaded nanoparticles (UA-NPs) with 4.75% drug loading were developed (10). Therefore, new formulation strategies should be considered to enhance drug loading and reduce the excipients percentage.
Nanotechnology-based drug delivery systems, which can be divided into matrix nanoparticles and nanocrystals, are effective methods to increase the solubility and dissolution rate of water-insoluble compounds. Matrix nanoparticles, such as polymeric nanoparticles, nanoemulsions, nanostructured lipid carriers and solid lipid nanoparticles, consist of drug and either polymer or lipid (11). Not only drug loading of matrix nanoparticles is usually low but also some matrixes used in the system could induce negative side effects. Conversely, nanocrystals consist of pure drug crystal particles with small amounts of stabilisers adsorbed onto the surface of the particles, in which the drug-loading capacity could reach 100% (12). Since nanocrystals were invented in 1990, they have been widely studied, and there are many nanocrystal products on the market, including Rapamune®, Emend®, Tricor® and Megace ES®. Many clinical trials have indicated that nanocrystals could effectively increase the bioavailability and reduce food-related effects as well as the individual variance of poorly water-soluble drugs (13).
There are three basic techniques for the production of the nanocrystals: milling, homogenisation and precipitation. Among these, milling and homogenisation are top–down approaches and are generally used to produce commercial products (14). However, those methods have some disadvantages, e.g. residue from milling media or high-energy output during the homogenisation process. For drugs that can be dissolved in a specific solvent, precipitation is a simple method to produce nanocrystals (15). In the present study, stable UA nanocrystals were first prepared by the anti-solvent precipitation method. The effects of stabiliser, the ratio of organic phase to aqueous solution and the concentration of drug on both the particle size and polydispersion index (PdI) were investigated to obtain a stable formulation. Secondly, the UA nanocrystals were characterised in terms of morphology, crystallinity, dissolution rate and short-term physical stability. Finally, the effects of nanosizing on the cytotoxicity, cellular uptake and cell cycle analysis on MCF-7 breast cancer cells were compared with those of a solubilised UA solution.
MATERIALS AND METHODS
Materials
UA (purity, > 98%) was purchased from International Laboratory (South San Francisco, USA). Glycyrrhetinic acid (GA) was purchased from the National Institute for The Control of Pharmaceutical and Biological Products (Beijing, China). Polyvinylpyrrolidone K90 (PVP K90) and Polysorbate 80 (Tween 80) were obtained from Sigma-Aldrich (Saint Louis, USA). Methocel A15 premium LV hydroxypropyl methylcellulose (HPMC A15) and Methocel E3 premium LV hydroxypropyl methylcellulose (HPMC E3) were purchased from Shanghai Colorcon Coating Technology Limited (Shanghai, China). Sodium dodecyl sulfate (SDS), methanol and acetonitrile (ACN) were obtained from Merck (Darmstadt, Germany). Methyl thiazolyl tetrazolium (MTT) and poloxamer 188 (F68) were purchased from Sigma-Aldrich (Steinheim, Germany). Dulbecco’s modified Eagle’s medium and fetal bovine serum (FBS) was purchased from Life Technologies (New York, USA). A BCA protein assay kit was obtained from Thermo Scientific (Waltham, USA). The MCF-7 human breast cancer cell line was purchased from the American Type Culture Collection (Rockville, USA). Milli-Q water was obtained from a Millipore Direct-Q ultra-pure water system (Millipore, Bedford, USA). Absolute ethanol was purchased from Tianjin Kaitong Chemical Reagent Co. Ltd. (Tianjin, China).
Methods
Preparation of the UA Nanocrystals
The UA nanocrystals were prepared by the anti-solvent precipitation method. To study the effects of different stabilisers on the particle size, PdI and zeta potential, each stabiliser (F68, SDS, Tween80, PVP K90, HPMC E3 and HPMC A15) was dissolved in Milli-Q water to obtain aqueous solutions at the same concentration (0.05%, w/v). To investigate how the ratio of organic phase to aqueous solution influence the particle size, equal amounts of UA (6 mg) were dissolved in different volumes of ethanol (2–4 mL) to form organic phases. In addition, different amounts of UA were dissolved in 2 mL of absolute ethanol to obtain a series of organic phases containing UA at different concentrations (1–5 mg/mL), which were used to study the effect of the UA concentration on particle size. All of the solutions were filtered through 0.20 μm filters to remove insoluble particles. The organic solutions were then injected into 20 mL of aqueous solution either with or without stabilisers via syringe under stirring at about 1,000 rpm for 5 min at room temperature.
Characterisation of the UA Nanocrystals
Particle Size, Size Distribution and Zeta Potential
The particle size, PdI and zeta potential of the UA nanocrystals were measured by dynamic laser light scattering (Nano-Zetasizer, Malvern, UK). Measurements were carried out in triplicate for each sample, and all the experiments were performed in triplicate.
Transmission Electron Microscopy
A drop of the UA nanocrystals was placed on the surface of a copper grid, and the excess liquid was drained onto filter paper. The copper grid was then stained for 2 min in 2% phosphotungstic acid. Transmission electron microscopy (TEM) graphs were obtained using a transmission electronic microscope (JEM 1400, JEOL, Japan) operated at 120 kV.
Differential Scanning Calorimetry Analysis
The phase transition of the UA crystals was analysed by differential scanning calorimetry (DSC; DSC-60A, SHIMADZU, Japan) at a heating rate of 10°C/min from 50°C to 300°C. This procedure was operated under a nitrogen atmosphere, and Al2O3 was used as a reference.
Dissolution Studies
Using SDS solution (0.5%, w/v) as medium (pH = 7.0), dissolution study was performed by dissolution-tester 700 (Erweka, Germany) at 37 ± 0.5°C with the paddle speed at 100 rpm. Compared with the UA nanocrystals formulation, equivalent UA (30 mg), ethanol (10 mL) and Milli-Q (100 mL) water were mixed to obtain coarse suspension. Then 25 mL of nanocrystals or coarse suspension (equal to 6.8 mg UA) were added into 900 mL of dissolution media, respectively. Samples of 5 mL were withdrawn at predetermined time intervals and filtered through 0.20 μm filters. After each withdrawal, an equal volume of the dissolution medium was added to maintain the volume constant. The content of dissolved UA was determined by HPLC (Agilent, USA). The analytical column was ZORBAX SB-C18 (4.6 × 250 mm, 5 μm), and the mobile phase consisted of ACN and 0.1% (v/v) phosphoric acid (85:15, v/v). The flow rate was maintained at 1 mL/min, and the detection was performed at 210 nm. Results showed the method accuracy ranged from 96.2% to 102.0%, with an RSD of 0.72–2.06%. The RSD of both the intra- and inter-day precision was less than 3%, which indicated that the method was reliable. All of the dissolution experiments were performed in triplicate.
In Vitro Evaluation of Free UA and UA Nanocrystals in MCF-7 Cells
Cytotoxicity of UA Solution and UA Nanocrystals
The effects of the free UA and the UA nanocrystals on cell viability and proliferation were determined by MTT method. Briefly, MCF-7 cells were seeded in 96-well plates at a density of 5 × 103 cells per well and incubated for 24 h at 37°C in a 5% CO2 humidified incubator. The culture media were then removed and replaced with 100 μL of fresh media (blank), different concentrations of the UA solution (UA was dissolved in DMSO to obtained UA stock solution which was diluted to pre-determined concentrations with cell culture medium and the highest concentration of DMSO (v/v) in medium was less than 0.5%) or the UA nanocrystals (UA nanosuspension was diluted to pre-determined concentration with cell culture medium and the highest concentration of ethanol (v/v) in medium was ∼1%). After incubation for 12, 24 and 48 h, the medium in each well was substituted with 100 μL of MTT solution (1 mg/mL in 0.5% FBS medium). After incubation at 37°C for 4 h, the mixtures in the wells were removed, and then 100 μL of DMSO was added to each well and shaken at 100 rpm for 10 min. Absorbance was measured using a Spectra MaxM5 Microplate Reader (Molecular Devices, USA) at 570 nm. The viability rate (in per cent) was calculated as ((sample reading/control reading) × 100). All assays were performed in triplicate. To determine whether the ethanol in the nanocrystal system influenced cell viability, cytotoxicity studies were performed using the same method described above for blank medium containing different concentrations of ethanol ranging from 0.015% to1.2% (v/v).
Cellular Uptake Study
The cellular uptake study was designed and performed using Li’s method as a reference (16). MCF-7 cells (200, 000 cells/mL) were seeded into each 12-well plate (Orange Scientific, Belgium) and allowed to attach for 48 h. Next, the cell culture media were replaced with fresh media containing different concentrations of free UA or the UA nanocrystals (5 and 10 μmol/L). After incubation for predetermined time intervals (10 min, 30 min, 1 h, 3 h and 6 h), the cells were washed three times with cold PBS (4°C). The cells were then lysed by incubating with cell lysis buffer (0.1 mL/cell, Beyotime Institue of Biotechnology, China) on ice. After incubation for 30 min, the cell lysates were collected into 1.5 mL centrifuge tubes and centrifuged (15,000×g × 30 min) at 4°C. The resulting supernatants were processed to determine the UA contents by LC-ESI-MS/MS.
To determine the levels of cellular uptake of both UA nanocrystals and free UA, 20 μL of GA solution (49.5 ng/mL), which was used as an internal standard, was added into 80 μL of cell lysate sample and mixed for 60 s. Then, ACN (400 μL) was added to the sample to precipitate the protein by vortexing (120 s) followed by centrifugation (15,000×g × 20 min) at room temperature. The supernatant was transferred to clean vials for sample injection. A calibration curve was prepared with 70 μL blank cell lysate samples and 10 μL UA methanol solutions at different concentrations by the same method. The protein levels were assayed using a BCA protein assay kit according the kit protocol. The results were expressed as the amount (micrograms) of UA per milligram of total cell protein.
The LC-ESI-MS/MS analysis was performed on an Agilent 1200SL series (Agilent Technologies, USA) liquid chromatographer coupled with an Api-4000 Q-trap mass spectrometer (ABSciex, Foster City, CA), and a ZORBAX SB-C18 (4.6 × 50 mm, 5 μm, Agilent, USA) column was adopted for chromatographic separation. The isocratic mobile phase consisted of ACN and water (70:30, v/v) and was delivered at a rate of 0.5 mL/min. The injection volume was 4 μL, and the column was maintained at 35°C. The mass spectrometer was operated in the negative multiple reaction monitoring mode. The method validation was carried out in accordance with internationally accepted criteria (17). The linearity was evaluated using external calibration curves with more than seven calibration levels for each analyte prepared in triplicate. The limit of detection and the limit of quantitation of UA were 3.9 (S/N = 3) and 7.8 ng/mL (S/N = 10), respectively, and the linearity range was 7.8–2,000 ng/mL.
Cell Cycle Assay by Flow Cytometry Analysis
MCF-7 cells were incubated with either free UA or UA nanocrystals over a range of concentrations. After 24 h of treatment, the cells were harvested and fixed in 75% ethanol overnight at 4°C. The cells were then washed with cold PBS and incubated with RNase and propidium iodide for cell cycle analysis.
Statistical Analysis
Results were presented as the mean ± standard deviation. The statistical significance of the results was analysed using the two-tailed independent sample t test. Values of p < 0.05 were considered to be statistically significant.
RESULTS
Preparation and Characterisation of UA Nanocrystals
Effects of Process Parameters on Particle Size, PdI and Zeta Potential of the UA Nanocrystals
The particle size and stability of the UA nanocrystals were influenced by the type of the stabiliser applied. To select a suitable type of stabiliser to inhibit the ripening of the UA nanocrystals, six stabilisers were introduced into the aqueous solution at the same concentration (0.05%, w/v). As shown in Fig. 1a, the mean particle size of the freshly prepared nanocrystals were all smaller than 300 nm, and the PdI values were less than 0.25, which suggested that the systems were homogeneous. The order of the particle sizes (descending) were as follows: obtained with non-ionic stabilisers (∼190 to 240 nm), no stabiliser (∼185 nm), and obtained with SDS (∼120 nm). Figure 1b shows that the highest zeta potential value was observed in the UA nanocrystals obtained with SDS (−35 mV), and the zeta potential values of the UA nanocrystals obtained with non-ionic stabilisers were all lower than UA nanocrystals without stabiliser (−23 mV). The PdI value of a nanosuspension indicates the homogeneity of nanosuspension particle size. As shown in Fig. 1a, PdI values of samples obtained with SDS, F68, Tween 80 or HPMC E3 increased after storage at 4°C for 24 h, which did not correlated with an increase in particle size. We believe this increase reflected the instability of nansuspension most probably due to Ostwald ripening, which will happen when small crystals grow into larger ones. Though the time required for this process was not enough to be determined by dynamic light scattering (DLS), it was observed that nanosuspension with SDS as stabiliser precipitated after 3-day storage. However, the UA nanocrystals without stabiliser were stable after storage at 4°C for 7 weeks. Therefore, to eliminate the influence of stabilisers on the subsequent experiments, no stabilisers were used to prepare the UA nanocrystals.
Fig. 1.
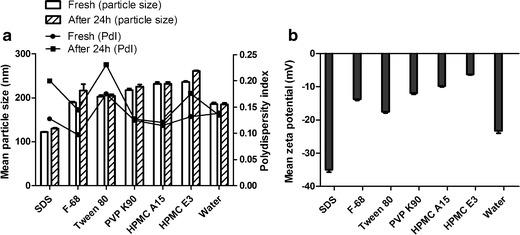
Effects of different stabilisers on (a) particle size and PdI, and (b) the zeta potential of the UA nanocrystals
The particle size of the UA nanocrystals was also influenced by the volume ratio of organic phase to aqueous solution (data not shown). Same amount of UA (6 mg) was respectively dissolved in 2, 3 and 4 mL ethanol and the organic phases were injected into 20 mL aqueous solution. When the ratio of organic phase to aqueous solution increased from 2:20 to 4:20, the particle size of sample significantly increased from 0.2 to 2.5 μm. These results indicated that the appropriate volume ratio of organic phase to aqueous solution was 2:20.
The effect of the UA concentration on both particle size and PdI was also investigated. Various amounts of UA were dissolved in 2 mL of ethanol to form a series of concentration range of UA (1–5 mg/mL). As shown in Fig. 2, the particle size increased from about 120 to 210 nm when the concentration of UA increased from 1 to 5 mg/mL, which indicated that the particle size was dependent on the drug concentration. After storage for 24 h at room temperature, there was no significant difference among the samples with UA concentration at 1–3 mg/mL in the organic phase, whereas both the particle size and PdI values increased when the UA concentration in organic phase was greater than 4 mg/mL. To prepare stable UA nanosuspension with high drug content in formulation, the UA concentration in the organic phase was selected at 3 mg/mL.
Fig. 2.

Effects of the UA concentrations on the particle size and PdI values of the UA nanocrystals
Based on the mentioned experimental results, stable UA nanocrystals without stabilisers were prepared as follows: UA was dissolved in ethanol to form organic phase with the UA concentration at 3 mg/mL, followed by injection of 2 mL of the organic phase into 20 mL of Milli-Q water at room temperature under stirring at 1,000 rpm for 5 min.
Characterisation of the UA Nanocrystals
DLS was used to determine the particle size and PdI of the UA nanocrystals. The mean particle size of the UA nanocrystals was 188.0 ± 4.4 nm and the PdI value was 0.154 ± 0.022, suggesting that the system was homogenous with a narrow particle size distribution. The mean zeta potential was −25.0 ± 5.9 mV. After storage for 7 weeks at 4°C, the particle size was 199.5 ± 9.2 nm, and the PdI was 0.169 ± 0.059, which suggested the samples were physically stable. The morphology of the UA nanocrystals was studied by TEM (Fig. 3). The UA nanocrystals were generally spherical in shape with a mean diameter of around 120–150 nm.
Fig. 3.
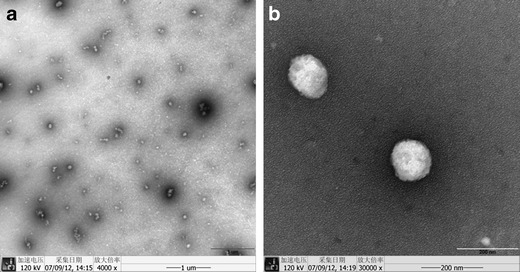
TEM images of the UA nanocrystals (a ×4,000 and b ×30,000)
Differential Scanning Calorimetry
DSC was used to assess the crystallinity of both the UA raw material and the nanocrystals (the powder obtained after freeze drying). As shown in Fig. 4, the endothermic peak of the UA raw material was 286.49°C, which was corresponding to the melting point of UA. However, the endothermic peak of the UA nanocrystals was slightly shifted to 281.97°C, and the enthalpy changed from 94.86 ± 2.60 J/g (n = 5) to 79.96 ± 5.13 J/g (n = 4), indicating that the crystallinity of UA nanocrystals reduced to 84% compared with UA raw material. There was an exothermic peak at 185–190°C in the DSC profile for both the raw material and formulated nanocrystals. To investigate the reason behind this exothermic peak, the UA raw material was heated to 220°C and held for 2 min followed by cooling to room temperature. This heat-treated UA powder was divided into two parts for DSC analysis and Fourier transform infrared spectroscopy (FTIR). The observed exothermic peak in the UA raw material was absent in the DSC profile of the heat-treated powder (data not shown). Compared with the raw material, the FTIR peak shift of the heat-treated powder can be clearly observed from 3,445 to 3,401 cm−1, and the peak of 1,700 cm−1 changed from single peak to triple peak. A weight loss of 5.39% (w/w) from the thermo-gravimetric analyser (TGA) curve was observed for the UA raw materials. Therefore, these results may suggest that the UA raw material exists in solvate crystalline form, which was recrystallised to a non-solvate crystalline form during the heating process. Further studies are necessary to elucidate the crystal structure of the UA and its possible polymorphism in solvents.
Fig. 4.

DSC curves of the UA raw material and the UA nanocrystals
Drug Release
When UA raw material was added into mixed solvent of water and ethanol to obtain coarse suspension, the UA powder floated on the solution, which suggested UA had poor wettability. After processed by anti-solvent precipitation, the UA nanocrystals uniformly distributed in solution, which indicated that the aqueous dispersibility of UA had been enhanced. Because UA is poorly dissolved in water, SDS was introduced into the dissolution medium to enhance the drug release (18). The dissolution profiles of the UA nanocrystals and UA raw material were compared in Fig. 5. The dissolution rate of the UA nanocrystals was much faster than that of raw material. Nearly 100% of the drugs from the UA nanocrystals were dissolved in 120 min, which is approximately twofold faster compared with those from the raw material groups under the same conditions.
Fig. 5.
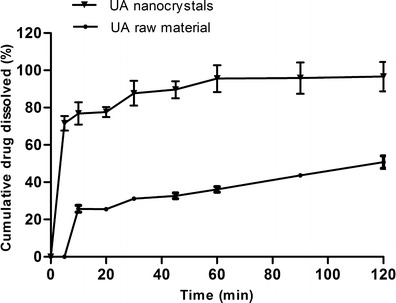
Dissolution profiles of the UA nanocrystals and the physical mixture
In Vitro Evaluation of Free UA and UA Nanocrystals in MCF-7 Cells
Cytotoxicity of the UA Nanocrystals and UA Solution
The cytotoxicity of UA solution and the UA nanocrystals was compared using an MTT assay with the MCF-7 cell line. As shown in Fig. 6, the cell cytotoxicity increased when either the concentration of UA was increased or the incubation time was prolonged for both the free UA groups and the UA nanocrystals groups. These data indicated that the cytotoxicity of UA against MCF-7 cells occurred in a concentration- and time-dependent manner. As shown in Table I, the IC50 values of the UA nanocrystals at 12 h, 24 h and 48 h were significantly lower than that of the free UA, which suggested that the cytotoxicity of the UA nanocrystals was significantly higher than that of free UA (P < 0.05).
Fig. 6.

Dose- and time-dependent growth inhibition of MCF-7 cells by free UA and the UA nanocrystals: a 12, b 24 and c 48 h. *p < 0.05 between free UA and the UA nanocrystals; **p < 0.01 between free UA and the UA nanocrystals
Table I.
IC50 Comparison of UA Solution and UA Nanocrystals
| Time (h) | UA solution (μmol/L) | UA nanocrystals (μmol/L) |
|---|---|---|
| 12 | 15.42 ± 1.19 | 7.90 ± 1.11 |
| 24 | 9.70 ± 1.05 | 5.39 ± 1.05 |
| 48 | 6.84 ± 1.03 | 3.17 ± 1.10 |
Cellular Uptake of the UA Nanocrystals and UA Solution
The effect of nanosizing on the uptake of UA was studied in MCF-7 cells. The cells were treated with either different concentrations of UA for the same incubation time or the same concentration of UA for the different incubation time. Figure 7 shows that the cells that were treated with a higher concentration of UA or with longer incubation time had a significant enhancement of the uptake of UA with both the UA nanocrystals and the UA solutions. Moreover, there was a significant difference in the cellular uptake between the UA nanocrystals and the UA solution (P < 0.05). The cellular uptake of the UA solution was approximately twofold higher than that of the UA nanocrystals under the same conditions.
Fig. 7.
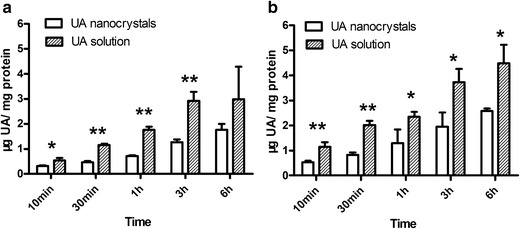
Cellular uptake of free UA and the UA nanocrystals by MCF-7 cells (a 5 and b 10 μmol/L). *p < 0.05 between free UA and UA nanocrystals; **p < 0.01 between free UA and UA nanocrystals
Cell Cycle Assay by Flow Cytometry Analysis
To confirm that the effects of UA on the proliferation of MCF-7 cells were mediated through arresting the cell cycle, the cell cycle phases were analysed by flow cytometry (Fig. 8). After treatment with free UA for 24 h, the percentage of G0/G1 phase cells markedly decreased while the percentage of cells within G2/M phase (14.05–14.59%) markedly increased compared with that of the control cells. Increasing the UA concentration had an additional effect on the distribution of MCF-7 cells in the cell cycle. As shown in Fig. 8, the UA nanocrystals induced higher cell cycle arrest in contrast to treatment with free UA at the same concentration. These results indicated that UA could arrest MCF-7 cells in the G2/M phase, and that the UA nanocrystals induced a higher percentage of cells to be arrested (25.44–29.36%) compared with free UA. Additionally, this arresting occurred in a concentration-dependent manner.
Fig. 8.

Effect of free UA and the UA nanocrystals on the proliferative cycle of MCF-7 cells: a Control, b 2 μM UA solution, c 5 μM UA solution, d 2 μM UA nanocrystals and e 5 μM UA nanocrystals
DISCUSSIONS
Generally, a stabiliser (either an ionic or non-ionic polymer) is an essential component for obtaining stable nanocrystals formulation. The stabiliser could inhibit agglomeration of the particles by electrostatic repulsion or/and steric stabilisation (19). In this study, the mean particle size of formulations obtained with non-ionic polymers increased whereas the absolute zeta potential values decreased compared with UA nanocrystals without any stabilisers. It appeared that the non-ionic polymers were effectively adsorbed to the surface of the UA nanocrystals to reduce the electrostatic repulsion. The same phenomenon was also observed in the preparation of camptothecin nanocrystals (20). Conversely, when SDS was used in the nanocrystals, the particle size decreased and the absolute zeta potential increased. It was suggested that the use of an ionic surfactant could effectively prevent the particle agglomerating by increasing electrostatic repulsion. However, it was reported that SDS had solubilising capacity for UA (21). Considering that the Ostwald ripening would be augmented in this system, this may explain why the system was unstable with a significantly increased PdI value after 24 h storage. Moreover, UA nanocrystals with good short-term physical stability without stabilisers have been successfully prepared; the stabilisation mechanisms of this process need to be elucidated in future studies.
In this study, the mean particle size increased when the ratio of organic phase to aqueous solution increased. When the ethanol proportion in the system increased, the aqueous solubility of UA was also increased, which may also enhance the Ostwald ripening process; as a consequence, the particle size increased. The particle size also increased when the drug concentration increased because more nuclei were formed at the interface of the two phases, which resulted in more straightforward aggregation. A similar result was also reported in the preparation of cefuroxime axetil nanoparticles (22).
There were an exothermic peak at around 190°C and an endothermic peak at around 280°C in the DSC curve of UA raw material, which was consistent with the report (18). Tong reported that the methanol and ethanol solvates of oleanolic acid (OA) underwent phase transformation during heating to a new crystalline phase that was similar to OA non-solvate at about 190–195°C (23). As UA and OA are isomers, their physical properties should be similar, and the structure of UA ethanol solvate has been reported (24). Therefore, it is expected that the exothermic peak of UA may be related to the recrystallisation of UA solvate into UA non-solvate. Further studies are necessary for confirmation of this hypothesis. Although the endothermic peak of the UA raw materials slightly shifted from 286.49°C to 281.97°C after being formulated into nanocrystals, the majority of the UA powders (84%) were maintained in a crystalline state, which was calculated by the enthalpy changes from 94.86 ± 2.60 J/g (n = 5) to 79.96 ± 5.13 J/g (n = 4) for the raw materials and nanocrystals, respectively (25). The reduction of Tm values in NP compared with raw material has been also reported previously, e.g. the Tm of nitrendipine nanocrystals (157.56°C) was lower than that of raw material (160.42°C) (26), and the Tm of spironolactone nanoparticle (203.4°C) was lower than that of raw material (207.9°C) (27). This reduced Tm may be due to the rapid appearance of abundant crystal nucleus upon solvent injection into anti-solvent, which did not allow the formation of perfect crystals.
The free UA and UA nanocrystals inhibited MCF-7 cell growth in both a concentration- and time-dependent manner, in which the IC50 value of free UA was approximately twofold higher than that of the UA nanocrystals. To eliminate the influence of ethanol in the UA nanocrystals, we attempted to remove the ethanol by separating the nanocrystals from the solution using centrifugal filters (10,000 MWCO). However, the UA nanocrystals agglomerated after the centrifugation process. Although the ethanol was not removed from the system, the MTT results showed that cell viability was ∼ 90% after incubation with medium containing the highest ethanol percentage (∼1.2%) to the lowest (∼0.015%) for 12 h, which demonstrated that the ethanol in nanocrystal system had no significant effect on the cytotoxicity (data not shown). Therefore, the higher cytotoxicity efficiency of the UA nanocrystals should be attributed to the nanosizing effect but not the ethanol residues. A similar phenomenon was also observed in tetrandrine-loaded nanoparticles against Lovo cells (28) and silybin nanosuspension against PC-3 cell (29), in which the anticancer activity of the drug nanocrystals were stronger than free drug.
To explain these results, a cellular uptake test was performed, because Zhang suggested that increased cellular uptake of drug nanoparticles may contribute to the stronger cytotoxicity compared with free drug (10). Contrary to our expectation, although the cells uptake for both of the UA nanocrystals and free UA were in a time- and concentration-dependent manner, the uptake of free UA was around twice higher than UA nanocrystals when the cells were treated with same concentration for the same incubation time. This phenomenon was different from experiments measuring camptothecin nanocrystals against MCF-7 cells (20) and oridonin nanosuspension against PANC-I cell (30), in which the uptake of the drug nanoparticles was higher than free drug. We hypothesised that because of the higher lipophilicity of UA (log P = 6.46) (31), it could be passively transported in a solubilised molecule, resulting in a percentage of UA embedding in the phospholipid bilayer of cell membrane and unable to contribute to the cytotoxicity in MCF-7 cells. However, drug in nanocrystals and kept in a crystal state may be taken up through endocytosis as nanoparticles (32). As a consequence, UA delivered as the nanocrystals could transport across the cell membrane and may be located within cells to produce cytotoxicity. Mo also suggested that before quantitative analysis of cellular uptake, one should investigate how the nanoparticles and free drug distributed in the cells to confirm that the nanoparticles and molecular drug have entered into the cells but not only adhesion or located on the cell membrane, as this phenomenon would interfere with the quantitative analysis of drug uptake (33). As UA nanocrystals have no fluorescence and could not be labelled by fluorescent marker because of a lack of polymer or lipid in the nanocrystals to encapsulate the marker, it is difficult to confirm the distribution of both free UA and the UA nanocrystals in the cells by confocal laser scanning microscopy.
Subsequently, we investigated whether the mechanism of the UA nanocrystals inhibiting the growth of MCF-7 cells was different from that of the free UA. Consistent with the MTT results, the data showed the induction of growth inhibition of MCF-7 cells by both of free UA solution and UA nanocrystals is due to the cell cycle arrest mechanism, in which UA nanocrystals could induce higher cell cycle arrest in contrast to the free UA. It has also been demonstrated previously the UA could also induce apoptosis to several cancer cell lines, such as HT-29 cells (34), SMMC-7721 (35) and A549 (7). It has been demonstrated that UA could induce cell cycle arrest and apoptosis on MCF-7 cells in a concentration-dependent manner via modulating glucocorticoid receptor and activator protein-1 (3,36).
CONCLUSIONS
In this study, a UA nanocrystal formulation without any stabilisers was prepared by the anti-solvent precipitation method. The stability test showed that the UA nanocrystals were physically stable after storage at 4°C for 7 weeks, despite the lack of stabiliser for preventing nanoparticle growth and/or agglomeration. This indicated that UA might be an appropriate model compound to investigate the effects of processing parameters on its particle size and stability, as well as how the nanocrystals interact with cells without interference of the excipient. Furthermore, the in vitro cytotoxicity assay demonstrated that the anti-proliferative efficiency of the UA nanocrystals was twofold higher than that of free UA although the cellular uptake of the UA nanocrystals was remarkably lower than that of free UA. In summary, a stable UA nanocrystal formulation without stabiliser has been successfully developed, which may be used as a potential delivery system for UA with enhanced aqueous dispensability, dissolution velocity and anticancer activity.
ACKNOWLEDGEMENTS
This work was supported by the Macao Science and Technology Development Fund (no. 044/2011/A2) and the research grant by University of Macau (MRG002/ZY/2012/ICMS). We would like to thank Professor Albert Chow, Dr. SF Chow and Dr. XR Zhang at the Chinese University of Hong Kong, and Dr. XP Chen and Mr. JB Sun at our institute for their valuable advice on the project and help with the TGA analysis.
Abbreviations
- UA
Ursolic acid
- HPMC
Hydroxypropyl methylcellulose
- SDS
Sodium dodecyl sulfate
- Tween 80
Polysorbate 80
- F68
Poloxamer 188
- PVP
Polyvinylpyrrolidone
- TEM
Transmission electron microscope
- DSC
Differential scanning calorimetry
- PdI
Polydispersity index
- DLS
Dynamic light scattering
References
- 1.Liu J. Pharmacology of oleanolic acid and ursolic acid. J Ethnopharmacol. 1995;49:57–68. doi: 10.1016/0378-8741(95)90032-2. [DOI] [PubMed] [Google Scholar]
- 2.Achiwa Y, Hasegawa K, Udagawa Y. Regulation of the phosphatidylinositol 3-kinase-Akt and the mitogen-activated protein kinase pathways by ursolic acid in human endometrial cancer cells. Biosci, Biotechnol, Biochem. 2007;71:31–37. doi: 10.1271/bbb.60288. [DOI] [PubMed] [Google Scholar]
- 3.Kassi E, Sourlingas TG, Spiliotaki M, Papoutsi Z, Pratsinis H, Aligiannis N, et al. Ursolic acid triggers apoptosis and Bcl-2 downregulation in MCF-7 breast cancer cells. Cancer Invest. 2009;27:723–733. doi: 10.1080/07357900802672712. [DOI] [PubMed] [Google Scholar]
- 4.Zheng QY, Jin FS, Yao C, Zhang T, Zhang GH, Ai X. Ursolic acid-induced AMP-activated protein kinase (AMPK) activation contributes to growth inhibition and apoptosis in human bladder cancer T24 cells. Biochem Bioph Res Co. 2012;419:741–747. doi: 10.1016/j.bbrc.2012.02.093. [DOI] [PubMed] [Google Scholar]
- 5.Song YH, Jeong SJ, Kwon HY, Kim B, Kim SH, Yoo DY. Ursolic acid from Oldenlandia diffusa induces apoptosis via activation of caspases and phosphorylation of glycogen synthase kinase 3 beta in SK-OV-3 ovarian cancer cells. Biol Pharm Bull. 2012;35:1022–1028. doi: 10.1248/bpb.b110660. [DOI] [PubMed] [Google Scholar]
- 6.Li J, Liang X, Yang X. Ursolic acid inhibits growth and induces apoptosis in gemcitabine-resistant human pancreatic cancer via the JNK and PI3K/Akt/NF-kappaB pathways. Oncol Rep. 2012;28:501–510. doi: 10.3892/or.2012.1827. [DOI] [PubMed] [Google Scholar]
- 7.Huang CY, Lin CY, Tsai CW, Yin MC. Inhibition of cell proliferation, invasion and migration by ursolic acid in human lung cancer cell lines. Toxicol In Vitro. 2011;25:1274–1280. doi: 10.1016/j.tiv.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 8.Xiong XJ, Chen W, Li K, Xu XQ. Study on the experiment of the poison of ursolic acid. J Yichun Med. 2001;13:54. [Google Scholar]
- 9.Zhou XJ, Hu XM, Yi YM, Wan J. Preparation and body distribution of freeze-dried powder of ursolic acid phospholipid nanoparticles. Drug Dev Ind Pharm. 2009;35:305–310. doi: 10.1080/03639040802302165. [DOI] [PubMed] [Google Scholar]
- 10.Zhang H, Li X, Ding J, Xu H, Dai X, Hou Z, et al. Delivery of ursolic acid (UA) in polymeric nanoparticles effectively promotes the apoptosis of gastric cancer cells through enhanced inhibition of cyclooxygenase 2 (COX-2) Int J Pharm. 2013;441:261–268. doi: 10.1016/j.ijpharm.2012.11.034. [DOI] [PubMed] [Google Scholar]
- 11.Müller RH, Gohla S, Keck CM. State of the art of nanocrystals—special features, production, nanotoxicology aspects and intracellular delivery. Eur J Pharm Biopharm. 2011;78:1–9. doi: 10.1016/j.ejpb.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Müller RH, Shegokar R, Gohla S, Keck CM. Nanocrystals: production, cellular drug delivery, current and future products. Intracell Deliver. 2011;5:411–432. doi: 10.1007/978-94-007-1248-5_15. [DOI] [Google Scholar]
- 13.Junghanns JU, Müller RH. Nanocrystal technology, drug delivery and clinical applications. Int J Nanomed. 2008;3:295–309. doi: 10.2147/ijn.s595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keck CM, Müller RH. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation. Eur J Pharm Biopharm. 2006;62:3–16. doi: 10.1016/j.ejpb.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 15.Chan HK, Kwok PC. Production methods for nanodrug particles using the bottom-up approach. Adv Drug Deliver Rev. 2011;63:406–416. doi: 10.1016/j.addr.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 16.Li W, Das S, Ng KY, Heng PW. Formulation, biological and pharmacokinetic studies of sucrose ester-stabilized nanosuspensions of oleanolic acid. Pharm Res. 2011;28:2020–2033. doi: 10.1007/s11095-011-0428-3. [DOI] [PubMed] [Google Scholar]
- 17.US FDA. Bioanalytical method validationin: guidance for Industry. Silver Spring, MD: FDA; 2001.
- 18.de Oliveira Eloy J, Saraiva J, de Albuquerque S, Marchetti JM. Solid dispersion of ursolic acid in gelucire 50/13: a strategy to enhance drug release and trypanocidal activity. AAPS Pharmscitech. 2012;13:1436–1445. doi: 10.1208/s12249-012-9868-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu L, Zhang J, Watanabe W. Physical and chemical stability of drug nanoparticles. Adv Drug Deliver Rev. 2011;63:456–469. doi: 10.1016/j.addr.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 20.Zhang H, Hollis CP, Zhang Q, Li T. Preparation and antitumor study of camptothecin nanocrystals. Int J Pharm. 2011;415:293–300. doi: 10.1016/j.ijpharm.2011.05.075. [DOI] [PubMed] [Google Scholar]
- 21.Jin IJ, Kim YM, Han SK. Solubilization of oleanolic acid and ursolic acid by cosolvency. Arch Pharm Res. 1997;20:269–274. doi: 10.1007/BF02976156. [DOI] [PubMed] [Google Scholar]
- 22.Zhang JY, Shen ZG, Zhong J, Hu TT, Chen JF, Ma ZQ, et al. Preparation of amorphous cefuroxime axetil nanoparticles by controlled nanoprecipitation method without surfactants. Int J Pharm. 2006;323:153–160. doi: 10.1016/j.ijpharm.2006.05.048. [DOI] [PubMed] [Google Scholar]
- 23.Tong HH, Wu HB, Zheng Y, Xi J, Chow AH, Chan CK. Physical characterization of oleanolic acid nonsolvate and solvates prepared by solvent recrystallization. Int J Pharm. 2008;355:195–202. doi: 10.1016/j.ijpharm.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 24.Simon A, Delage C, Saux M, Chulia AJ, Najid A, Rigaucl M. Sturcture of ursolic acid ethanol solvate. Acta Crystallogr, Sect C: Cryst Struct Commun. 1992;48:726–728. doi: 10.1107/S0108270191012581. [DOI] [Google Scholar]
- 25.Ali HS, York P, Ali AM, Blagden N. Hydrocortisone nanosuspensions for ophthalmic delivery: a comparative study between microfluidic nanoprecipitation and wet milling. J Control Release. 2011;149:175–181. doi: 10.1016/j.jconrel.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 26.Xia D, Quan P, Piao H, Piao H, Sun S, Yin Y, et al. Preparation of stable nitrendipine nanosuspensions using the precipitation -ultrasonication method for enhancement of dissolution and oral bioavailability. Eur J Pharm Sci. 2010;40:325–334. doi: 10.1016/j.ejps.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 27.Dong Y, Ng WK, Shen S, Kim S, Tan RB. Preparation and characterization of spironolactone nanoparticles by anti-solvent precipitation. Int J Pharm. 2009;375:84–88. doi: 10.1016/j.ijpharm.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Zhen D, Lu X, Xu H, Shao Y, Xue Q, et al. Enhanced cytotoxicity and activation of ROS-dependent c-Jun NH2-terminal kinase and caspase-3 by low doses of tetrandrine-loaded nanoparticles in Lovo cells–a possible Trojan strategy against cancer. Eur J Pharm Biopharm. 2010;75:334–340. doi: 10.1016/j.ejpb.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 29.Zheng D, Wang Y, Zhang D, Liu Z, Duan C, Jia L, et al. In vitro antitumor activity of silybin nanosuspension in PC-3 cells. Cancer Lett. 2011;307:158–164. doi: 10.1016/j.canlet.2011.03.028. [DOI] [PubMed] [Google Scholar]
- 30.Qi X, Zhang D, Xu X, Feng F, Ren G, Chu Q, et al. Oridonin nanosuspension was more effective than free oridonin on G2/M cell cycle arrest and apoptosis in the human pancreatic cancer PANC-1 cell line. Int J Nanomed. 2012;7:1793–1804. doi: 10.2147/IJN.S29483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Claude B, Morin P, Lafosse M, Andre P. Evaluation of apparent formation constants of pentacyclic triterpene acids complexes with derivatized β- and γ-cyclodextrins by reversed phase liquid chromatography. J Chromatogr, A. 2004;1049:37–42. [PubMed] [Google Scholar]
- 32.Zhao F, Zhao Y, Liu Y, Chang X, Chen C, Zhao Y. Cellular uptake, intracellular trafficking, and cytotoxicity of nanomaterials. Small. 2011;7:1322–1337. doi: 10.1002/smll.201100001. [DOI] [PubMed] [Google Scholar]
- 33.Mo L, Hou L, Guo D, Xiao X, Mao P, Yang X. Preparation and characterization of teniposide PLGA nanoparticles and their uptake in human glioblastoma U87MG cells. Int J Pharm. 2012;436:815–824. doi: 10.1016/j.ijpharm.2012.07.050. [DOI] [PubMed] [Google Scholar]
- 34.Shan JZ, Xuan YY, Zheng S, Song Q, Zhang SZ. Ursolic acid inhibits proliferation and induces apoptosis of HT-29 colon cancer cells by inhibiting the EGFR/MAPK pathway. J Zhejiang Univ Sci B. 2009;10:668–674. doi: 10.1631/jzus.B0920149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu YX, Gu ZL, Yin JL, Chou WH, Kwok CY, Qin ZH, et al. Ursolic acid induces human hepatoma cell line SMMC-7721 apoptosis via p53-dependent pathway. Chinese Med J-Peking. 2010;123:1915–1923. [PubMed] [Google Scholar]
- 36.Wang JS, Ren TN, Xi T. Ursolic acid induces apoptosis by suppressing the expression of FoxM1 in MCF-7 human breast cancer cells. Med Oncol. 2012;29:10–15. doi: 10.1007/s12032-010-9777-8. [DOI] [PubMed] [Google Scholar]