

Abstract
Aprepitant (APR), a neurokinin 1 receptor antagonist, is an approved treatment for chemotherapy-induced nausea and vomiting and for post-operative nausea and vomiting. However, it has poor water solubility. This study was performed to optimize the capsule formulation of an inclusion complex of APR with sulfobutyl ether-β-cyclodextrin (SBE-β-CD), and to evaluate its water solubility, dissolution rate, and bioavailability. The complex was prepared through the saturated-aqueous solution method and then characterized by Fourier transform infrared spectroscopy, x-ray powder diffraction, and differential scanning calorimetry. Subsequently, a pharmacokinetic study was performed using liquid chromatography–tandem mass spectrometry. Emend, which features an innovative formulation that incorporates drug nanoparticles with high bioavailability, was used as a reference for comparison with the optimized formulation. As a result, the dissolution rates and extent of release of the test formulation in various media were enhanced relative to those of Emend. The bioavailability of the drug complex was comparable to that of Emend. In summary, the SBE-β-CD complexation could provide a practical and cost-effective option for enhancing the solubility and bioavailability of APR according to our research.
KEY WORDS: aprepitant, bioavailability, dissolution rate, inclusion complex, sulfobutyl ether-β-cyclodextrin
INTRODUCTION
Aprepitant (APR), an antiemetic drug that blocks the neurokinin 1 receptor, is used in preventing chemotherapy-induced nausea and emesis (1–3). Previous studies have demonstrated that APR is generally safe and well tolerated. It has been shown to offer benefits over other similar drugs in terms of reduction of nausea and sexual dysfunction (4). Early clinical data suggest that the projected efficacious human dose for APR is relatively high; thus, development of a more bioavailable formulation for APR could potentially reduce its required dose (5). At present, APR capsules of 40, 80, and 125 mg dosages are manufactured by Merck & Co., Inc., and are commercially available in the USA.
APR is a white to off-white, crystalline, non-hygroscopic solid with a melting point of 254°C (Fig. 1). As a weak basic compound, APR is relatively lipophilic (log P at pH 7 is 4.8) and poorly water soluble (3–7 μg/mL) at pH 2–10 (6). Early screening of the salt form has shown that all salts examined rapidly are disproportionate in water and have poor chemical stability (5). Because of its poor solubility, it is difficult to apply APR as a systemic and effective therapy using conventional formulations (7). Merck & Co., Inc. used particle-size reduction methodologies to increase the available surface area and enhance its rate of solubilization. The bioavailability of Emend (Merck & Co., Inc.) is about 65%. Despite the high bioavailability of Emend, efforts to increase the solubility and dissolution rate of APR have never been stopped because of the high cost of this drug.
Fig. 1.
Structural formula of aprepitant (molecular weight, MW = 534)
In the last decades, several methods have been applied to enhance the solubility of poorly soluble drugs. These methods include the use of surfactants (8) and solid dispersions (9), hot-melt extrusion technique (10), and cyclodextrin inclusion complexation (11). Various strategies may be formulated on the basis of the physicochemical characteristics of drug compounds to overcome the problem of low-water solubility. A feasible and commercially viable approach among the above methods is cyclodextrin (CD) inclusion complexation, which has been extensively used to enhance drug solubility, convert liquid drugs into microcrystalline powders, prevent drug–drug or drug–additive interactions, reduce gastrointestinal or ocular irritation, and reduce or eliminate unpleasant taste and smell in many pharmaceutical preparations (12). The inclusion complex involves molecular encapsulation of the guest molecule by the host CD molecule, which results in modification of the physicochemical properties, such as enhanced solubility and stability of the guest molecules (13).
Sulfobutyl ether-β-cyclodextrin (SBE-β-CD), a derivative of β-cyclodextrin (β-CD), has attracted a growing interest because of its high intrinsic solubility, which confers improved abilities to form complexes. In particular, SBE-β-CD has a high affinity for drugs that contain nitrogen or easily form positive charges because of its negatively charged side chain. Compared with β-CD and hydroxypropyl-β-cyclodextrin, SBE-β-CD appears to be a parenterally safer material with low kidney toxicity and low tendency to cause hemolysis. However, the formation of a complex for various CDs in water involves a process similar to the replacement of water molecules within the lipophilic central cavity by the drug molecule (14,15). Since its approval as a pharmaceutical excipient by the US Food and Drug Administration (US FDA), SBE-β-CD has been widely applied in various dosage forms such as injection, tablet, and capsule. The inclusion complex of ziprasidone with SBE-β-CD and its injectable preparation have been successfully developed and marketed by Pfizer.
The objective of this study was to optimize the oral formulation of the APR–SBE-β-CD complex and to compare the dissolution rate and bioavailability of its inclusion complex with those of Emend on the basis of a previous study by Ridhurkar et al. Fourier transform infrared (FTIR) spectroscopy, differential scanning calorimetry (DSC), and x-ray powder diffraction (XRPD) were used to characterize the complex. We also conducted a dissolution experiment using the dissolution medium required by the US FDA, as well as four other media, namely, aqueous HCl (pH 1.0), sodium acetate–acetic acid buffer solution (NaAc–HAc buffer, pH 4.5), phosphate-buffered solution (PBS, pH 6.8), and water. The bioavailability of the formulation in healthy beagles was also determined.
MATERIALS AND METHODS
Materials
APR used in the study was synthesized by our team (batch number: 20110619, 99.65% of purity). SBE-β-CD (degree of substitution = 7; molecular weight (MW) = 2,241) was provided by Macrocyclic Pharmaceutical Technology Development Co. (Nanjing, Jiangsu Province, China). Emend (the APR reference formulation; Merck & Co., Inc., NJ, USA) was procured from the market. Sodium dodecyl sulfate (SDS) and microcrystalline cellulose were purchased from Colorcon (Shanghai, China). All other chemicals and reagents were of analytical grade. Freshly prepared distilled water was used throughout the study.
Preparation and Optimization of the Inclusion Complex
The inclusion complex of APR with SBE-β-CD was prepared using the saturated-aqueous solution method. An accurately weighed amount of SBE-β-CD was dissolved in distilled water to obtain a colorless, transparent solution (10%, w/v). An equal volume of APR in ethanol (1.2%, w/v) was then added slowly to the solution. The pH of the resulting solution was adjusted to 2 using 1 M HCl and 0.1 M HCl. The solution was stirred (800 rpm) at 55°C for 7 h, after which, it became clearer and transparent. Finally, the solution was vacuum-dried to a constant weight at 50°C and about −0.1 MPa in a ZK-82BB vacuum-drying oven (Shanghai, China). An off-white and tasteless crystalline powder was obtained. All process parameters in the synthesis of the inclusion complex were optimized to obtain a powder with high-embedding ratio (ER, ER = ma/m0, ma is the amount of drug determined by high-performance liquid chromatography (HPLC) or UV–vis, m0 is the amount of drug added before the preparation of drug complex.). The test formulation in this method for a batch size of 1,000 capsules is summarized in Table I.
Table I.
Composition of Aprepitant Capsules (40 mg)
| Ingredient | Quantity (mg/capsule) | |
|---|---|---|
| Emend | Test formulation | |
| Aprepitant | 40 | 40 |
| SBE-β-CD | × | 336 |
| Microcrystalline cellulose | √ | 45 |
| Sodium dodecyl sulfate | √ | 20 |
| Sucrose | √ | × |
| Hydroxypropyl cellulose | √ | × |
| Capsule shell size | 1# | 0# |
√ the ingredient was added; × the ingredient was not added
The initial conditions for optimization were as follows: 50°C temperature, 5-h reaction time, 50% (v/v) concentration of aqueous ethanol (dissolution medium), 6.71% (w/v) concentration of aqueous SBE-β-CD, 1:4 APR/SBE-β-CD feed ratio, and constant pH value. When one of these variables was studied, the other variables were fixed. All of the conditions above were based on the structure and physicochemical properties of APR, as well data from various references. The feed ratio was investigated last as other variables were optimized.
Phase–Solubility Study
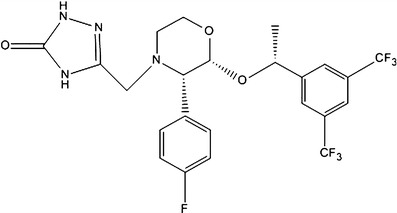
The phase–solubility study was carried out in distilled water according to the method of Higuchi and Connors (16). An excess amount of APR (40 mg) was mixed in an aqueous solution (10 mL) containing specific amounts of SBE-β-CD (0–10.0 mM). The mixtures were shaken for 24 h at 37°C in an oven-controlled oscillator (THZ-22, Taicang, Jiangsu, China). After the samples were allowed to attain equilibrium, they were filtered through a 0.45-μm membrane filter (Shanghai, China). The amount of solubilized APR was determined using a validated method based on HPLC (17). The apparent stability constant (Ks) of the complex was calculated from the slope of the linear portion of the phase–solubility diagram according to Eq. (1):
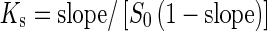 |
1 |
where S0 is the water solubility of APR. The complexation efficiency (CE) based on the drug (D)/cyclodextrin ratio was determined using Eqs. (2) and (3), as detailed below (16,18):
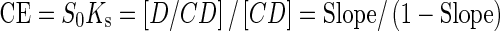 |
2 |
 |
3 |
where [D/CD] is the concentration of the drug complex and [CD] is the concentration of CD in the complex. The slope was obtained from the phase–solubility plot.
HPLC Analysis of the APR Content of the Complex
A suitable HPLC analytical method was developed for quantitating the amount of APR. The chromatographic system consisted of a quaternary pump (Shimadzu LC-20A intelligent pump with degasser), a variable-wavelength UV spectrophotometric detector (SPD-M20A), and a chromatographic data control and acquisition system (LC-PDA). A reversed-phase, Kromasil C18 column (particle size 5 μm, 250 × 4.6 mm i.d.) was used.
The mobile phase consisted of acetonitrile and PBS (70:30, v/v; 1.36-g KH2PO4 dissolved in 1,000-mL distilled water, adjusted to pH 7 with 1-M NaOH). It was filtered (0.45-μm polyvinylidene difluoride (PVDF) filter) and deaerated prior to use. The absorbance was monitored at 215 nm. The flow rate was 1.0 mL/min and the injection volume was 20 μL. The retention time of APR was found to be 5.34 ± 0.1 min.
FTIR Spectroscopy
The infrared (IR) spectrum of the inclusion complex was obtained according to the KBr disc method, using a Shimadzu FTIR-8 900 spectrometer (Shimadzu, Japan). For comparison, the IR spectra of pure APR, SBE-β-CD, as well as their physical mixture (APR/SBE-β-CD, 1:2 molar ratio, certain amounts of APR and SBE-β-CD were weighted and then mixed immediately with physical method) were also obtained through the same procedure. The scans were executed at a resolution of 4 cm−1 from 4,000 to 400 cm−1. Data acquisition was performed by Spectrum software version 5.0.2 (PerkinElmer Corp., USA).
XRPD Studies
The solid-state properties of APR, SBE-β-CD, the physical mixture, and the drug complex were studied by XRPD, using a Philips vertical scanning diffractometer (Type PW1729, Philips Electronic Instruments, Mount Vernon, NY, USA). A software package for the diffractometer was used to calculate the peak heights of all diffraction patterns. Samples were irradiated with monochromatized Cu Kα radiation and analyzed between 2θ angles of 3° and 40°. The analysis was carried out with an x-ray diffractometer at 40-kV voltage, a 40-mA current, and a scanning rate of 0.02°/min. The analysis was performed at room temperature under ambient conditions, and duplicate determinations were made for each of the samples.
DSC
DSC curves of APR, SBE-β-CD, the physical mixture, and drug complex were recorded on a Mettler TA 3000 differential scanning calorimeter (DSC 20; Greifensee, Switzerland). About 5–10 mg of sample was placed in a perforated, sealed 50-μL aluminum sample pan, and the temperature was increased from 40°C to 280°C at 10°C/min. An empty sealed pan was used as a reference. During the analysis, an inert environment was maintained using nitrogen gas. The instrument was periodically calibrated using pure metals with known melting points and heat of fusion (e.g., indium).
Dissolution Study
A USP type II apparatus (Paddles) was used to study the rate and extent of drug dissolution of the test and reference formulations at a stirring rate of 100 rpm. Degassed media (900 mL) were quantitatively transferred to individual bowls (n = 6). All of the samples were collected and then filtered (0.45 μm PVDF filter) at recommended sampling times (5, 10, 15, 20, 30, 45 and 60 min), and analyzed using the validated HPLC method. The temperature of the bath was maintained at 37°C. Apart from 2.2% aqueous SDS solution, HCl aqueous solution (pH 1.0), NaAc–HAc buffer (pH 4.5), PBS (pH 6.8), and water were also used as dissolution media at different stirring rates (50, 75, 100 rpm) in our research.
Evaluation of Pharmacokinetic Parameters
Protocol for the Animal Experiments
Eight purpose-bred beagle dogs housed in a USDA-approved facility in accordance with AAALAC guidelines were used to study the bioavailability of the drug complex and test formulation (19). The weights of all dogs ranged from 10 to 12 kg at the beginning of the studies. An equal high-calorie dinner was given to each dog 0.5 h before dosing. Standard laboratory feed and water were offered ad libitum 4 h after dosing. All studies were conducted using a randomized crossover design to eliminate the variability of absorption among the dogs with a wash-out period of 1 week. A total of 12 (1 × 5 mL) blood samples were collected from each dog according to the method of Wu et al. (19). The pre-dose (0.00 h) blood samples, which were obtained within 1.5 h prior to drug administration, and the post-dose samples at 15, 30 min, 1, 2, 4, 6, 8, 12, 24, 48, and 72 h were obtained by venipuncture. Blood was immediately transferred to a heparinized blood collection tube and stored in ice until use. The samples were centrifuged at 2,500 rpm at 5°C for 15 min. Afterwards, the upper plasma was transferred to polypropylene tubes and the samples were stored at −20°C until analysis by liquid chromatography–tandem mass spectrometry (LCMS/MS).
Bioanalytical Methods
A validated LCMS/MS method for the determination of APR in dog plasma was used for the analysis of the unknown samples. The analytical method for the determination of APR concentrations was based on liquid–liquid extraction of analyte and internal standard (IS) from the basified biological matrix using methyl t-butyl ether. The analog of APR (purchased from NICPBP; Fig. 2) was selected as IS. The organic extracts were collected and the extraction solvents were removed under a gentle flow of nitrogen. Chromatography was performed on a Keystone Scientific Prism RP analytical column (3 μm, 50 × 2 mm, Bellefonte, PA). The mobile phase for HPLC consisted of 40% acetonitrile and a 60% aqueous solution of 0.1% trifluoroacetic acid at pH 3. The flow rate of the mobile phase was 0.5 mL/min, and the total run time was 10 min. The retention times of APR and IS were 4.3 and 3.2 min, respectively. All analyte peaks were well separated from the baseline.
Fig. 2.
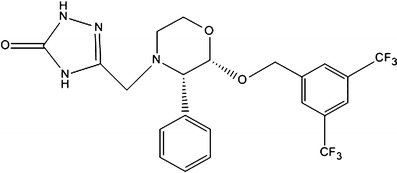
Structural formula of the internal standard (MW = 502)
A Sciex (API 3000) triple quadrupole mass spectrometer was interfaced with the HPLC system via a Sciex HN probe. The HN probe was maintained at 500°C and gas-phase chemical ionization was effected by positive ion atmospheric pressure chemical ionization using a corona discharge needle (+4 μA). The nebulizing gas (N2) pressure was set at 80 psi, and the sampling orifice potential was +35 V. The dwell time was 400 ms, and the temperature of the interface heater was set at 60°C. All mass analyzers were operated at unit mass resolution. For the triple quadrupole instrument peak areas, ratios obtained from multiple reaction monitoring of the analytes (m/z 535 → 277)/(503 → 259) were utilized for the construction of calibration curves for APR. Weighted (1/x2) linear least-squares regression of the plasma concentrations and measured peak area ratios were used for the quantification of both analytes. Data acquisition and analysis were performed by using the Analyst software version 1.4.2 (Applied Biosystems, Foster City, CA, USA).
Data analysis
The pharmacokinetic data were fitted using WinNonlin v.5.2.1 to calculate the area under the concentration–time curve (AUC), peak plasma concentration (Cmax), and peak time (Tmax). Statistical significance was set at P < 0.05. All of the other analyses were performed using Origin software version 8.6.
Stability Studies
Studies on the stability and performance of the test formulation were performed by storing samples at room temperature (25°C ± 2°C/60% ± 5% relative humidity (RH)) and at accelerated conditions (40°C ± 2°C/75% ± 5% RH) for 3 months. Sixty capsules (each contains 40-mg APR) in commercial packaging were used for each condition.
RESULTS AND DISCUSSION
Preparation and Optimization of the Inclusion Complex
As shown in Fig. 3, the best conditions for the preparation of the inclusion complex were 55°C temperature, 7-h reaction time, 50% (v/v) concentration of aqueous ethanol solution (dissolution media), 10% (w/v) concentration of aqueous SBE-β-CD solution, pH 2, and of 1:2 APR/SBE-β-CD feed ratio.
Fig. 3.
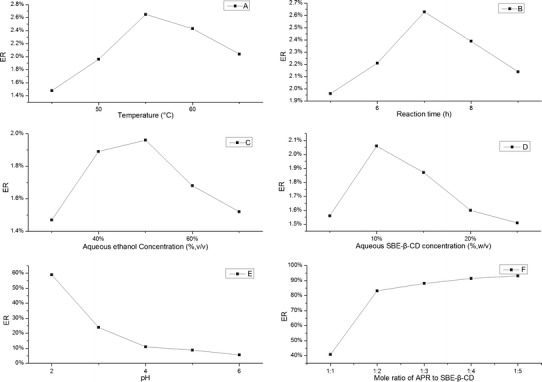
Optimization of the complex formulation
The most important factor in our experiment was the pH. It significantly affected the ER of APR, and it was rather difficult to obtain the dried powder at pH <2. Other variables such as temperature, reaction time, concentration of aqueous ethanol, and concentration of aqueous SBE-β-CD were secondary factors, but they were also important especially in mass production. The required amount of SBE-β-CD added should be as low as possible in order for it to be applicable to conventional formulations such as tablet and capsule. To achieve this goal, the feed ratio was investigated last as other variables were optimized. Not as the inclusion ratio, the feed ratio of CD/drug was usually too high and even reached 10:1. This was a great challenge for producing the pharmaceutical preparation. The SBE-β-CD/APR feed ratios varied widely (1:1, 3:1, 5:1, 10:1, and 15:1) in the preliminary experiment of our study. The ERs of APR only slightly increased when the SBE-β-CD/APR feed ratios were >3:1. Consequently, we adjusted the feed ratio (results of the adjustment are shown in Fig. 3f). The optimized process above could significantly enhance the utilization rate of SBE-β-CD and indirectly reduce its amount. With our method, the aqueous solubility of APR was increased to 924 μg/mL, which is more than 130 times its original solubility. Ridhurkar et al. have demonstrated that the water solubility of APR could be little improved by inclusion complex with β-CD. However, SBE-β-CD could achieve this goal and provide convenience for subsequent pharmaceutical preparation of APR.
Phase–Solubility Study
A phase–solubility study was performed by gently shaking centrifugal tubes containing an excess amount of APR and a SBE-β-CD solution (0 to 10.0 mM). The study allowed us to follow the inclusion phenomena and evaluate the apparent stability of the complex. Figure 4 shows the phase–solubility diagram of APR and SBE-β-CD after analyzing the filtrate of the mixture supernatant. The solubility of APR clearly increased with the increase in the SBE-β-CD concentration. The phase–solubility profile of APR-SBE-β-CD resulted in an Ap-type Higuchi phase–solubility diagram. The positive curvatures of the diagram indicate the existence of soluble complexes with an order >1 in CD. In other words, at the higher concentrations of SBE-β-CD, complexation between more than one SBE-β-CD molecule and one guest molecule (i.e., APR) was likely to have occurred. The slope of the linear portion of the diagram (slope was always lower than 1) and S0 were 5.66667 × 10−4 (r2 = 0.9905) and 0.01602 mM, respectively. The value of Ks and CE calculated from Eqs. (1) and (2) were 35.4 ± 1.2 M−1 and 5.67 × 10−4, respectively. Such result is reasonable according to previous studies (20).
Fig. 4.
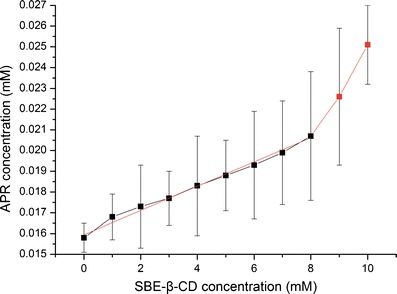
Phase–solubility diagram of APR–SBE-β-CD
FTIR Spectroscopy
In Fig. 5, the FTIR spectra of the four samples (APR, SBE-β-CD, physical mixture, and inclusion complex) are stacked for visual comparison. APR was identified by characteristic peaks at 1,703 cm−1 (C=O stretch); 1,510 cm−1 (C=C stretch); and 1,288 cm−1 (C–F stretch). As a derivative of β-CD, SBE-β-CD-produced absorption peaks at 3,421 cm−1 (O–H stretch), 2,938 cm−1 (C–H stretch), and 1,648 cm−1 (C=O stretch), which were similar to those of β-CD. The spectra of the physical mixture show no significant differences from the respective spectra of each of the pure components. However, there were varying degrees of reduction in characteristic peak intensity of APR, especially the absorption peaks due to C=O and C–F stretching vibrations. These changes suggest that some rings of APR were probably entrapped in the host cavities during the inclusion complexation. All of the results indicate that the vibration and bending of the APR molecule was restricted because of the formation of the inclusion complex. The inclusion complex APR–SBE-β-CD did not produce any new peaks, indicating that no chemical bonds were created in the formed complexes.
Fig. 5.
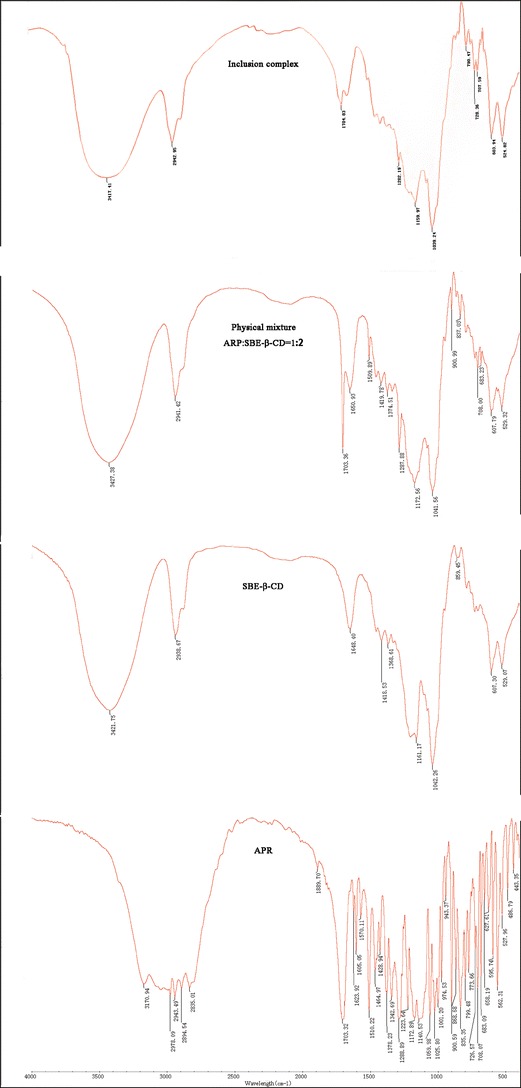
FTIR spectra of APR, SBE-β-CD, the physical mixture, and the drug complex APR–SBE-β-CD
XRPD Studies
XRPD patterns for APR, SBE-β-CD, the physical mixture, and the drug complex are depicted in Fig. 6. The diffraction pattern corresponding to APR is typical of crystalline materials, as it is characterized by a large number of sharp diffraction peaks. In contrast, the SBE-β-CD pattern shows only a weak peak, which is characteristic of an amorphous material. Some peaks in the physical mixture that could be attributed to the drug are visible, but their intensity is much lower because of the dilution process. The pattern for the drug complex reveals that the drug was no longer present in a crystalline form, consistent with the complete absence of its characteristic reflection. These results are in agreement with previous studies, and demonstrate that XRPD was a practical method to characterize the complexes.
Fig. 6.
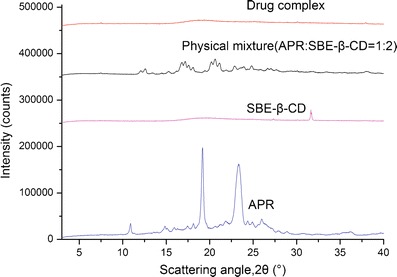
XRPD thermograms of APR, SBE-β-CD, the physical mixture, and the drug complex APR–SBE-β-CD
DSC Analysis
DSC could be used to characterize the inclusion complexes. When guest molecules were embedded into the CD cavities, their melting, boiling, and sublimating points generally shifted to different temperatures or disappeared (21). All of the DSC thermograms are presented in Fig. 7. APR produced a significant endothermic peak at 257.2°C, which is close to its melting point. The trace of SBE-β-CD shows a slight endothermic peak at 277.0°C and an inconspicuous broad endothermic effect (85°C–150°C) corresponding to dehydration. Both characteristic peaks of water loss in CD and drug melting are also visible in the thermograms of the physical mixture, but the melting endotherms are slightly shifted to 256.2°C and 275.4°C for APR and SBE-β-CD, respectively. These shifts may be due to an interaction between the two species during their mixing. During conversion to the inclusion complex, there was no sharp endothermic peak produced in the temperature range investigated. The amorphous character of the inclusion complex suggests that the crystalline APR molecule was contained within the ringlike cavity of the SBE-β-CD molecule (22).
Fig. 7.
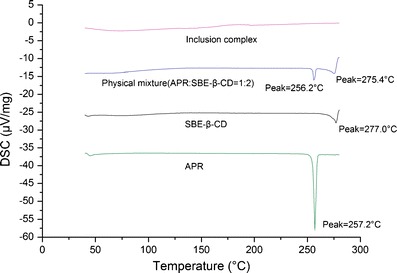
DSC patterns of APR, SBE-β-CD, the physical mixture, and the drug complex APR–SBE-β-CD
Dissolution Study
The dissolution profiles of the reference product and test formulation in various dissolution media (2.2% SDS solution, pH 1.0 HCl solution, pH 4.5 NaAc–HAc buffer, pH 6.8 PBS and water) are presented in Figs. 8, 9, 10, 11, and 12. The dissolution rate of the test formulation was enhanced relative to that of the reference product in 2.2% SDS solution and HCl solution. It even exceeded 85% in 15 min under stirring at 50 rpm (Figs. 8 and 9). This result is due to the solubilizing effect of the surfactant, the acidic conditions, and the complexation. Previous studies have demonstrated the effect of food on the absorption of APR. However, these studies ignored the significant difference in function of the gastrointestinal tracts of people of different ages; some patients produce insufficient amounts of gastric acid and bile (19). Therefore, we studied the dissolution profiles in various media at different stirring rates. Both the rate and extent of dissolution of the test formulation markedly increased compared with that of the reference product in NaAc–HAc buffer (Fig. 10); these changes in the dissolution profile may be attributed to the inclusion of APR in SBE-β-CD. The incomplete release of both test formulation and reference product in PBS (pH 6.8) and water are depicted in Figs. 11 and 12. This phenomenon is due to the partial degradation of the inclusion complex in the near-neutral pH environment of PBS and water. Even though the drug complex was released faster, the advantages (enhanced dissolution rate and extent) were not obvious. Higher stability of the drug complex may not be better; on the contrary, excessive stability may not be conducive to drug absorption. The five figures (Figs. 8, 9, 10, 11, and 12) describing the dissolution also suggests that food or other factors that could stimulate the secretion of gastric acid and bile would enhance the absorption of the drug. In addition, the influence of stirring rate weakened at lower pH; this trend suggests that, to some degree, the effect of intestinal peristalsis is more important than that of peristole for the absorption of APR. Actually, the results of dissolution profiles (Figs. 8, 9, 10, and 11) well suggested that the bioavailability of test formulation was really higher than that of the reference product. It should be noted that the above conclusions are based on the dissolution profiles rather than on bioequivalence studies.
Fig. 8.
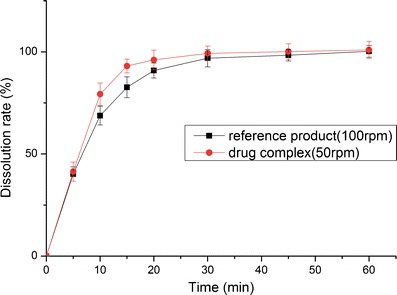
Dissolution profile of the test formulations and the reference product in 2.2% SDS solution
Fig. 9.
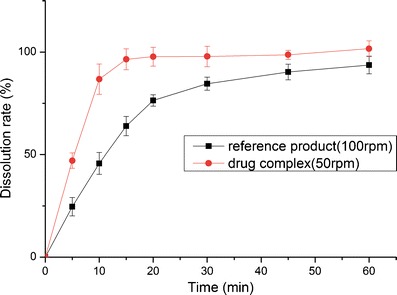
Dissolution profile of the test formulations and the reference product in HCl solution (pH 1.0)
Fig. 10.
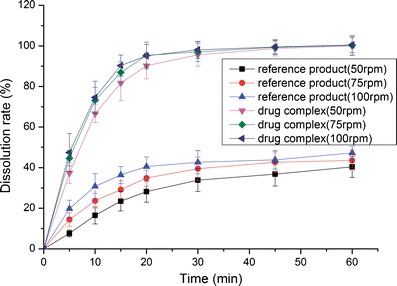
Dissolution profile of the test formulations and the reference product in NaAc–HAc buffer (pH 4.5)
Fig. 11.
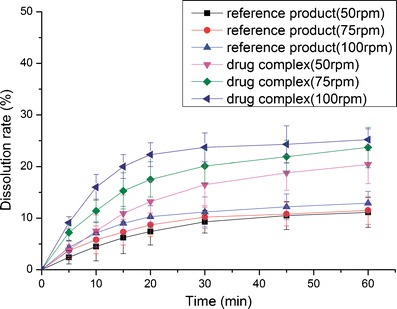
Dissolution profile of the test formulations and the reference product in PBS (pH 6.8)
Fig. 12.
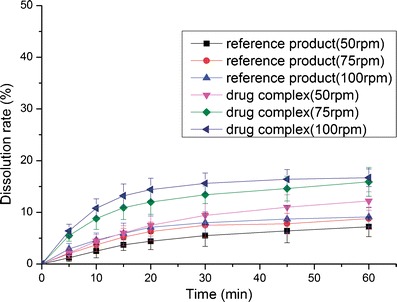
Dissolution profile of the test formulations and the reference product in water
Evaluation of Pharmacokinetic Parameters
Figure 13 shows the concentration–time curve following the oral administration of the reference product (Emend, 40 mg) and the test formulation. The corresponding pharmacokinetic (PK) parameters are summarized in Table II. Noncompartmental analysis was performed on the data. The mean extent of absorption, as indicated by AUC0–t and AUC0–∞, were 53,516 and 57,741 ng · h/mL, respectively, for the reference product. The mean AUC0–t and AUC0–∞ were 53,020 and 57,705 ng · h/mL, respectively, for the test formulation; these results indicate that the AUC0–t of the test formulation was comparable to that of the reference product. The Cmax of the reference product and test formulation were 2,383 and 2,413 ng/mL, respectively. These data further suggest that the drug had good gastrointestinal permeability, and that the absorption of the drug molecules was rapid as long as they were solubilized. The test/reference ratio for Cmax and AUC0–t (90% confidence interval) were within 80–125, which demonstrates the bioequivalence of the two formulations.
Fig. 13.
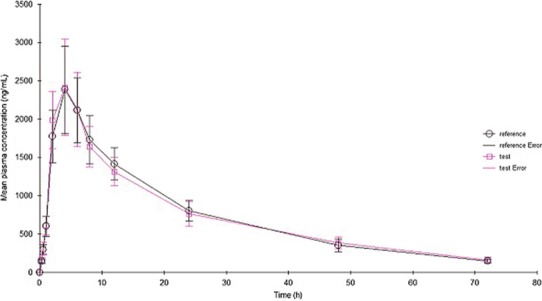
Mean plasma concentration–time profiles following oral administration of the drug complex and Emend (40 mg). Each value represents the mean ± SD (n = 8)
Table II.
In Vivo Pharmacokinetic (PK) Parameters Following Oral Administration of the Reference Product and Test Formulation in Healthy Male and Female Beagles
| Bioequivalence study (n = 8; 4 males and 4 females) | ||||||
|---|---|---|---|---|---|---|
| PK parameter | AUC0–t (ng · h/mL) | AUC0–∞ (ng · h/mL) | C max (ng/mL) | T max (h) | T 1/2 (h) | Kel (1/h) |
| Reference product | ||||||
| Mean | 53,516.01 | 57,741.37 | 2,383.70 | 5.17 | 23.15 | 0.043 |
| SD | 13,796.42 | 17,064.20 | 571.37 | 1.92 | 31.88 | 22.65 |
| Test formulation | ||||||
| Mean | 54,020.00 | 57,705.06 | 2,413.61 | 5.29 | 20.34 | 0.049 |
| SD | 14,389.63 | 21,858.65 | 626.60 | 2.44 | 45.27 | 36.41 |
AUC area under curve; C max peak concentration; T max peak time; T 1/2 half-life; Kel elimination rate constant; SD standard deviation
Stability Studies
The stability and dissolution profiles of the test formulation before and after storage are described in Table III. The content, amounts of related substances, and dissolution profiles of the product under both conditions were clearly in accordance with relevant standards. This indicates that the formulation was stable under the accelerated conditions.
Table III.
Stability and Dissolution Profiles of Test Formulation Following Storage at Room Temperature and Accelerated Conditions
| Parameter | Initial | 25°C ± 2°C/60% ± 5% RH | 40°C ± 2°C/75% ± 5% RH | ||||
|---|---|---|---|---|---|---|---|
| 1 month | 2 months | 3 months | 1 month | 2 months | 3 months | ||
| Assay (mg) | 40.34 | 40.12 ± 0.39 | 40.01 ± 0.47 | 39.76 ± 0.55 | 40.03 ± 0.51 | 39.87 ± 0.59 | 39.54 ± 0.66 |
| Diastereoisomer I | 0.07 | 0.07 ± 0.01 | 0.07 ± 0.02 | 0.07 ± 0.01 | 0.07 ± 0.02 | 0.07 ± 0.02 | 0.07 ± 0.02 |
| Diastereoisomer II | 0.03 | 0.03 ± 0.01 | 0.03 ± 0.01 | 0.03 ± 0.01 | 0.03 ± 0.01 | 0.03 ± 0.01 | 0.03 ± 0.02 |
| Diastereoisomer III | 0.09 | 0.09 ± 0.02 | 0.09 ± 0.02 | 0.09 ± 0.02 | 0.09 ± 0.01 | 0.09 ± 0.03 | 0.09 ± 0.03 |
| Single unknown maximum (M) | 0.18 | 0.18 ± 0.01 | 0.18 ± 0.01 | 0.18 ± 0.02 | 0.18 ± 0.01 | 0.18 ± 0.02 | 0.18 ± 0.02 |
| Total impurities | 0.37 | 0.37 ± 0.02 | 0.37 ± 0.02 | 0.37 ± 0.02 | 0.37 ± 0.02 | 0.37 ± 0.03 | 0.37 ± 0.03 |
| % Dissolved drug at 30 min (in 2.2% SDS) | 99.30 | 99.02 ± 3.38 | 98.27 ± 2.94 | 99.46 ± 4.21 | 98.15 ± 5.22 | 98.43 ± 4.27 | 98.03 ± 4.83 |
CONCLUSION
Instrumental analyses (FTIR spectroscopy, XRPD, and DSC) confirmed the existence of the inclusion complex of APR with SBE-β-CD prepared through the saturated-aqueous solution method. Furthermore, results of the analyses confirm encapsulation of APR in the cavity of SBE-β-CD rather than occurrence of chemical reactions. The pH value strongly influenced the ability of SBE-β-CD to form an inclusion complex with the drug. The phase–solubility study demonstrated that the SBE-β-CD/APR complexation ratio was greater than 1:1 when the concentration of CD was increased. Dissolution profiles suggest significant advantages (enhanced dissolution rate and extent in vitro) of the formulation of the inclusion complex over the reference product, especially for the elderly and patients with gastrointestinal dysfunction. The PK parameters Cmax and extent of drug absorption (as indicated by the AUC) for both formulations were comparable, according to the results of studies on systemic exposure in beagles. The stability studies under accelerated conditions demonstrated that the complex was stable. The content, amounts of related substances, and dissolution profiles of the product under both conditions complied with relevant standards. The results from our study demonstrate that the inclusion complex with SBE-β-CD could be used as a viable approach to develop a conventional dosage form with enhanced solubility, dissolution rate, and comparable bioavailability with the marketed product. However, we also found that it was difficult to achieve a high-dose drug administration with the APR–SBE-β-CD complex in conventional formulations such as tablet and capsule because of the high molecular weight of SBE-β-CD, and this is a great challenge for pharmaceutical preparation.
Acknowledgments
The Pharmaceutical Institute of Jiangsu Province (China) is acknowledged for its technical and instrumental assistance.
References
- 1.Cocquyt V, Van Belle S, Reinhardt RR, Decramer ML, O’Brien M, Schellens JH, et al. Comparison of L-758, 298, a prodrug for the selective neurokinin-1 antagonist, L-754, 030, with ondansetron for the prevention of cisplatin-induced emesis. Eur J Cancer. 2001;37:835–842. doi: 10.1016/S0959-8049(00)00416-0. [DOI] [PubMed] [Google Scholar]
- 2.Navari RM, Reinhardt RR, Gralla RJ, Kris MG, Hesketh PJ, Khojasteh A, et al. Reduction of cisplatin-induced emesis by a selective neurokinin-1-receptor antagonist. L-754, 030 Antiemetic Trials Group. N Engl J Med. 1999;340:190–195. doi: 10.1056/NEJM199901213400304. [DOI] [PubMed] [Google Scholar]
- 3.De Wit R, Hesketh PJ, Warr D, Petty K, Carides AD, Evans JK, et al. The oral NK1 antagonist aprepitant for prevention of nausea and vomiting in patients receiving highly emetogenic chemotherapy: a review. Am J Cancer. 2005;4:35–48. doi: 10.2165/00024669-200504010-00003. [DOI] [Google Scholar]
- 4.Keller M, Montgomery S, Ball W, Morrison M, Snavely D, Liu GH, et al. Lack of efficacy of the substance P(Neurokinin 1 Receptor) antagonist aprepitant in the treatment of major depressive disorder. Biol Psychiatry. 2006;59:216–223. doi: 10.1016/j.biopsych.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 5.Olver I, Shelukar S, Thompson KC. Nanomedicines in the treatment of emesis during chemotherapy: focus on aprepitant. Int J Nanomedicine. 2007;2:13–18. doi: 10.2147/nano.2007.2.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shono Y, Jantratid E, Kesisoglou F, Reppas C, Dressman JB. Forecasting in vivo oral absorption and food effect of micronized and nanosized aprepitant formulations in humans. Eur J Pharm Biopharm. 2010;76:95–104. doi: 10.1016/j.ejpb.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Grunberg SM, Deuson RR, Mavros P, Geling O, Hansen M, Cruciani G, et al. Incidence of chemotherapy-induced nausea and emesis after modern antiemetics. Cancer. 2004;100:2261–2268. doi: 10.1002/cncr.20230. [DOI] [PubMed] [Google Scholar]
- 8.Müller RH, Jacobs C, Kayser O. Nanosuspensions as particulate drug formulations in therapy: rationale for development and what we can expect for the future. Adv Drug Deliv Rev. 2001;47:3–19. doi: 10.1016/S0169-409X(00)00118-6. [DOI] [PubMed] [Google Scholar]
- 9.Urbanetz NA, Lippold BC. Solid dispersions of nimodipine and polyethylene glycol 2000: dissolution properties and physico-chemical characterisation Original Research Article. Eur J Pharm Biopharm. 2005;59:107–118. doi: 10.1016/j.ejpb.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 10.Singhal S, Lohar VK, Arora V. Hot melt extrusion technique. WebmedCentral Pharm Sci. 2011;2(1).
- 11.Zingone G, Rubessa F. Preformulation study of the inclusion complex warfarin-β-cyclodextrin. Int J Pharm. 2005;291:3–10. doi: 10.1016/j.ijpharm.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 12.Loftsson T, Brewster ME. Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J Pharm Sci. 1996;85:1017–1025. doi: 10.1021/js950534b. [DOI] [PubMed] [Google Scholar]
- 13.Robert O, Williams III, Mahaguna V, Sriwongjanya M. Characterization of an inclusion complex of cholesterol and hydroxypropyl-β-cyclodextrin. Eur J Pharm Biopharm. 1998;46:355–360. doi: 10.1016/S0939-6411(98)00033-2. [DOI] [PubMed] [Google Scholar]
- 14.Waleczek KJ, Cabral Marques HM, Hempel B, Schmidt PC. Phase solubility studies of pure (-)-α-bisabolol andcamomile essential oil with β-cyclodextrin. Eur J Pharm Biopharm. 2003;55:247–251. doi: 10.1016/S0939-6411(02)00166-2. [DOI] [PubMed] [Google Scholar]
- 15.Liu LX, Zhu SY. Preparation and characterization of inclusion complexes of prazosin hydrochloride with β-cyclodextrin and hydroxypropyl-β-cyclodextrin. J Pharm Biomed Anal. 2006;40:122–127. doi: 10.1016/j.jpba.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 16.Higuchi T, Connors KA. Phase solubility techniques. Adv Anal Chem Instrum. 1965;4:117–212. [Google Scholar]
- 17.Ridhurkar DN, Ansari KA, Kumar D, Kaul NS, Krishnamurthy T, Dhawan S, Pillai R. Inclusion complex of aprepitant with cyclodextrin: evaluation of physico-chemical and pharmacokinetic properties. Drug Dev Ind Pharm. 2013;39(11):1783–1792. doi: 10.3109/03639045.2012.737331. [DOI] [PubMed] [Google Scholar]
- 18.Loftsson T, Hreindótter D, Másson M. The complexation efficiency. J Incl Phenom Macrocycl Chem. 2007;57:545–552. doi: 10.1007/s10847-006-9247-2. [DOI] [Google Scholar]
- 19.Wu YH, Loper A, Landis E, Hettrick L, Novak L, Lynn K, et al. The role of biopharmaceutics in the development of a clinical nanoparticle formulation of MK-0869: a Beagle dog model predicts improved bioavailability and diminished food effect on absorption in human. Int J Pharm. 2004;285:135–146. doi: 10.1016/j.ijpharm.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 20.Fernandes CM, Teresa Vieira M, Veiga FJ. Physicochemical characterization and in vitro dissolution behavior of nicardipine-cyclodextrins inclusion compounds. Eur J Pharm Sci. 2002;15:79–88. doi: 10.1016/S0928-0987(01)00208-1. [DOI] [PubMed] [Google Scholar]
- 21.Liu LX, Zhu SY. A study on the supramolecular structure of inclusion complex of β-cyclodextrin with prazosin hydrochloride. Carbohydr Polym. 2007;68:472–476. doi: 10.1016/j.carbpol.2006.11.007. [DOI] [Google Scholar]
- 22.Loukas YL, Vraka V, Gregoriadis G. Drugs in cyclodextrins, in liposomes: a novel approach to the chemical stability of drugs sensitive to hydrolysis. Int J Pharm. 1998;162:137–142. doi: 10.1016/S0378-5173(97)00421-3. [DOI] [Google Scholar]