

Abstract
Streptococcus pneumoniae is a major human pathogen, and a leading cause of disease and death worldwide. Pneumococcal invasive disease is triggered by initial asymptomatic colonization of the human upper respiratory tract. The pneumococcal serine-rich repeat protein (PsrP) is a lung-specific virulence factor whose functional binding region (BR) binds to keratin-10 (KRT10) and promotes pneumococcal biofilm formation through self-oligomerization. We present the crystal structure of the KRT10-binding domain of PsrP (BR187–385) determined to 2.0 Å resolution. BR187–385 adopts a novel variant of the DEv-IgG fold, typical for microbial surface components recognizing adhesive matrix molecules adhesins, despite very low sequence identity. An extended β-sheet on one side of the compressed, two-sided barrel presents a basic groove that possibly binds to the acidic helical rod domain of KRT10. Our study also demonstrates the importance of the other side of the barrel, formed by extensive well-ordered loops and stabilized by short β-strands, for interaction with KRT10.
Keywords: bacterial virulence factor, PsrP, Streptococcus pneumoniae, adhesion, MSCRAMM, keratin-10
2. Introduction
Streptococcus pneumoniae (pneumococcus) is a human-adapted, Gram-positive commensal bacterium that colonizes the upper respiratory tract in about 10% of healthy adults and up to 60% of children. Although normally not causing any symptoms, pneumococcus is a major human pathogen, and a leading cause of disease and death worldwide [1]. Streptococcal antigenicity is determined to a large extent by the structure and contents of the outermost layer of the cell, including a variety of proteins with differing functions localized within the polysaccharide capsule [1,2]. Surface-associated adhesins play a pivotal role for pneumococcal colonization of the nasopharynx and for the development of infectious pneumococcal disease through interactions with specific cellular surface structures in the host [3].
The pneumococcal serine-rich repeat protein (PsrP) is an important lung-specific virulence factor that is present in 60% of strains capable of causing pneumonia in children [2]. The C-terminal cell wall anchoring domain of PsrP contains an LPxTG motif that is covalently anchored to the peptidoglycan by Sortases [4]. A characteristic feature of the serine-rich repeat protein (SRRP) family is the presence of a long, highly repetitive and glycosylated C-terminal serine-rich repeat (SRR) region (figure 1a) that can vary between 400 and 4000 residues [5]. The size of the possibly super-helical SRR region might correlate with the capsule thickness of each species, extending the highly basic functional binding region (BR) domain of each SRRP out of the capsule [5–8]. The sequence of the BR domain, which includes the N-terminal SRR1 and the longer C-terminal SRR2 regions, is extremely variable among all known SRRPs, which could account for the broad range of targets bound by this adhesin family [5,8]. Pneumococcal PsrP promotes both biofilm formation through self-oligomerization and adherence to keratin 10 (KRT10)-expressing lung epithelial cells. These disparate functions are facilitated by two distinct regions within the surface-exposed BR domain [7,9].
Figure 1.

Crystal structure of the KRT10-binding region of PsrP (BR187–385). (a) PsrP is organized into five domains, comprising the N-terminal signal sequence (S) for export of PsrP to the extracellular surface, two serine-rich repeat regions (SRR1 and SRR2), the binding region (BR) domain and the cell wall (CW) domain. The basic BR domain harbours two distinct subregions for KRT10-binding (black bar, residues 273–341) and self-oligomerization (black bar, residues 122–166), respectively. BR187–385 contains only the KRT10-binding subregion. (b) Two orientations of the BR187–385 crystal structure are presented as ribbon diagrams with β-strands, α-helices and loops in red, green and white, respectively. While one side of the compressed barrel of BR187–385 is created by an extended and twisted antiparallel β-sheet, the other side is formed by well-ordered loops and two sets of β-sheet belts.
Keratins (KRTs) are intermediary filament (IF) proteins that are mainly regarded as intracellular constituents of the cytoskeleton [10]. More than 50 distinct human KRT genes are expressed in a highly cell-type- and cell-differentiation-state-dependent manner [11]. All KRTs exhibit a tripartite structure characterized by a long α-helical rod domain flanked by an amino- and a carboxy-terminal non-α-helical end domain. The secondary structure of the rod domain, which is highly conserved among IF proteins, is divided into four heptad-repeat-containing helical segments called 1A, 1B, 2A and 2B, which are interrupted by three short linker sequences L1, L12 and L2 [10]. The heptad-repeat-containing segments form the structural basis for the heteromeric assembly of KRT filaments [12]. For example, the acidic type-I KRT-10 and the basic type-II KRT-1 form an obligate heterodimer that is the main building block in filament assembly.
It has been previously demonstrated that KRTs are also readily available on the surface of epithelial cells, acting as potential surface-accessible docking sites for microbial adhesins. While the Staphylococcus aureus-derived adhesin clumping factor B (ClfB) interacts with KRT10 and possibly KRT8 on the surface of desquamated stratified squamous epithelial cells isolated from human nares [13,14], the Streptococcus agalactiae-derived SRR-1 protein interacts with KRT4 on the surface of human laryngeal carcinoma-derived Hep2-cells [15].
In the structurally and mechanistically well-described ‘dock, lock and latch’ binding mode of the microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) ClfB to KRT10, a peptide derived from the tail of KRT10 ‘docks’ into a binding trench localized between the two homologous subdomains N2 and N3 of ClfB, and undergoes a disorder-to-order transition by complementing a β-sheet within N3. The C-terminal extension of N3 is thereafter redirected in order to ‘lock’ the KRT10 peptide in place and to form a ‘latch’ through β-sheet complementation with N2 [16,17].
Both the N2 and N3 domains of ClfB display the DE-variant of the IgG fold (DEv-IgG) that has been described for the A-region of the S. aureus-derived CNA protein [18,19]. Interestingly, CnaA subdomains with similar topology have been identified in the two other available crystal structures of SRRPs, Fap1 and GspB, derived from Streptococcus parasanguinis and Streptococcus gordonii, respectively [6,8]. The presence of a CnaA-subdomain has also been predicted for the Streptococcus agalactiae-derived KRT4-binding SRRP SRR-1 [8]. However, the topology for the BR domain of PsrP could not be predicted owing to missing sequence homology. In this study, the crystal structure of the KRT10-binding region of PsrP (BR187–385) was determined, revealing a novel fold distantly related to CnaA subdomains. While one face of the compressed, two-sided barrel of BR187–385 is created by an extended β-sheet that presents a highly basic binding groove, extensive well-ordered loop regions distort the other face of the barrel, forming a paperclip-like substructure. In vitro alanine substitution of residues localized within this paperclip structure efficiently disrupted BR187–385/KRT10 complex formation.
3. Results and discussion
3.1. The crystal structure of monomeric BR187–385 presents a compressed β-barrel fold with two remarkably different faces
BR187–385 crystallized in two crystal forms of the space groups P43212 and P4122, with differing unit cell parameters (table 1). Single anomalous dispersion (SAD) data, collected from a seleno-methionine derivative of the P43212 crystal form that diffracted to 2.25 Å, was used to solve the phase problem. Three BR187–385 polypeptide chains were placed in the asymmetric unit, and refined to R- and Rfree-values of 18.6% and 21.5%, respectively. The native P4122 dataset that diffracted to 2.0 Å was solved using BR187–385 from P43212 as a template for molecular replacement (MR). A single BR187–385 molecule was found in the P4122 asymmetric unit, and a model comprising residues L203–S378 was refined to R- and Rfree-values of 17.7% and 20.1%, respectively. The structural deviation between the P4122-BR187–385 monomer and each of the three P43212-BR187–385 molecules was minimal, with root mean square deviations (r.m.s.d.) of 0.6 Å and less than 0.4 Å, following superposition of the P4122 monomer on the P43212 chains A and B/C, respectively.
Table 1.
X-ray data collection and refinement statistics.
| SeMet | native | |
|---|---|---|
| data collection | ||
| space group | P43212 | P4122 |
| unit cell parameters (a, b, c in Å; α, β, γ in degree) | a = 105.6; b = 105.6; c = 120. 5; α = β = γ = 90 | a = 74.5; b = 74.5; c = 121.2; α = β = γ = 90 |
| X-ray source | ESRF ID-29 | ESRF ID-29 |
| detector | Pilatus 6M | Pilatus 6M |
| temperature (K) | 100 | 100 |
| resolution limits (Å) | 48.35–2.25 (2.31–2.25) | 48.32–2.00 (2.05–2.00) |
| wavelength (Å) | 0.979 | 0.977 |
| no. of observations | 283775 (20035) | 296169 (21744) |
| no. of unique reflections | 61680 (4563) | 44053 (3252) |
| redundancy | 4.6 (4.4) | 6.7 (6.7) |
| completeness (%) | 100 (100) | 100 (100) |
| I/σ | 14.83 (3.18) | 18.18 (2.96) |
| Rsym (%) | 6.5 (48.7) | 6.3 (69.5) |
| Rmeas (%) | 7.3 (55.5) | 6.9 (75.4) |
| Rmrgd-F (%) | 10.6 (52.8) | 7.4 (53.5) |
| phasing | ||
| software | SHELX [20] | |
| resolution limit | 20–2.25 | |
| no. of sites found | 12 | |
| CC all/weak | 60.46/40.35 | |
| FOM after SHELXE | 0.707 | |
| FOM after DM | 0.904 | |
| structure refinement | ||
| PDB entry | 3ZGI | 3ZGH |
| molecules in ASU | 3 | 1 |
| mask estimated solvent (%) | 56 | 68 |
| Rwork (%) | 18.6 | 17.7 |
| Rfree (%) | 21.5 | 20.1 |
| no. of residues | ||
| protein | 522 | 177 |
| water | 146 | 114 |
| small molecule | 6 | 2 |
| other | 1 | 4 |
| no. of atoms | 4171 | 1494 |
| mean isotropic B-value (Ų) | 51.4 | 42.5 |
| Wilson B-factor (Ų) | 36.1 | 35.3 |
| r.m.s.d. from ideal bond lengths (Å) | 0.01 | 0.012 |
| r.m.s.d. from ideal bond angle (°) | 1.47 | 1.417 |
| molprobity | ||
| Ramachandran | ||
| outliers (%) | 1.4 | 0.6 |
| favoured (%) | 96 | 95 |
| all atom clash score (percentile) | 6.44 (98th) | 8.44 (89th) |
| molprobity score (percentile) | 1.65 (97th) | 1.87 (82nd) |
The overall three-dimensional structure of BR187–385, composed of 43% β-strands, 2% α-helices, 17% turns and 38% loop regions, can be described as a compressed barrel with two remarkably different faces (figure 1b). While one side of the barrel forms an extended and twisted antiparallel β-sheet that comprises the six strands A1, A, B, E, D and D1, the other side mainly consists of well-ordered loops, each stabilized by two sets of β-sheet belts, comprising strands D2, D3, C2, F1, G and D4, C1, F2, respectively. Furthermore, the highly ordered loops are also stabilized by several β-turns and hairpin motifs (data not shown).
Crystal packing analysis revealed that two symmetry-related molecules in the P4122 crystal formed an intermolecular β-sheet resulting in an interface area of 585 Å2. The same interface was also observed for chains B and C in the P43212 crystal form. However, a single population with a sedimentation coefficient of 1.85 S corresponding to a monomer with a hydrodynamic radius of 25 Å was clearly assessed using analytical ultracentrifugation (AUC; figure 2a). A similar hydrodynamic radius value was also derived from the retention volume of the BR187–385 monomer using size exclusion chromatography (SEC; data not shown). Finally, small angle X-ray scattering (SAXS) analysis of BR187–385 revealed a monomer in solution with a molecular weight estimated from the forward scattering I(0) to 18 ± 2 kDa and from the Porod volume to 23 ± 2 kDa (expected at 22 kDa; figure 2b; electronic supplementary material, table S1). While the radii of gyration Rg obtained from the Guinier approximation and from the distance distribution function p(r) were 22.7 ± 1.2 Å and 22.5 ± 2.0 Å, respectively, the Dmax value was 77.0 ± 8.0 Å. It should be noted that the extended p(r) function suggested a partially unfolded protein. Furthermore, fitting of the experimental data using the ensemble optimization method (EOM) also indicated the formation of a globular envelope with N- and C-terminal extensions (see electronic supplementary material, figure S1). The ensemble of 18 monomer models yielded a theoretical average sedimentation coefficient of 1.94 ± 0.06 S, in good agreement with the AUC analysis (figure 2a).
Figure 2.
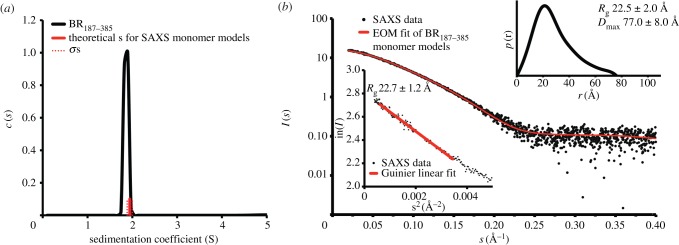
BR187–385 is a monomer in solution. (a) The continuous distribution of sedimentation coefficients reveals a single population for the BR187–385 monomer with a sedimentation coefficient of 1.85 S. The averaged theoretical sedimentation coefficients calculated for the SAXS-derived BR187–385 monomer models are in agreement with the experimentally determined value. The averaged value and the standard deviation are in red and dotted red, respectively. (b) The theoretical scattering curve obtained for an ensemble of 18 BR187–385 monomer models (see also electronic supplementary material, figure S1) fits the SAXS profile of BR187–385 with a χ2-value of 0.58. The radius of gyration Rg obtained from the Guinier approximation was 22.7 ± 1.2 Å (mean ± s.d.). The distance distribution function p(r) gave Rg and Dmax values of 22.5 ± 2.0 Å and 77.0 ± 8.0 Å, respectively.
In conclusion, the crystal structure of the BR187–385 monomer takes a compressed barrel fold with one face formed by an extended and twisted β-sheet, and the other face mainly consisting of well-ordered loops. We believe, at the present stage, that the formed pseudo-complexes are probably owing to crystal packing.
3.2. The KRT10-binding region domain of PsrP adopts a novel MSCRAMM fold-variant
A search for structural homologues revealed that BR187–385 adopts an MSCRAMM-related DEv-IgG fold (figure 3). The typical DEv-IgG fold topology can be described as a compressed barrel composed of two opposing β-sheets that are formed by β-strands ABED (sheet I) and CFG (sheet II) [19]. The insertion of two extra strands between strands D and E distinguishes the DEv-IgG variant from the IgG-constant domain [22]. BR187–385 takes a novel DEv-IgG fold variant with one side of the barrel distorted by loops and β-turns, as well as extensive insertions of shorter strands and loops between strands D and E (figure 3).
Figure 3.

BR187–385 adopts a fold that is distantly related to the MSCRAMM-typical DEv-IgG fold. Both CnaA-subdomains of Fap1 (PDB: 2X12) and GspB (3QD1), as well as the N3 subdomain of ClfB (3AU0), superimpose to BR187–385 with r.m.s.d. values of 3.0, 3.2 and 3.9 Å, despite sequence identities of only 15%, 8% and 5% (see electronic supplementary material, figure S2 and supplemental movies), respectively. The fold of BR187–385 is distantly related to the canonical DEv-IgG fold displayed by the N3 domain of ClfB. Topology diagrams of the DEv-IgG fold variants are designated and colour-coded according to [8,19,21].
The first nine structural homologues, identified using Dali [23], belonged to the MSCRAMM or SRRP family (see electronic supplementary material, table S2). Although they shared only 5–17% sequence identity with BR187–385, they all superimposed to BR187–385 with r.m.s.d. values stretching from 3.0 to 4.7 Å. In particular, both crystal structures of the CnaA-subdomains of the SRRPs Fap1 and GspB superimposed to BR187–385 with r.m.s.d. values of 3.0 and 3.2 Å, despite sequence identities of only 15% and 8%, respectively (see electronic supplementary material, table S2 and figure S2). Compared with GspB, the CnaA subdomains of Fap1 and BR187–385 are more distantly related to the canonical DEv-IgG fold displayed by ClfB (figure 3). While both BR187–385 and Fap1 bind using their CnaA-like subdomains to their cognate ligands KRT10 and saliva-coated hydroxylapatite, respectively, GspB binds the carbohydrate ligand sialyl T-antigen via its Siglec subdomain [6,8]. The S. aureus-derived ClfB was also chosen for comparison with BR187–385 (see electronic supplementary material, table S2 and figure S2) because it binds to a KRT10-derived linear peptide motif (LPM) via the ‘dock, lock and latch’ mechanism [16,17]. Here again, despite a sequence identity of only 5%, the N3 subdomain of ClfB superimposed to BR187–385 with an r.m.s.d. value of 3.9 Å (figure 3; electronic supplementary material, figure S2). However, superimposition of ClfB and BR187–385 also revealed that BR187–385 can probably not bind to KRT10 via the same ‘dock, lock and latch’ binding mode, because KRT10 is bound on different sites of the two proteins (see electronic supplementary material, figure S3).
3.3. The KRT10-binding region of BR187–385 resembles a paperclip
The KRT10-binding region of PsrP that comprises residues 273–341 corresponds to a region involving most of strand E, as well as strands C2, D, D1, D2, D3 and D4, all connected by the loops LC2/D, LC1/C2, LD1/D2, LD2/D3 and LD3/D4 (figure 4a). A substructure within this region takes a paperclip form with back- and front-loops formed by residues 268–295 and 305–324, respectively, which provides a possible explanation to previous experimental observations (figure 4a) [9]. Indeed, while pre-incubation of KRT10+-A549 cells with a BR-construct comprising residues 273–341 (front- and back-loops) blocked binding of pneumococcal TIGR4, pre-incubation with a shorter BR construct stretching from residues 291 to 325 (front-loop only) resulted in binding to KRT10, but did not block binding of TIGR4 bacteria to A549 cells.
Figure 4.

BR187–385 comprises a paperclip-like region and a basic binding groove for interaction with KRT10. (a) The putative paperclip region provides an explanation for previous experimental observations [9]. The back- and front-loops of the clip, formed by residues 268–295 and 305–324, are coloured red and orange, respectively. Boxes highlight this specific region. Binding of KRT10 most probably requires conformational re-arrangements of the three loops LC1/C2, LD1/D2 and LD3/D4 (see electronic supplementary material, figure S4). (b) Hydrophobic patches on BR187–385 are displayed on a surface hydrophobicity distribution plot. Regions coloured red and blue are hydrophobic (positive values) and hydrophilic (negative values), respectively. Two hydrophobic patches are localized within the KRT10-binding region. While hydrophobic patch-1 is localized proximally to strand D4, hydrophobic patch-2 is localized underneath the tip of the front-loop of the clip. Hydrophobicity is plotted on a relative scale. (c) Analysis of the surface electrostatic potential of BR187–385 reveals a highly positively charged binding groove formed by the extended antiparallel β-sheet I that could accommodate the acidic helical rod domain of the KRT10/KRT1 heterodimer (see electronic supplementary material, figure S5). The electrostatic potential is plotted in 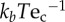 with the Boltzmann's constant kb, the charge of an electron ec at a temperature of 298 K.
with the Boltzmann's constant kb, the charge of an electron ec at a temperature of 298 K.
We hypothesized that binding of KRT10 may require conformational re-arrangements of the three loops LC1/C2, LD1/D2 and LD3/D4 (figure 4a). Analysis of the distribution of B-factor values revealed a high mobility of LC1/C2 and LD3/D4, as well as of LD2/D3 localized at the tip of the front-loop, compared with the rest of the structure (see electronic supplementary material, figure S4a). Rigidity analysis of BR187–385 confirmed that LC1/C2 did not belong to the single large rigid cluster formed by almost the entire BR187–385 structure (see electronic supplementary material, figure S4b). Our analysis indicated that LD3/D4 could be uncoupled by breakage of a single hydrogen bond interaction between the backbone oxygen of the asparagine residue N321 and the hydroxyl group of the serine residue S308. Furthermore, removal of three and four hydrogen bond interactions between the front- and back-loop regions would uncouple LD1/D2 and the entire front-loop region from the rigid cluster, respectively.
Hydrophobic interactions represent an essential mechanism for binding to intrinsically disordered protein regions [24] such as KRT10-associated glycine loops. Two distinct solvent-accessible hydrophobic pockets, localized proximally to strand D4 and underneath the front-loop, could possibly act as initial anchor points for interaction (figure 4b). Furthermore, inspection of the electrostatic surface of BR187–385 revealed the presence of a highly basic groove with a solvent-accessible surface area of 180 Å2 (figure 4c) that could easily accommodate the elongated and negatively charged helical rod domain of KRT10 (see electronic supplementary material, figure S5). The complementary charges of the basic BR187–385 and the acidic rod domains of KRT10/KRT1 (with theoretical isoelectric points of 4.6 and 4.8, respectively) could play an important role in initial complex formation, because electrostatic interactions are dominant long-range forces for protein associations [25]. Interestingly, the functional binding domain of the S. agalactiae-derived SRR-1 probably contains a CnaA-like domain, as predicted by sequence homology [8]. This CnaA subdomain with a theoretical isoelectric point of 4.7 binds to the carboxy-terminal domain of keratin-4 (KRT4) that belongs to basic type-II IFs [15]. This suggests that the surface of the two hitherto known keratin-binding SRRPs, PsrP and SRR-1, could be charge-optimized for efficient binding to oppositely charged IF protein ligands.
In conclusion, our structural analysis suggests that the KRT10-minimal binding region of BR187–385 resembles a paperclip that could allow for binding to KRT10 following conformational rearrangements of the clip-associated loops. Furthermore, the extended β-sheet on one side of the compressed barrel may provide a basic binding groove that could accommodate parts of the highly negatively charged helical rod domains of the KRT10/KRT1 heterodimer.
3.4. Binding of BR187–385 to keratin-10 is disrupted by alanine substitution of several residues within the paperclip region
The interaction between BR187–385 and KRT10 was confirmed in a pull-down experiment in which Strep-Tag-II BR187–385 (STII-BR187–385) bound to Ni–NTA bead-immobilized full-length KRT10 (figure 5a). For ELISA assays, three shorter KRT10 constructs comprising the head and tail end regions (including the associated helical segments 1A and 2B from the rod domain, as well as the entire rod domain of KRT10) were produced. While STII-BR187–385 bound with similar capacity to both the KRT10-full-length (KRT10-FL) and the KRT10-tail-rod-2B (KRT10-TRD) domains with a shared EC50 value of 345 nM, binding to the KRT10-rod domain (KRT10-ROD) was significantly reduced, with an estimated EC50 value of 1.8 μM. Finally, binding of STII- BR187–385 to the KRT10 head-rod-1A domain (KRT10-HRD) was at a very low level (figure 5b).
Figure 5.

BR187–385 binds to the tail-rod-2B end of KRT10. (a) BR187–385 bound to KRT10-FL immobilized on Ni-NTA magnetic agarose beads (+), and a small fraction remained associated after washing, while BR187–385 incubated with empty beads (−) was completely removed following the fourth wash step. Beads were analysed using SDS–PAGE before incubation with BR187–385 (0), after 10 min wash using magnetic PBS buffer (1), 1 min wash using magnetic washing buffer (2) and three times of a 10 min wash using magnetic washing buffer (3–5). The supernatant of BR187–385 after incubation with the beads (SN) confirms that equal amounts of protein were used for the assay. (b) A scheme of the tripartite structure of the intermediate filament protein keratin-10 (KRT10) shows the glycine loops localized at both end domains that are separated by a α-helical rod domain. The KRT10-FL, KRT10-HRD, KRT10-TRD and KRT10-ROD constructs comprise the stretches of residues 1–584, 1–179, 385–579 and 137–448, respectively. While BR187–385 bound to both KRT10-FL and KRT10-TRD with a common EC50-value of 345 nM (256–460 nM at 95% CI, R2 of 0.82), binding to the KRT10-rod domain (KRT10-ROD) was significantly reduced with an estimated EC50-value of 1.8 μM (1.4–2.3 μM at 95% CI, R2 of 0.74). Binding to the KRT10 head-region-domain was at a much lower level (KRT10-HRD). Values are given as mean with 95% CI. EC50 values were derived using a single four-parameter logistic nonlinear regression model. The mean normalized immobilization levels for KRT10-FL, KRT10-HRD, KRT10-TRD and KRT10-ROD as determined by HRP-coupled anti-His antibodies were 1.0 ± 0.2 (mean ± s.d.), 1.1 ± 0.4, 0.7 ± 0.2 and 1.1 ± 0.2, respectively.
Both head and tail domains are composed of glycine-rich loops, anchored to stacked arrays of aromatic and/or large apolar residues [10,26–28]. It has been previously suggested that, in contrast to the head domain, the tail of KRT10 is composed of fewer but larger (and therefore potentially more flexible) loop regions [27], which could play a key role in interactions with other proteins. For example, ClfB complements a KRT10 tail-derived LPM into a β-sheet, enforcing disorder-to-order transition of the peptide [16,17]. The peptide-binding site prediction server PepSite [29] identified two clip-associated binding sites in BR187–385 where several KRT10-derived LPMs could fit (see electronic supplementary material, figure S6). While binding site-1 was located within the first surface-accessible hydrophobic patch and localized proximally to strand D4, binding site-2 was localized underneath the front-loop region of the clip.
Ten residues located within or in the near vicinity of the two predicted paperclip-associated binding sites of BR187–385 were substituted to alanine (figure 6a), and all mutated proteins were tested for binding to KRT10-TRD using ELISA. Residue K231, which is not located within the predicted binding sites, was selected as a negative control. The interaction levels were reduced by at least 80% compared with WT-BR187–385 and control K231A-BR187–385 for seven of 10 mutations, including residues F329, M325 and I294 localized in site-1, as well as residues V290, M277, Y317 and W319 localized in site-2 (figure 6a,b). Furthermore, the Y305A and M310A substitutions within binding sites-1 and -2 reduced binding to KRT10-TRD by 15% and 60%, respectively. Finally, the N321A substitution did not affect BR187–385-binding to KRT10-TRD, probably due to the fact that its side chain points towards the solvent instead of the predicted binding site. Importantly, the mutations affected neither the expression and solubility levels of the mutated proteins nor their retention volumes in SEC (data not shown). Furthermore, comparative circular dichroism (CD) spectra analysis of WT- and mutated BR187–385 proteins indicated that their overall secondary structure was not significantly affected by the introduced substitutions (see electronic supplementary material, figure S7). However, it cannot be excluded that the introduced substitutions may induce small conformational changes that were not detected by CD spectroscopy, but could affect binding to KRT10-TRD.
Figure 6.

KRT10-TRD is bound in two contiguous paperclip-associated binding sites within BR187–385. (a) The location of each substituted residue is indicated. The centre of mass of each residue is displayed as a sphere and coloured according to its importance for binding to KRT10-TRD (red and blue for low and high binding intensities, respectively). The introduced substitutions did not significantly alter the overall secondary structure of the mutated BR187–385 proteins (see electronic supplementary material, figure S7). (b) Normalized intensity values were determined for binding of WT BR187–385 and each mutated variant to KRT10-TRD coated in wells of ELISA plates. Values are given as mean with standard deviation. WT and mutated BR187–385 variants were classified into groups a–d using the Tukey test at a significance level of p < 0.05.
In conclusion, our results demonstrate that BR187–385 binds to KRT10-TRD using two contiguous paperclip-associated binding sites. Furthermore, binding of KRT10 to site-2 probably requires conformational adaptations by the front-loop.
3.5. Concluding remarks
In this study, the crystal structure of the KRT10-binding region domain (BR187–385) of PsrP was determined, revealing the compressed barrel fold as a member of the MSCRAMM family of adhesin proteins despite very low sequence identity. Our results suggest that electrostatic interactions may play an important role in initial complex formation. Indeed, the acidic helical rod domain of KRT10 could fit in the highly basic groove of BR187–385, created by an extended β-sheet on one side of the compressed barrel. Structural analysis and in vitro binding data also indicate the importance of the other face of the barrel that resembles a paperclip for binding to the tail-rod-2B region of KRT10. Future crystal structure determination of BR187–385 in complex with a KRT10-derived binding motif is required to elucidate the exact binding mechanisms, possibly confirming the importance of electrostatic interactions for initial complex formation.
4. Experimental procedures
4.1. Cloning
All protein constructs were cloned into the pET21d expression vector (Novagen) using the ligation-independent FastCloning method [30]. The coding sequence for the full-length basic region of PsrP (residues 2–395) was prepared from S. pneumoniae TIGR4 chromosomal DNA as described before [31] and used as template for PCR amplification to generate expression constructs comprising residues 187–385 of PsrP (BR187–385) with N-terminal poly-His (HHHHHH) and STII (SAWSHPQFEK) tags, respectively. Mutated expression constructs of BR187–385 were generated following previously described protocols [32].
The coding sequence for full-length KRT10 (clone ID HsCD00045373, Uniprot ID P13645) obtained from the DNASU repository [33] was PCR amplified to generate expression constructs of the head-rod-1A domain (KRT10-HRD: residues 1–179), tail-rod-2B domain (KRT10-TRD: residues 385–579), the rod domain (KRT10-ROD: residues 137–448, a M150L mutation was essential to prevent second translation initiation at ATG codon) and a full-length version (FL: 1–584) with N-terminal poly-His tag followed by a TEV cleavage site (HHHHHHENLYFQG; figure 5). All coding sequences of the protein-expression constructs were confirmed by DNA sequencing and are listed in the electronic supplementary material.
4.2. Expression, purification and optimization of protein constructs
Several poly-histidine-tagged constructs spanning different parts of the binding-region (BR) domain and with short length variations (of about six to eight residues) at both the N- and the C-termini were designed based on the overall domain organization of PsrP. Protein expression and solubility levels were checked using a small-scale expression test (http://tinyurl.com/EMBL-Heidelberg). The subdomain BR187–385 with higher crystallization probability was identified as the most promising construct using a limited proteolysis approach [34]. Protein expression was induced at OD 0.4–0.7 using 400 µM IPTG and performed overnight at 25°C. Expression of Seleno-methionine (Se-Met)-substituted protein was performed using Se-Met medium complete (Molecular Dimensions, UK) and Met-auxotroph E. coli B834 cells (EMBL, Hamburg, Germany).
Poly-histidine-tagged BR187–385 was purified using immobilized metal affinity (IMAC) and cation exchange chromatography (CEC; HisTrap FF and HiTrap SPFF; GE Healthcare, Sweden). STII-tagged BR187–385 was purified using affinity chromatography on a Strep-Tactin superflow high-capacity column (IBA, Germany). Monomeric BR187–385 was eluted using SEC on Superdex 75 or 200 columns (GE Healthcare). The purity of poly-His- and STII-tagged BR187–385 constructs were assessed by SDS–PAGE to be at least 99% (see electronic supplementary material, figure S8a–c).
The soluble poly-His-tagged KRT10-TRD was purified using IMAC and anion exchange chromatography (AEC) on a 1 ml HiTrap Q HP (GE Healthcare). The poly-His-tagged KRT10-FL and KRT10-ROD were purified as inclusion bodies as described previously [35] and further purified in the presence of 6 M urea using AEC on a HiTrap Q HP 1 ml. KRT10-HRD was purified in the presence of 6 M urea using IMAC and CEC on a HiTrap Q SPFF 1 ml column (GE Healthcare). KRT10-FL, KRT10-ROD and KRT10-HRD were thereafter dialysed against urea-free buffer. The final purity of KRT10-TRD, KRT10-HRD, KRT10-ROD and KRT10-FL were estimated as 99%, 99%, 99% and more than 90% (see electronic supplementary material, figure S8b), respectively.
4.3. Crystallization of BR187–385
The BR187–385 monomer was concentrated to 20 mg ml−1 in 20 mM sodium citrate, 500 mM NaCl, 10% (v/v) glycerol, pH 5.5. Well-diffracting crystals of wild-type and Se-Met-BR187–385 were obtained in 0.2 M lithium sulfate, 0.1 M sodium acetate trihydrate pH 4.6, 25% PEG4000 (w/v) using the sitting drop vapour-diffusion method followed by micro-seeding. Crystals were cryo-protected by soaking in mother liquor supplemented with 25% glycerol and flash-frozen in liquid nitrogen.
4.4. Data collection and determination of the crystal structure of BR187–385
X-ray diffraction data from crystals of native and Se-Met-substituted BR187–385, both collected at beam line ID29 at the synchrotron radiation facility at the ESRF (Grenoble, France), were processed using the XDS program package [36] (table 1). The SAD dataset of a P43212 Se-Met-substituted BR187–385 crystal diffracting to 2.25 Å was used to determine the crystal structure of BR187–385, based on the SAS protocol from Auto-Rickshaw [37]. Almost complete models for three BR187–385 molecules were obtained that were complemented through automatic rebuilding in Buccaneer [38]. Coot was used for all subsequent model building [39].
An MR search was performed using Phaser [40], and a single BR187–385 molecule was located in the asymmetric unit of the native crystal. Initial rigid body and restrained refinement rounds were performed in CCP4 Refmac [38] followed by model refinement using Phenix [41] with individual isotropic ADP factors and TLS refinement of the entire chain. A single Ramachandran plot outlier was found in the final model corresponding to residue T271, located in a β-turn motif with weak electron density. Finally, the side-chain atoms Oγ1 and Cγ2 of residue T378 were not built as a result of poor electron density.
The structural model was used to further refine the model corresponding to the anomalous dataset, using a simulated annealing protocol with subsequent LBFGS minimization with individual isotropic ADP factors, whole-chain TLS group refinement and NCS Cartesian restraints. At later stages, simulated annealing was omitted and NCS Cartesian restraints were altered to NCS torsion restraints. Crystal packing analysis revealed that the overall mobility of chain A (residues I204-S377) was relatively higher compared with the mobility of chains B and C (both comprising residues N203–S376), as reflected by higher overall B-factor values and lower map correlation coefficients (data not shown).
4.5. Structural analysis of BR187–385
The hydrophobicity of BR187–385 was assessed using the program package VASCo 1.0.2 [42]. PDB2PQR 1.7.1 [43] and APBS 1.3 in PyMOL [44] were used to calculate the electrostatic surface potentials. All figures were created using PyMOL version 1.3.0 [45]. Further programs used for structural analysis are listed in the electronic supplementary material.
4.6. Analytical ultracentrifugation analysis and sedimentation velocity experiments
Sedimentation velocity experiments were carried out on an analytical ultracentrifuge XLI (Beckman Coulter, Palo Alto, CA) with a rotor speed of 50 000 rpm, at 20°C, using a rotor Anti-50, and double-sector cells of optical path length 12 or 3 mm equipped with sapphire windows. Acquisitions were made using absorbance at 280 nm. Two samples of BR187–385 in 20 mM sodium citrate, 250 mM NaCl, 2.5% glycerol pH 5.5 were investigated. Solvent density of 1.017 g ml−1 and viscosity of 1.11 mPa s were measured at 20°C on density-meter DMA 5000 and viscosity-meter AMVn (Anton Paar), and the partial specific volume was estimated to 0.721 ml g−1 with the program SEDNTERP. The analysis was carried out in terms of distribution of sedimentation coefficients, c(s), and non-interacting species, with SEDFIT software, version 14.0c. The c(s) distributions showed a species at 1.85 S contributing to 97% of the total signal for two different sample concentrations of 45 and 22.5 µM. Their analysis in terms of one non-interacting species gave independent values for the molar mass of 23 and 20.5 kDa at the two concentrations, close to the theoretical value of 22.1 kDa. The theoretical sedimentation coefficients for 20 models from the generated EOM ensemble (see below) were calculated by the atomic-type/shell-model calculation in HYDROPRO [46] with a radius of the atomic elements of 2.9 Å.
4.7. Small angle X-ray scattering data processing and analysis
Synchrotron radiation X-ray scattering data were collected from five solute concentrations of BR187–385 in the range 1.1–8.7 mg ml−1 in 20 mM sodium citrate, 250 mM NaCl, 2.5% glycerol pH 5.5 were collected on the X33 camera of the EMBL on storage ring DORIS III (DESY, Hamburg, Germany) [47]. Data were collected using a photon counting Pilatus 1M detector at a sample–detector distance of 2.7 m and a wavelength of λ = 1.5 Å, the range of momentum transfer 0.01 < s < 0.6 Å−1 was covered (s = 4π sinθ/λ, where 2θ is the scattering angle). The forward scattering I(0), the radius of gyration Rg along with the pair distribution function of the particle p(r) and the maximum dimension Dmax were computed by the automated SAXS data analysis pipeline [48]. The molecular mass (MM) of BR187–385 was evaluated by comparison of the forward scattering with that from a reference solution of bovine serum albumin (MM = 66 kDa). The excluded volume of the hydrated protein was computed with the program AUTOPOROD [49]. For globular proteins, the hydrated volumes in Å3 are about 1.6 times the MMs in Dalton. To assess the flexibility of BR187–385, the EOM [50] was used. See the electronic supplementary materials for details about data collection and analysis.
4.8. Pull-down assay
KRT10-FL was immobilized to 30 µl Ni–NTA magnetic agarose beads (Qiagen, Germany) in 10 mM potassium phosphate, pH 7.2, 250 mM NaCl, 0.05% Triton X-100 (magnetic PBS buffer). Beads not loaded with KRT10-FL protein were used as negative control. Beads were washed using 10 mM potassium phosphate, pH 7.8, 300 mM NaCl, 20 mM imidazole, 8% glycerol, 0.2% Triton X-100 (magnetic washing buffer) for 30 min. A volume of 100 µl of 25 µM STII-BR187–385 was incubated with the beads in 20 mM HEPES, 50 mM NaCl, 10% glycerol, pH 7.5 for 1 h. Beads were analysed using SDS–PAGE before incubation with BR187–385, after 10 min wash using magnetic PBS buffer, 1 min wash and three times of a 10 min wash using magnetic washing buffer.
4.9. ELISAs
ELISAs were performed using Nunc C96 MicroWell plates. PBS-T (PBS with 0.05% Tween-20) was used as washing buffer. Conjugates were detected using TMB liquid substrate system (Sigma Aldrich, USA). Coating levels of KRT10 constructs were detected using HRP-conjugated anti-His antibodies (anti-His AB-HRP, ab1187; Abcam, UK) and adjusted to the coating levels of KRT10-TRD incubated at a concentration of approximately 5 μg ml−1. 2% BSA (w/v) in PBS was used as blocking agent. STII-BR187–385 to KRT10 construct binding assays were performed in PBS with STII-BR187–385 concentrations ranging from 23 nM to 3 μM. For the second assay, WT-BR187–385 and mutated variants were used at a concentration of 1 μM for the BR187–385/KRT10-TRD interaction. Binding of STII-BR187–385 to KRT10 was detected using 250 ng μl−1 Strep-Tactin HRP conjugate in PBS-T (stock of IBA, Germany). Data were averaged and normalized as described in the electronic supplementary material.
Supplementary Material
Acknowledgements
We gratefully acknowledge excellent technical assistance by Åke Engström, fruitful discussions with Tatyana Sandalova, Frederico Fogilari and Peter Mellroth, as well as access to synchrotron beamline ID29 at ESRF (Grenoble, France). This work was facilitated by the Protein Science Facility at Karolinska Institutet/SciLifeLab (http://psf.ki.se).
Data accessibility
PDB and structure factor files have been deposited under PDB ID codes 3ZGI and 3ZGH. Data that have not been deposited in a public repository will be made available upon request.
Funding statement
This work was supported by grants from the Swedish Research Council (A.A., B.H.-N., S.N.), the Knut and Alice Wallenberg Foundation (A.A., B.H.-N., S.N.), the Wenner Gren Foundation (T.S.), the European Community's Seventh Framework Programme (FP7/2007–2013) under BioStruct-X (grant agreement no. 283570), and used the platforms of the Grenoble Instruct Centre (ISBG; UMS 3518 CNRS-CEA-UJF-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB).
References
- 1.Van der Poll T, Opal SM. 2009. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet 374, 1543–1556. (doi:10.1016/S0140-6736(09)61114-4) [DOI] [PubMed] [Google Scholar]
- 2.Muñoz-Almagro C, Selva L, Sanchez CJ, Esteva C, de Sevilla MF, Pallares R, Orihuela CJ. 2010. PsrP, a protective pneumococcal antigen, is highly prevalent in children with pneumonia and is strongly associated with clonal type. Clin. Vaccine Immunol. 17, 1672–1678. (doi:10.1128/CVI.00271-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gamez G, Hammerschmidt S. 2012. Combat pneumococcal infections: adhesins as candidates for protein-based vaccine development. Curr. Drug Targets 13, 323–337. (doi:10.2174/138945012799424697) [DOI] [PubMed] [Google Scholar]
- 4.Spirig T, Weiner EM, Clubb RT. 2011. Sortase enzymes in Gram-positive bacteria. Mol. Microbiol. 82, 1044–1059. (doi:10.1111/j.1365-2958.2011.07887.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lizcano A, Sanchez CJ, Orihuela CJ. 2012. A role for glycosylated serine-rich repeat proteins in Gram-positive bacterial pathogenesis. Mol. Oral Microbiol. 27, 257–269. (doi:10.1111/j.2041-1014.2012.00653.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramboarina S, et al. 2010. Structural insights into serine-rich fimbriae from gram-positive bacteria. J. Biol. Chem. 285, 32 446–32 457. (doi:10.1074/jbc.M110.128165) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanchez CJ, Shivshankar P, Stol K, Trakhtenbroit S, Sullam PM, Sauer K, Hermans PWM, Orihuela CJ. 2010. The pneumococcal serine-rich repeat protein is an intra-species bacterial adhesin that promotes bacterial aggregation in vivo and in biofilms. PLoS Pathog. 6, e1001044 (doi:10.1371/journal.ppat.1001044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pyburn TM, et al. 2011. A structural model for binding of the serine-rich repeat adhesin GspB to host carbohydrate receptors. PLoS Pathog. 7, e1002112 (doi:10.1371/journal.ppat.1002112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shivshankar P, Sanchez C, Rose LF, Orihuela CJ. 2009. The Streptococcus pneumoniae adhesin PsrP binds to keratin 10 on lung cells. Mol. Microbiol 73, 663–679. (doi:10.1111/j.1365-2958.2009.06796.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parry DAD, Steinert PM. 1999. Intermediate filaments: molecular architecture, assembly, dynamics and polymorphism. Q. Rev. Biophys. 32, 99–187. (doi:10.1017/S0033583500003516) [DOI] [PubMed] [Google Scholar]
- 11.Moll R, Divo M, Langbein L. 2008. The human keratins: biology and pathology. Histochem. Cell Biol. 129, 705–733. (doi:10.1007/s00418-008-0435-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee C-H, Kim M-S, Chung BM, Leahy DJ, Coulombe PA. 2012. Structural basis for heteromeric assembly and perinuclear organization of keratin filaments. Nat. Struct. Mol. Biol. 19, 707 (doi:10.1038/nsmb.2330) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Brien LM, Walsh EJ, Massey RC, Peacock SJ, Foster TJ. 2002. Staphylococcus aureus clumping factor B (ClfB) promotes adherence to human type I cytokeratin 10: implications for nasal colonization. Cell. Microbiol. 4, 759–770. (doi:10.1046/j.1462-5822.2002.00231.x) [DOI] [PubMed] [Google Scholar]
- 14.Haim M, Trost A, Maier CJ, Achatz G, Feichtner S, Hintner H, Bauer JW, Önder K. 2010. Cytokeratin 8 interacts with clumping factor B: a new possible virulence factor target. Microbiology 156, 3710–3721. (doi:10.1099/mic.0.034413-0) [DOI] [PubMed] [Google Scholar]
- 15.Samen U, Eikmanns BJ, Reinscheid DJ, Borges F. 2007. The surface protein Srr-1 of Streptococcus agalactiae binds human keratin 4 and promotes adherence to epithelial HEp-2 cells. Infect. Immun. 75, 5405–5414. (doi:10.1128/IAI.00717-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ganesh VK, et al. 2011. Structural and biochemical characterization of Staphylococcus aureus clumping factor B/ligand interactions. J. Biol. Chem. 286, 25 963–25 972. (doi:10.1074/jbc.M110.217414) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiang H, Feng Y, Wang J, Liu B, Chen Y, Liu L, Deng X, Yang M. 2012. Crystal structures reveal the multi-ligand binding mechanism of Staphylococcus aureus ClfB. PLoS Pathog. 8, e1002751 (doi:10.1371/journal.ppat.1002751) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Symersky J, et al. 1997. Structure of the collagen-binding domain from a Staphylococcus aureus adhesin. Nat. Struct. Mol. Biol. 4, 833–838. (doi:10.1038/nsb1097-833) [DOI] [PubMed] [Google Scholar]
- 19.Deivanayagam CCS, Wann ER, Chen W, Carson M, Rajashankar KR, Hook M, Narayana SVL. 2002. A novel variant of the immunoglobulin fold in surface adhesins of Staphylococcus aureus: crystal structure of the fibrinogen-binding MSCRAMM, clumping factor A. EMBO J. 21, 6660–6672. (doi:10.1093/emboj/cdf619) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheldrick GM. 2010. Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D, Biol. Crystallogr. 66, 479–485. (doi:10.1107/S0907444909038360) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bork P, Holm L, Sander C. 1994. The immunoglobulin fold: structural classification, sequence patterns and common core. J. Mol. Biol. 242, 309–320. (doi:10.1006/jmbi.1994.1582) [DOI] [PubMed] [Google Scholar]
- 22.Vengadesan K, Narayana SVL. 2011. Structural biology of Gram-positive bacterial adhesins. Protein Sci. 20, 759–772. (doi:10.1002/pro.613) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holm L, Kääriäinen S, Rosenström P, Schenkel A. 2008. Searching protein structure databases with DaliLite v.3. Bioinformatics 24, 2780–2781. (doi:10.1093/bioinformatics/btn507) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mészáros B, Tompa P, Simon I, Dosztányi Z. 2007. Molecular principles of the interactions of disordered proteins. J. Mol. Biol. 372, 549–561. (doi:10.1016/j.jmb.2007.07.004) [DOI] [PubMed] [Google Scholar]
- 25.Schreiber G, Haran G, Zhou H-X. 2009. Fundamental aspects of protein−protein association kinetics. Chem. Rev. 109, 839–860. (doi:10.1021/cr800373w) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mack JW, Torchia DA, Steinert PM. 1988. Solid-state NMR studies of the dynamics and structure of mouse keratin intermediate filaments. Biochemistry 27, 5418–5426. (doi:10.1021/bi00415a006) [DOI] [PubMed] [Google Scholar]
- 27.Steinert PM, Mack JW, Korge BP, Gan SQ, Haynes SR, Steven AC. 1991. Glycine loops in proteins: their occurrence in certain intermediate filament chains, loricrins and single-stranded RNA binding proteins. Int. J. Biol. Macromol. 13, 130–139. (doi:10.1016/0141-8130(91)90037-U) [DOI] [PubMed] [Google Scholar]
- 28.Strnad P, Usachov V, Debes C, Gräter F, Parry DAD, Omary MB. 2011. Unique amino acid signatures that are evolutionarily conserved distinguish simple-type, epidermal and hair keratins. J. Cell. Sci. 124, 4221–4232. (doi:10.1242/jcs.089516) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petsalaki E, Stark A, García-Urdiales E, Russell RB. 2009. Accurate prediction of peptide binding sites on protein surfaces. PLoS Comput. Biol. 5, e1000335 (doi:10.1371/journal.pcbi.1000335) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li C, Wen A, Shen B, Lu J, Huang Y, Chang Y. 2011. FastCloning: a highly simplified, purification-free, sequence- and ligation-independent PCR cloning method. BMC Biotechnol. 11, 92 (doi:10.1186/1472-6750-11-92) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mellroth P, Daniels R, Eberhardt A, Rönnlund D, Blom H, Widengren J, Normark S, Henriques-Normark B. 2012. LytA, major autolysin of Streptococcus pneumoniae, requires access to nascent peptidoglycan. J. Biol. Chem. 287, 11 018–11 029. (doi:10.1074/jbc.M111.318584) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Li C, Xiao W, Yuan D, Wan G, Ma L. 2008. Site-directed mutagenesis by combination of homologous recombination and DpnI digestion of the plasmid template in Escherichia coli. Anal. Biochem. 373, 389–391. (doi:10.1016/j.ab.2007.10.034) [DOI] [PubMed] [Google Scholar]
- 33.Cormier CY, et al. 2010. Protein structure initiative material repository: an open shared public resource of structural genomics plasmids for the biological community. Nucleic Acids Res. 38, D743–D749. (doi:10.1093/nar/gkp999) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koth CM, Orlicky SM, Larson SM, Edwards AM. 2003. Use of limited proteolysis to identify protein domains suitable for structural analysis. In Macromolecular crystallography, part C (eds CW Carter Jr, RM Sweet), pp. 77–84. San Diego, CA: Academic Press. [DOI] [PubMed] [Google Scholar]
- 35.Hatzfeld M, Weber K. 1990. Tailless keratins assemble into regular intermediate filaments in vitro. J. Cell. Sci. 97, 317–324. [DOI] [PubMed] [Google Scholar]
- 36.Kabsch W. 1993. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26, 795–800. (doi:10.1107/S0021889893005588) [Google Scholar]
- 37.Panjikar S, Parthasarathy V, Lamzin VS, Weiss MS, Tucker PA. 2005. Auto-Rickshaw: an automated crystal structure determination platform as an efficient tool for the validation of an X-ray diffraction experiment. Acta Crystallogr. D, Biol. Crystallogr. 61, 449–457. (doi:10.1107/S0907444905001307) [DOI] [PubMed] [Google Scholar]
- 38.CCP4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, Biol. Crystallogr. 50, 760–763. (doi:10.1107/S0907444994003112) [DOI] [PubMed] [Google Scholar]
- 39.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D, Biol. Crystallogr. 60, 2126–2132. (doi:10.1107/S0907444904019158) [DOI] [PubMed] [Google Scholar]
- 40.McCoy AJ. 2007. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D, Biol. Crystallogr. 63, 32–41. (doi:10.1107/S0907444906045975) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adams PD, et al. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D, Biol. Crystallogr. 58, 1948–1954. (doi:10.1107/S0907444902016657) [DOI] [PubMed] [Google Scholar]
- 42.Steinkellner G, Rader R, Thallinger GG, Kratky C, Gruber K. 2009. VASCo: computation and visualization of annotated protein surface contacts. BMC Bioinformatics 10, 32 (doi:10.1186/1471-2105-10-32) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. 2004. PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667. (doi:10.1093/nar/gkh381) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl Acad. Sci. USA 98, 10 037–10 041. (doi:10.1073/pnas.181342398) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schrödinger L. 2010. PyMOL molecular graphics system. See http://www.pymol.org. [Google Scholar]
- 46.García De La Torre J, Huertas ML, Carrasco B. 2000. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J. 78, 719–730. (doi:10.1016/S0006-3495(00)76630-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blanchet CE, et al. 2012. Instrumental setup for high-throughput small- and wide-angle solution scattering at the X33 beamline of EMBL Hamburg. J. Appl. Crystallogr. 45, 489–495. (doi:10.1107/S0021889812013490) [Google Scholar]
- 48.Franke D, Kikhney AG, Svergun DI. 2012. Automated acquisition and analysis of small angle X-ray scattering data. Nucl. Instrum. Methods Phys. Res. A 689, 52–59. (doi:10.1016/j.nima.2012.06.008) [Google Scholar]
- 49.Petoukhov MV, et al. 2012. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350. (doi:10.1107/S0021889812007662) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bernadó P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. 2007. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 129, 5656–5664. (doi:10.1021/ja069124n) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
PDB and structure factor files have been deposited under PDB ID codes 3ZGI and 3ZGH. Data that have not been deposited in a public repository will be made available upon request.