

Abstract
Campylobacter jejuni is an important cause of human foodborne gastroenteritis; strategies to prevent infection are hampered by a poor understanding of the complex interactions between host and pathogen. Previous work showed that C. jejuni could bind human histo-blood group antigens (BgAgs) in vitro and that BgAgs could inhibit the binding of C. jejuni to human intestinal mucosa ex vivo. Here, the major flagella subunit protein (FlaA) and the major outer membrane protein (MOMP) were identified as BgAg-binding adhesins in C. jejuni NCTC11168. Significantly, the MOMP was shown to be O-glycosylated at Thr268; previously only flagellin proteins were known to be O-glycosylated in C. jejuni. Substitution of MOMP Thr268 led to significantly reduced binding to BgAgs. The O-glycan moiety was characterized as Gal(β1–3)-GalNAc(β1–4)-GalNAc(β1–4)-GalNAcα1-Thr268; modelling suggested that O-glycosylation has a notable effect on the conformation of MOMP and this modulates BgAg-binding capacity. Glycosylation of MOMP at Thr268 promoted cell-to-cell binding, biofilm formation and adhesion to Caco-2 cells, and was required for the optimal colonization of chickens by C. jejuni, confirming the significance of this O-glycosylation in pathogenesis.
Keywords: Campylobacter jejuni, histo-blood group antigens, FlaA, major outer membrane protein, O-glycosylation, biofilm
2. Introduction
Campylobacter jejuni is the most common causative agent of bacterial gastroenteritis reported worldwide [1]. It is responsible for a spectrum of intestinal symptoms, including watery diarrhoea, bloody diarrhoea, vomiting, fever and abdominal cramps. Symptoms range from mild to severe and may lead to death in immunocompromised patients [2]. Acute-phase disease is sometimes followed by serious long-term sequelae, including reactive arthritis [3,4], the autoimmune neuropathies Guillain–Barré syndrome (GBS) and Miller–Fisher syndrome [5] and immunoproliferative small intestinal disease [6]. The biochemical structures of gangliosides play a crucial role in the pathogenesis of GBS. Infection with C. jejuni is responsible for 35% of all types of GBS and 20–30% of GBS-linked antibodies are directed against gangliosides [7].
Prevention and treatment of the primary and secondary consequences of Campylobacter infection are hampered by a poor understanding of the detailed molecular interplay between cells of the gastrointestinal lining and the pathogen. Following ingestion, C. jejuni infect and invade the epithelium of the small intestine and colon. Infection is dependent on motility mediated by polar flagella and outer membrane adhesins, including PEB1a, PEB3, JlpA, MOMP, CapA and CadF [8–13]. Thus, surface-exposed bacterial ligands play major roles in mediating mucosal adhesion and invasion. Campylobacter jejuni harbours both N- and O-linked glycosylation systems [14,15]; both have been implicated in adhesion to, and invasion of, epithelial cells in vitro [16] and host colonization and virulence in vivo [17,18]. The only target of the O-linked glycosylation system described to date is flagellin [19], whereas the N-linked system has a variety of targets, including periplasmic and outer membrane proteins [20].
Ruiz-Palacios and co-workers [21,22] showed that fucosylated sugar components in human breast milk could inhibit C. jejuni binding to HEp-2 cells in vitro and to human intestinal mucosa ex vivo, and also prevent C. jejuni infection of mice in vivo. Subsequent solid-phase assays confirmed the binding of C. jejuni strains, grown under host-like conditions, to a broad range of fucose (and galactose)-containing glycans, with different strains exhibiting differing binding profiles and avidities [21,23]; however, the identity of the C. jejuni lectin(s) responsible for this binding has not to date been determined. The common ABO histo-blood group antigens (BgAgs) comprise a complex and polymorphic group of fucosylated carbohydrates expressed on the surfaces of erythrocytes, but they are also highly expressed in the oro-gastro-intestinal (OGI) epithelium, as well as endothelial cells, and in secretions, such as milk, saliva and tears [24,25]. Their common denominators are the types I and II glycan core chains which may be fucosylated in the bone marrow by the H-II-(fucosyl)-transferase, before being absorbed into the surface of erythrocytes [26] or by the H-I-(fucosyl)-transferase in the mucosal glandular tissues, for example that along the OGI tract. Fucosylated glycans are substrates for further glycosylation reactions that give rise to the epitopes defining the A, B and Lewis BgAgs.
The ABO (or ABH) and Lewis BgAgs are epidemiologically associated with susceptibility to several infectious agents [26]. Among enteropathogenic bacteria, Helicobacter pylori bind the H-I and Leb antigens [27]. The H. pylori BabA (blood group antigen-binding) and SabA (sialic acid-binding adhesin) proteins are the best described ABO/Leb and sialyl-Lewis x (sLex) antigen-binding adhesins and predictors for the development of overt gastric disease [28,29].
Here, we identify the C. jejuni surface proteins FlaA and MOMP as BgAg-binding adhesins. Furthermore, we show that flagellins are not the only O-glycosylated proteins in C. jejuni; MOMP is also O-glycosylated, a modification that influences the MOMP conformation and thus BgAg interactions. MOMP O-glycosylation promoted biofilm formation and auto-aggregation, and was required for optimal adhesion to Caco-2 cells and colonization of chickens, confirming the significance of this post-translational modification in C. jejuni pathogenesis.
3. Results
3.1. Campylobacter jejuni binds a wide range of human histo-BgAgs
To confirm that C. jejuni binds BgAgs, the core-I, core-II, H-II, Leb, Ley and Lex BgAgs were adsorbed to the surface of amino-linked ELISA plates and incubated with digoxigenin (DIG)-labelled C. jejuni strain NCTC11168. The structures of these molecules are shown in the electronic supplementary material, table S1. NCTC11168 bound to each of the glycoconjugates tested (figure 1a). Importantly, the pre-incubation of NCTC11168 with soluble BgAgs inhibited binding in this assay (figure 1a). In addition, 45 clinical isolates of C. jejuni (see electronic supplementary material, table S2) also bound to many of the same BgAgs, albeit to a varied degree (see electronic supplementary material, table S3) confirming that BgAg binding is a widespread phenotypic trait among C. jejuni isolates.
Figure 1.

Binding of C. jejuni to BgAgs and identification of C. jejuni BgAg-binding adhesins. (a) Binding of digoxigenin (DIG)-labelled C. jejuni NCTC11168 to ELISA wells coated with BgAgs. Specific binding was calculated by subtracting the bovine serum albumin (BSA; negative control) values from the BSA–BgAg absorbance. Binding was significantly inhibited (p < 0.05) by the pre-incubation of bacteria with soluble glycoconjugates H-II, Leb or Ley prior to adding to the ELISA plate. NT, non-treated. Error bars: mean of triplicate values ± s.e.m. from three independent experiments. (b) Identification of BgAg-binding proteins from C. jejuni NCTC11168 using re-tagging (baits used: H-II; H-I and Leb). Two proteins were identified at sizes of 45 and 59 kDa, corresponding to MOMP and FlaA, respectively.
3.2. Campylobacter jejuni FlaA and MOMP mediate binding to human BgAgs
A technique employing a multi-functional cross-linker, termed as retagging [29], was used for the identification of C. jejuni BgAg-binding adhesins. NCTC11168 cells were incubated with BgAg conjugated to a light-activated cross-linker. Following photo-activation and subsequent exposure to reducing conditions to allow transfer of the reactive biotin moiety to molecules in close proximity to the BgAgs, bacteria were lysed and the biotin-tagged C. jejuni proteins purified with streptavidin-coated magnetic beads, as previously described [29]. Extracted biotin-tagged proteins were separated by SDS-PAGE and identified using MALDI-TOF. Two proteins were identified: the 45 kDa major outer membrane protein (MOMP) and the 59 kDa flagellin FlaA (figure 1b). FlaA is the major flagellin and is required for the expression of functional flagella. To confirm that this molecule contributed to the observed BgAg-binding activity, flaA was deleted in NCTC11168 and the mutant tested for its ability to bind to BgAgs in ELISA experiments. Deletion of flaA led to significantly reduced binding of the NCTC11168 mutant to all examined BgAgs (figure 2a). MOMP, encoded by porA, is an essential multi-functional porin [30]. As it was not possible to delete porA, we purified the MOMP under non-denaturing conditions from strain NCTC11168. The purified protein significantly inhibited binding of NCTC11168 to the H-II glycoconjugate (figure 2b).
Figure 2.
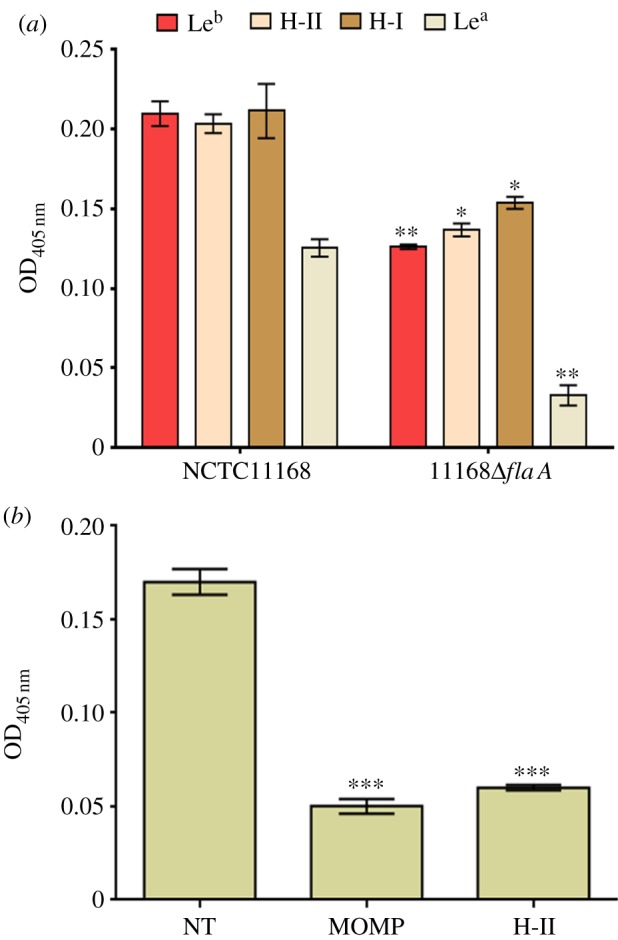
FlaA and MOMP are required for C. jejuni BgAg binding. (a) NCTC11168 and its ΔflaA mutant derivative were examined for binding to Leb, H-II, H-I and Lea. Binding of 11168ΔflaA was significantly reduced compared with the wild-type (*p < 0.05; **p < 0.01). Error bars: mean of triplicate values ± s.e.m. from three independent experiments. (b) Binding of NCTC11168 to H-II glycoconjugate in the absence of inhibitor (non-treated, NT) or after H-II coated plates were treated with purified NCTC11168 MOMP before the addition of bacteria or after NCTC11168 were pre-incubated with H-II glycoconjugate (H-II). ***significantly reduced compared with non-treated (p < 0.001). Error bars: mean of triplicate values ± s.e.m. from three independent experiments.
3.3. NCTC11168 MOMP is O-glycosylated
Both O- and N-glycosylation systems have been described in C. jejuni. FlaA is a known glycoprotein that is post-translationally modified with O-linked glycans that can constitute up to 10% of the molecule's mass [31]. Furthermore, an increasing number of periplasmic and outer membrane proteins of C. jejuni have been found to be N-glycosylated. Inhibition of key enzymatic steps in each pathway blocks the addition of glycans to target proteins. In the N-glycosylation (Pgl) pathway, the oligosaccharyltransferase PglB is critical for transfer of the final glycan to the target protein in the periplasm. Deletion of pglB had no detectable impact on bacterial binding to any of the examined BgAgs (figure 3a), indicating that the N-glycosylation system does not influence BgAg binding.
Figure 3.

O-Glycosylation but not N-glycosylation modulates C. jejuni BgAg binding. (a) NCTC11168 and 11168ΔpglB were examined for binding to core-II, core-I, H-I, H-II, Leb, Lex and Ley. Binding was not significantly reduced following pglB mutation. Error bars: mean of triplicate values ± s.e.m. from three independent experiments. (b) NCTC11168 and 11168ΔpseD were examined for binding to core-II, H-I, H-II, Leb and Ley. The binding of 11168ΔpseD was significantly reduced compared with the wild-type (***p < 0.001). Error bars: mean of triplicate values ± s.e.m. from three independent experiments. (c) Immunoblot analysis of purified MOMP from wild-type NCTC11168, 11168MOMP268T/G and 11168ΔpseD strains using anti-MOMP antibodies. MOMP from the latter two strains showed an apparent size shift compared with the wild-type protein indicating a loss of glycosylation.
The predominant O-glycans attached to the Campylobacter flagellum are derivatives of pseudaminic acid (Pse) or legionaminic acid (Leg), which are C9 sugars related to sialic acids [32]. Mutation of pseD, which encodes a putative PseAm transferase, results in a fully motile phenotype [19,33], but with a loss of PseAm from the flagellin [19]. Mutation of pseD resulted in a significant reduction in the binding of NCTC11168 to all examined BgAgs (figure 3b), suggesting that O-glycosylation of BgAg ligands is required for optimal BgAg binding. Intriguingly, when MOMPs purified from NCTC11168 and 11168ΔpseD were compared by immunoblotting, the latter exhibited an apparent size shift compared with MOMP from the wild-type strain, consistent with the loss of glycan modifications (figure 3c) and reminiscent of the size shift seen when comparing glycosylated and unglycosylated flagellin proteins [31]. We therefore hypothesized that, in addition to flagellin proteins, MOMP may also be O-glycosylated. To address this, MOMP from NCTC11168 was analysed by Nanoflow LC-MS/MS FT/ICR following in-gel protein digestion as described by Shevchenko et al. [34]. The MS/MS analysis showed that a peptide corresponding to amino acids 268–278 of NCTC11168 MOMP was glycosylated; specifically, the amino acid Thr268 was carrying Hex-(HexN-acetylamine)3 (where Hex can be glucose or galactose; figure 4a–c). To confirm that Thr268 was indeed the amino acid residue targeted by the O-glycosylation system, we substituted Gly for Thr268 yielding a MOMP268T/G derivative in NCTC11168. This substitution had no deleterious effect on in vitro growth (data not shown), but the apparent size of the 11168 MOMP268T/G protein was distinct from that of wild-type MOMP and indistinguishable from that of MOMP purified from 11168ΔpseD (figure 3c), confirming that Thr268 was glycosylated in the wild-type protein, the glycosylation was responsible for the altered electrophoretic mobility and that PseD was required for this modification. Nanoflow LC-MS/MS FT/ICR analysis of purified MOMP268T/G confirmed the absence of the Hex-(HexN-acetylamine)3 glycosylation.
Figure 4.

Overview of the LC-MS/MS analysis of NCTC11168 MOMP and confirmation of the role of Thr268 glycosylation in BgAg binding. (a) Base peak chromatogram: tryptic peptides are loaded on an online coupled C18 column and eluted into the mass spectrometer for analysis. (b) MS precursor scan of the double charged glycosylated peptide at m/z 978.91. (c) CID-MS/MS spectrum of the selected ion. (d) Substitution of the glycosylation site in NCTC11168 MOMP (Thr268 to Gly) was achieved and the binding to Leb, H-II and H-I was examined. Also included was a double mutant (DM) in which flaA was also inactivated and 11168-Kan, which contains the resistance cassette at the porA locus but without the substitution. Compared with wild-type, BgAg binding was significantly reduced in both 11168MOMP268T/G and 11168-DM strains. 11168-Kan showed no significant difference in BgAg-binding activity compared with NCTC11168. Error bars: mean of triplicate values ± s.e.m. from three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001.
3.4. O-Glycosylation of NCTC11168 MOMP effects BgAg binding
The ability of the MOMP268T/G mutant to bind to a range of BgAgs in ELISA assays was examined, along with a double mutant strain in which flaA had also been inactivated. The MOMP268T/G substitution significantly reduced BgAg binding (figure 4d). To rule out polar effects linked to the insertion of a kanamycin resistance cassette at the end of porA (used to facilitate selection for the MOMP268T/G substitution), we also constructed 11168-Kan, which contains the resistance cassette at the porA locus but without the substitution. This strain showed no significant difference in BgAg-binding activity compared with NCTC11168 (figure 4d).
3.5. NCTC11168 MOMP glycosylation is an O-linked trimeric form of T-antigen
Lectin binding was used for further characterization of the O-glycan attached to NCTC11168 MOMP. Seven different lectins with overlapping specificities (see electronic supplementary material, table S4) were tested. Purified NCTC11168 MOMP showed significant binding to the lectins Jacalin [35] and, to a lesser extent, DSL and LEL (figure 5a). Among the galactose-specific lectins, Jacalin exhibits specificity for human tumour-specific Thomsen-Friedenreich disaccharide (T-antigen; Gal(β1–3)-GalNAcα1-Ser/Thr) [36]. To confirm that the Jacalin binding was the result of O-glycosylation of amino acid Thr268, MOMPs purified from NCTC11168 and NCTC11168 MOMP268T/G were probed with monoclonal anti-T-antigen in ELISA assays (figure 5b). Both the antibody and Jacalin lectin bound strongly to MOMP purified from NCTC11168; significant reduced binding was observed to MOMP268T/G (figure 5b). Combined with the previous MS/MS analysis suggesting a Hex-(HexN-acetylamine)3 glycan, our lectin-binding analysis indicates that the MOMP glycan in NCTC11168 is likely to be the O-linked trimeric form of T-antigen: Gal(β1–3)-GalNAc(β1–4)-GalNAc(β1–4)-GalNAcα1-Thr268 (electronic supplementary material, figure S1).
Figure 5.

Detection of glycan constituents of purified MOMP from NCTC11168 using biotinylated labelled lectins and antibodies against glycans. (a) Binding of the following glycans: GSL II, Griffonia (Bandeiraea) simplicifolia lectin II; DSL, Datura Stramonium lectin; ECL, Erythrina cristagalli lectin; LEL, Lycopersicon esculentum (tomato) lectin; STL, Solanum tuberosum (potato) lectin; VVA, Vicia villosa agglutinin; and Jacalin, Artocarpus integrifolia lectin to immobilized NCTC11168 MOMP was investigated using ELISA. Jacalin lectin showed the greatest degree of binding to NCTC11168 MOMP. Jacalin lectin specifically recognizes Gal(β1–3)-GalNAcα1-Ser/Thr (T-antigen). (b) ELISA confirmation using anti-T-antigen monoclonal antibody or Jacalin lectin to confirm that the glycan detected on NCTC11168 MOMP was T-antigen. 11168MOMP268T/G showed significantly reduced binding to either Jacalin lectin or anti-T antigen compared with NCTC11168 MOMP. Error bars: mean of triplicate values ± s.e.m. from three independent experiments. ***p < 0.001.
3.6. Modelling the structure of glycosylated and non-glycosylated NCTC11168 MOMP
Although Gram-negative bacterial porins show considerable diversity in their primary sequences, they are remarkably similar in their secondary and tertiary structure, in which beta strands combine to form a trans-membrane beta-barrel structure [37]. CD spectroscopy analysis demonstrated that the folded MOMP monomer mainly comprised β-sheet secondary structure [38]. Accordingly, two-dimensional crystallographic analysis showed that MOMP is structurally related to the family of trimeric bacterial porins [39]. Here, we utilized the beta-barrel structure of the anion-selective porins of Comamonas acidovorans (1E54.pdb) and Pseudomonas aeruginosa (2QTK.pdb) as templates to construct a model of C. jejuni NCTC11168 MOMP. The lowest energy structures of glycosylated and non-glycosylated MOMP at 300 K were calculated; these comprised nine loops and 18 beta strands (figure 6a). The structures of non-glycosylated MOMP and MOMP glycosylated at residue 268 were calculated and superimposed (figure 6b,c); the modification was predicted to result in major conformational changes affecting loops 4, 6 and 7 (corresponding to amino acids 169–200, 256–274 and 296–333, respectively); thus we hypothesized that the O-glycosylation of MOMP leads to conformation changes in the molecule which might favour BgAg binding. In addition, further simulations showed that the protein has a canal-like cavity, which is expected to be capable of accommodating very large molecules. However, non-glycosylated MOMP has a larger cavity compared with its glycosylated form (figure 6d,e).
Figure 6.

Three-dimensional model of MOMP. (a) Non-glycosylated (i) or glycosylated (ii) at Thr268 (loop 6) of NCTC11168 MOMP embedded in the outer membrane. Approximate boundaries of the hydrophobic regions of the proteins predicted to be embedded in the outer membrane are represented by horizontal lines. (b) The superimposed lowest energy structure of non-glycosylated MOMP on the lowest energy structure of glycosylated MOMP (magenta) with RMSD of 1.291. Loops are shown in the following colours: β strands are green; L1 (residues 41–60), red; L2 (residues 87–109), magenta; L3 (residues 128–147), orange; L4 (residues 169–200), yellow; L5 (residues 227–233), black; L6 (residues 256–274), blue; L7 (residues 296–333), grey; L8 (residues 360–379), cyan; L9 (residues 399–414), purple. (c) Superimposed surface representation of the structures of non-glycosylated MOMP (grey) on its glycosylated form (magenta) viewed from the external face. Surface representation of the structures of non-glycosylated MOMP (d) and its glycosylated form (e), viewed from the external face.
3.7. Modelling NCTC11168 MOMP–BgAg interactions
To further support our hypothesis, Leb (as an example of a type-1 Lewis antigen) and H-II (as an example of a type-2 antigen; electronic supplementary material, figure S2) were docked into the central channel-forming cavity of NCTC11168 MOMP predicted in the model structures of glycosylated MOMP (figure 7a and d, respectively) and the complexes computed in molecular dynamics (MD) [9] simulations. Interestingly, the predicted interaction between the galactose residues with Arg328 is broken in the complex of MOMP and H-II, in which the region containing the glycosylated residue moves towards loop 4 resulting in a new interaction with the residues Thr186 and Thr187. A similar conformational change was not predicted when the interaction between glycosylated MOMP and Leb was modelled. The average energies derived from MD simulations of complexes are listed in the electronic supplementary material, table S5. The introduction of the ligands leads to conformational changes affecting the longest of the predicted loops (loops 4 and 7) in particular. It was found that the predicted glycosylated MOMP structure was relatively stable compared with the non-glycosylated form, with only loop 7 predicted to undergo marked conformational changes when glycosylated MOMP is complexed with BgAgs. This prediction suggests that glycosylation enhances the stability of the protein.
Figure 7.

Influence of glycosylation on conformational changes induced by ligand binding. A stereo cartoon of the MOMP backbone viewed from the extracellular side is presented. Regions are shown in the following colours: green (β strands); red (L1); magenta (L2); orange (L3); yellow (L4); black (L5); blue (L6); grey (L7); cyan (L8); purple (L9). The complexes are with Leb (a) and H-II (d). In addition, hydrogen bonds shown in light blue correspond to amino acids residues, which are putatively involved in the interactions of glycosylated MOMP (b and e) and its non-glycosylated form (c and f) with Leb and H-II, respectively, in their active sites.
Figure 7 highlights the residues of glycosylated and non-glycosylated MOMP contributing to the interaction of these proteins with Leb (b and c, respectively) and H-II (e and f, respectively). It is apparent that the glycosylated MOMP has favourable interactions with Leb compared with the unmodified version. Specifically, the residues Arg352, Lys278 and Lys385 are predicted to be major contributors to the interaction of the glycosylated protein with Leb via hydrogen bonds, whereas only the residues Asn258 and Lys278 of non-glycosylated MOMP are predicted to interact with Leb. Residues Arg352 and Lys385 are both within loop 7, which is predicted to be more susceptible to conformation changes as a result of glycosylation or interaction with BgAgs in MD simulations (figure 7b). Incorporation of the glycosyl group with this loop thus appears to stabilize a conformation that favours interaction with Leb.
Calculations also show that the glycosylated protein has more favourable van der Waals interactions compared with the non-glycosylated protein: the residues Leu290, Tyr294, Phe395 and Ile337 are located over the hydrophobic surface of Leb in the complex of glycosylated MOMP but not in the complex with non-glycosylated MOMP (figure 7b and c, respectively). This is reflected in a 67 kcal mol−1 van der Waals energy difference between the two complexes. H-II is predicted to be bound to both versions of the protein in a similar manner to Leb. The residues Lys278, Arg352 and Arg381 are involved in the complex of both glycosylated and non-glycosylated MOMP with H-II (figure 7e and f, respectively). The two complexes differ in that residue Asp261 contributes to the interaction of H-II with non-glycosylated MOMP, but not glycosylated MOMP, whereas the reverse is true for Ser397. MD calculations also show that active sites of the glycosylated and non-glycosylated proteins have different conformational orientations with respect to the two ligands.
We attempted to substitute five amino acids (S262-G, K278-G, R352-G, R381-G and S397-Y) within the predicted binding site of NCTC11168 MOMP (singly or in various combinations) but were unable to do so, suggesting perhaps that these amino acid residues are important for MOMP stability and thus function. Overall, our modelling confirms that the O-glycosylation of MOMP favours BgAg binding.
3.8. O-Glycosylation of NCTC11168 MOMP effects auto-agglutination, biofilm formation, adherence to Caco-2 cells and chicken colonization
Loss of O-glycans, for example PseAm, from C. jejuni flagellins has previously been shown to affect the ability of the organism to auto-aggregate, form biofilms, adhere to and invade monolayers of INT407 cells and cause disease in in vivo models of infection [19]. We therefore hypothesized that loss of the MOMP O-glycan may also lead to similar effects. As expected, 11168MOMP268T/G had a significant reduced propensity to auto-agglutinate or form biofilms compared with wild-type (figure 8a and b, respectively). Furthermore, the addition of soluble BgAgs to the culture media led to reduced biofilm formation by NCTC11168, suggesting that BgAgs compete with biofilm determinants on flagellin and MOMP (see electronic supplementary material, figure S3). Additionally, pre-treatment of Caco-2 cells with purified glycosylated MOMP (but not MOMP268T/G) significantly inhibited the association of wild-type NCTC11168 to Caco-2 cells (figure 8c), which express H-I, H-II and Leb BgAgs [40]. Adherence of 11168MOMP268T/G to Caco-2 cells was significantly reduced compared with wild-type NCTC11168. Levels of 11168MOMP268T/G adhesion were unaffected by the pre-treatment of Caco-2 cells with either MOMP derivative (figure 8c).
Figure 8.

O-glycosylation of MOMP effects auto-agglutination, biofilm formation, adherence to Caco-2 cells and chicken colonization. (a) The ability of 11168MOMP268T/G to auto-agglutination was significantly reduced compared with wild-type 11168 (p < 0.0001). 11168ΔflaA was used as a negative control for aggregation. Error bars (too small to be seen) = mean of triplicate values on three occasions ± s.e.m. (b) Similarly, the ability of 11168MOMP268T/G to form biofilms was significantly reduced compared with wild-type NCTC11168. 11168ΔflaA was used as a negative control for biofilm formation. Error bars = mean of triplicate values on three occasions ± s.e.m. *p < 0.05; ***p < 0.001. (c) Binding of NCTC11168 and 11168MOMP268T/G to Caco-2 cells in the absence of inhibitor (non-treated, NT) or following pre-treatment of Caco-2 cells with purified MOMP or MOMP268T/G. *Significantly reduced compared with the binding of non-treated NCTC11168 (p < 0.05). Error bars: mean of triplicate values ± s.e.m. from three independent experiments. (d) Colonization of chickens by NCTC11168-O and 11168-O MOMP268T/G mutant 7 days post-infection. The geometric mean colonization levels in caeca are significantly reduced (p < 0.0001) and are below the limit of detection (102 cfu g−1) in 8/10 birds inoculated with 11168-O MOMP268T/G. Similarly, levels of 11168-O MOMP268T/G mutant in the liver were below the limit of detection in 10/10 birds 7 days post-infection.
Finally, the ability of 11168MOMP268T/G to colonize chickens was determined. To do this, six-week-old birds (n = 10 per group) were challenged with 3 × 103 cfu wild-type strain NCTC11168-O (a robust colonizing strain, derived from NCTC11168 by passage through chicken) or its 11168-O MOMP268T/G mutant by oral gavage. Caecal colonization levels were determined in birds from each group at 7 days post-challenge. The results showed a significant reduction in the geometric mean colonization levels in the caeca and liver in the MOMP268T/G group compared with the wild-type (figure 8d). These results confirm the importance and biological relevance of MOMP O-glycosylation in the establishment of colonization in vivo.
4. Discussion
There is currently a need for a better understanding of the pathogenic mechanisms underlying the infection of humans and colonization of other animals, particularly chickens, by C. jejuni. Here, we confirm that C. jejuni, like H. pylori, can bind to BgAgs and to the lacto series type I and II, a characteristic that is likely to contribute to the ability of this intestinal bacterium to interact with epithelial cells lining the gastrointestinal tract. The high degree of specificity exhibited by H. pylori in its BgAg-binding interactions [28,29] is in contrast to our findings, and those of Day et al. [23], showing that C. jejuni can bind to a wide range of related antigens. This may reflect the contrast between the very restricted host range of H. pylori, which infects only humans, and that of C. jejuni, which is able to establish infection in a wide range of birds and mammals. The latter may thus have gained an evolutionary advantage by broadening its specificity and thus maximizing its survival in different hosts. Whether C. jejuni can bind to non-human BgAgs, especially those expressed by chickens, remains to be experimentally confirmed, although our in vivo data suggest that this is a possibility since a C. jejuni derivative lacking the MOMP O-glycan (and hence exhibiting reduced BgAg-binding) showed a significantly reduced ability to colonize chickens.
Identification of both FlaA and MOMP as BgAg-binding adhesins was surprising as these proteins have little in common in terms of their primary sequence, predicted structure or known functions. FlaA is the major structural subunit of the flagellum and is thus required for motility. Intact and fully functional flagella are required for the colonization of chickens, invasion into human epithelial cells and translocation of human intestinal cell monolayers [41–43]. MOMP is a member of the trimeric bacterial porin family and forms voltage-sensitive cation-selective ion channels [44]. It is predicted to comprise outer membrane-spanning beta strands separated by short periplasmic and longer surface-exposed loops [30]. In addition to its porin functions, MOMP has been shown to bind to the surface of human epithelial cells and to the basement membrane protein fibronectin [45]. Interestingly, it shares this latter property with FlaA, as well as the outer membrane protein CadF [45]. Thus, both MOMP and FlaA appear to mediate binding to multiple host cell molecules, which now includes BgAgs. MOMP is encoded by the porA gene, which is extremely genetically diverse with most variation occurring in regions encoding surface loops (particularly loop 4), suggesting that immune selection strongly influences the diversity of the MOMP protein [46].
Campylobacter flagella and numerous surface proteins are post-translationally decorated with glycan molecules [47]. This led us to hypothesize that, like FlaA, MOMP is glycosylated, and that BgAg binding by these two diverse ligands may be influenced by the presence of surface glycans. Campylobacter jejuni harbours both N- and O-linked glycosylation systems [47]. The genes known to contribute to the O-glycosylation of C. jejuni flagellin are clustered in the flagellar glycosylation locus [19]. We have demonstrated that, like fla gellin, MOMP is also O-glycosylated, the first demonstration that a non-flagellin protein is glycosylated by this system. Our results also demonstrate that the MOMP O-glycan contains an epitope which mimics a GM1 ganglioside epitope: Gal(β1–3)-GalNAc(β1–4)-Gal-[Neu5Acα2–3]-(β1–4)-Glcβ1-ceramide. As MOMP is highly immunogenic, the expression of a glycosylated form of MOMP may play a hitherto underappreciated role in the development of autoimmune diseases, for example GBS. This potentially significant finding requires further experimental consideration.
Our finding that MOMP is O-glycosylated may provide an explanation for a recent observation made by Hitchen et al. [48]. They showed that the product of the phase variable gene Cj1295 in C. jejuni NCTC11168 was responsible for the modification of Pse residues present on flagellins with di-O-methyl-glyceric acid [48]. Immunoblots of whole-cell extracts using rabbit polyclonal anti-flagellin antibodies showed the differing electrophoretic mobilities of flagellin with or without this Pse modification; however the anti-flagellin antibodies also detected one additional protein: a 45 kDa protein, subsequently identified as MOMP [48]. The detection of MOMP using anti-flagellin was suggested to be due to non-specific binding [48]. However, in light of our data confirming the O-glycosylation of MOMP, we hypothesize that the cross-reactivity is due to the presence of shared epitopes, likely to be shared or similar O-glycan determinants. We are currently investigating possible similarities between the MOMP and flagellin O-glycan repertoires.
Having shown glycosylation, we identified T268 as the target for the O-glycosylation of MOMP in strain NCTC11168; this residue is within the sixth predicted surface-exposed loop, but is conserved in only 25 of the 46 sequences examined (see electronic supplementary material, figure S4). These included six clinical isolates tested for their ability to bind BgAgs: three high BgAg binders and three isolates with low BgAg-binding activity. T268 was conserved in all three high binders, but also in two of the three low binders. Previously, Seal et al. [49] reported that arginine was substituted for T268 in the MOMP sequence of the isolate A74/C compared with NCTC11168-PMSRU. The latter, a derivative of strain NCTC11168, was described as a poor chicken colonizing strain, while the former was described as a robust colonizer [49]. Changes in the isoelectric point of MOMP were among the differences reported between these two isolates, while the apparent molecular mass (as judged by electrophoretic mobility in SDS-PAGE gels) was indistinguishable between the two proteins. This is in contrast to our findings, where the substitution of glycine for T268 resulted in a distinct mobility shift that was indistinguishable from that observed in MOMP isolated from a pseD mutant. It is notable that the only other substitution in the A74/C MOMP sequence with respect to the NCTC11168 sequence was threonine for R367, a residue predicted to reside within the eighth surface-exposed loop according to the model proposed by Zhang et al. [30]. It is tempting to speculate that threonine at position 367 (numbering corresponding to the NCTC11168 sequence) may be an alternative site for O-glycosylation. Of the isolates examined, eight of the 46 (17%) harboured a threonine at this position and 30 (65%) had threonine at either amino acid 268 or 367. Whether threonine at this position or indeed at other positions within variant MOMP proteins are targets for the O-glycosylation system remains to be determined. All of the C. jejuni clinical isolates tested bound to at least some of the BgAgs, but the degree of binding was highly variable; variations in MOMP primary sequence and perhaps the presence of specific targets for the O-glycosylation system are likely to account for at least some of this variation. Likewise, variable O-glycosylation of FlaA may also account for variations in BgAg binding between strains.
It remains to be determined whether the bacterial O-glycans exert a direct (i.e. bacterial glycan to host glycan) or indirect effect on BgAg binding. However, based on our molecular modelling data, our favoured hypothesis is that the effect is indirect, whereby the presence of O-glycans results in conformational changes or stabilization of particular conformations of the bacterial ligands, which favour BgAg binding.
Identification of protein glycosylation profiles of C. jejuni, especially those related to outer membrane proteins, is fundamental for understanding the diverse pathogenicity of C. jejuni strains among different hosts. The model for C. jejuni interaction to Leb and H-II antigens mediated by MOMP generated here substantially increases our knowledge about the protein and its interactions. The identification of BgAgs as receptors for the adhesive function of MOMP and the mapping of the binding site could inform vaccine design. Attempts to develop vaccines have met with little success, and concerns about the possible induction of autoimmunity-associated pathologies, for example GBS, are a barrier to the development of whole-cell vaccines for use in humans [50]. An alternative approach is to use epitopes that play an important role in colonization; glycosylated peptides would constitute particularly attractive targets for such an approach as they may be under selective pressure to be maintained, and neutralizing antibodies may reduce the ability of the pathogen to colonize the host. Immunization experiments with adhesins of uropathogenic Escherichia coli demonstrate the potential in the generation of antibodies with adhesion inhibitory properties [51]. Also, Houpt et al. [52] described an effective vaccine against intestinal Entamoeba histolytica infection using amoebic Gal/GalNAc lectin, which blocks both invasive disease and parasite transmission. Though the potential for developing vaccines against C. jejuni for use in humans may be limited, vaccines targeted at animal hosts of C. jejuni, particularly chicken, represent a promising approach to the reduction of C. jejuni contamination of poultry, and thus indirectly human infection. In a study by Huang et al. [53], vaccination of chickens with recombinant MOMP reconstituted in proteoliposomes demonstrated conformation-dependent humoral responses to MOMP, which confirms our finding that any modification of MOMP has important implications for protein conformation and may alter MOMP immunogenicity. The stereochemical fit of the molecular interaction between microbial adhesins and host receptors determines in part tissue tropism during the colonization process. The possibility of interfering with such interactions provides new approaches to the development of antimicrobial strategies at least during the early stage of colonization. Anti-adhesive receptor analogues may prove to be a viable strategy for lowering colonization levels in poultry.
In summary, this work provides key new insights into C. jejuni pathogenesis and, importantly, illustrates how glycosylation of outer membrane proteins dynamically furnishes C. jejuni with features that are of importance for the bacterial–host crosstalk during infection in the human host.
5. Material and methods
5.1. Bacterial strains and growth conditions
Escherichia coli strain JM109 was used as a host strain for the construction of mutagenic constructs (see electronic supplementary material, table S6). E. coli strains were grown in Lysogeny broth supplemented with ampicillin (100 µg ml−1), kanamycin (50 µg ml−1) or chloramphenicol (20 µg ml−1). Campylobacter jejuni NCTC11168 and most of the clinical isolates used in this study were kindly provided by Prof. J. Ketley (University of Leicester, UK; electronic supplementary material, table S2), with the remaining clinical isolates being obtained from the Department of Clinical Microbiology, Nottingham University Hospitals NHS Trust, Nottingham, UK (see electronic supplementary material, table S2). All C. jejuni strains were grown under microaerophilic conditions at 37°C on modified CCDA agar (Oxoid), or in Mueller–Hinton broth (Oxoid) supplemented with kanamycin (50 µg ml−1) and chloramphenicol (20 µg ml−1) where appropriate.
5.2. Digoxigenin labelling of bacteria
DIG-labelling was carried using the Roche DIG-NHS Protein labelling kit, used as per manufacturer's instructions. Briefly, cells were harvested from an overnight plate and washed three times in sodium carbonate buffer (142 mM NaHCO3, 8 mM Na2SO3, pH 9.0). Bacteria were resuspended to an optical density at 600 nm (OD600) of 1.0 and then labelled with 10 µg DIG-NHS at room temperature for 1 h. Cells were then washed three times in PBS with 0.05% Tween 20 (PBS-T) and resuspended in PBS containing 1% bovine serum albumin (1% BSA/PBS).
5.3. ELISA
BSA–BgAg conjugates were obtained from IsoSep, Tullinge, Sweden. Coupling of BgAgs to 96-well plates (NUNC Immobilizer Amino) was carried out by the addition of 100 µl BSA–BgAg (5 µg ml−1 unless stated otherwise) in sodium carbonate buffer to each well. Plates were incubated at room temperature for 2 h before unbound reagent was removed by washing three times in PBS-T. All wells were blocked by the addition of 100 µl 1% BSA/PBS and incubated for 2 h at room temperature. After further washing in PBS-T, 100 µl of DIG-tagged bacteria (at OD600 of 0.05 unless stated otherwise) were added to each well and incubated overnight at 4°C. Plates were washed three times in PBS-T before 100 µl anti-digoxigenin-POD solution (Roche Diagnostics; 1 in 5000 diluted in 1% BSA/PBS) was added and incubated for 1 h at room temperature. Plates were again vigorously washed in PBS-T and colour developed by adding 100 μl ABTS substrate (Roche). Plates were read with an ELISA reader (Biotek EL800) at an absorbance of 405 nm. Specific binding was determined by subtracting the binding of each strain to BSA (typically OD405 0.07–0.09) from the binding to each BSA–BgAg conjugate. Statistical significance was determined using Student's t-test. Inhibition assays were carried out as above but after the removal of the blocking solution, 100 µl purified MOMP (5 µg ml−1 in 1% BSA/PBS) was added to each well and incubated at 4°C overnight. Alternatively, DIG-labelled C. jejuni were pre-incubated for 4 h with BgAgs (20 μg ml−1) in 1% BSA/PBS before being added to each well.
5.4. Re-tagging
NCTC11168 (1 ml of OD600 0.25) was incubated for 5 h with BSA conjugated to Leb, H-II or H-I (20 µg) (or BSA alone as control) to which the Sulfo-SBED cross-linker (Pierce) had been conjugated according to the manufacturer's recommendations. Following washing in PBS-T, the photo-reactive cross-linker group was activated by 10 min exposure to ultraviolet irradiation (365 nm). Samples were then incubated under reducing conditions (50 mM dithiothreitol) for 10 min, washed with PBS-T and boiled three times for 5 min with 10 min intervals between boiling. Biotin-(re)tagged proteins were then purified with streptavidin-coated magnetic beads as previously described [28]. Extracted biotin-tagged proteins were separated by SDS-PAGE and bands digested with sequencing-grade trypsin (Promega) and analysed using a Micromass Tof-Spec E (Micromass).
5.5. Mutagenesis
To mutate flaA, a 5.58 kb fragment encompassing the flaA locus was amplified from C. jejuni NCTC11168 using primers FLAAF1 and FLAAR1 (see electronic supplementary material, table S7) using the Expand High Fidelity PCR system (Roche). The resulting amplicon was ligated into pGEM-T Easy according to the manufacturer's protocol to yield pNJO83 (see electronic supplementary material, table S6). This was subjected to inverse PCR using primers FLAAR2 and FLAAINV5 (see electronic supplementary material, table S7) resulting in an amplicon in which the flaA gene was deleted and containing a unique BamHI site which was used to introduce the chloramphenicol resistance cassette from pAV35 in place of the deleted gene. The resulting plasmid, pJS101, was confirmed by sequencing before being used to mutate C. jejuni by electro-transformation and allelic exchange as described previously [54]. To mutate pglB, chromosomal DNA extracted from C. jejuni 81–176ΔpglB (obtained from Prof. J. Ketley, University of Leicester, UK) was used to mutate NCTC11168 by natural transformation and allelic exchange as described previously [54]. To mutate pseD, a 3.9 kb fragment encompassing the pseD gene was amplified from NCTC11168 template DNA using primers Cj1333_F and Cj1333_R2 (see electronic supplementary material, table S7). The resulting amplicon was ligated into pGEM-T Easy to yield plasmid pKG352 (see electronic supplementary material, table S6). The pseD gene was deleted from this plasmid by inverse PCR amplification using primers Cj1333_M1 and Cj1333_M3 (see electronic supplementary material, table S7) and the resulting amplicon digested with BglII and self-ligated to produce plasmid pKG353 (see electronic supplementary material, table S6). Plasmid pKG353 was digested with BglII and ligated to a kanamycin resistance cassette obtained by the digestion of pJMK30 (see electronic supplementary material, table S6) with BamHI. A resulting plasmid containing the kanamycin resistance cassette in the reverse orientation to the deleted pseD gene was designated pKG354 (see electronic supplementary material, table S6) and used to mutate NCTC11168 as described above. To construct MOMP268T/G, a 2 kb region spanning porA was amplified from C. jejuni NCTC11168 using MOMPF4 and MOMPR4 (see electronic supplementary material, table S7) and cloned into pGEM-T Easy to yield pNJO76 (see electronic supplementary material, table S6). This construct was used as template for inverse PCR using MOMPF3 and MOMPR3, which introduced a unique BglII site in the intergenic region between porA and dnaJ into which the kanamycin cassette from pJMK30 could be introduced. The resulting plasmid, pNJO78, was confirmed by restriction digestion and sequencing before being used to mutate NCTC11168 by electro-transformation and allelic exchange to create 11168-Kan. pNJO78 was also used as template for the QuikChange Site-Directed mutagenesis kit (Stratagene). SDMOMP1 and SDMOMP2 primers (see electronic supplementary material, table S7) were designed to substitute the threonine at position 268. The resulting plasmid, pSD, was confirmed by sequencing before being used to mutate C. jejuni by electro-transformation and allelic exchange. Mutants lacking flaA in the MOMP268T/G genetic background were created by electro-transforming this mutant with pJS101 (see electronic supplementary material, table S6). All mutants were confirmed by PCR and DNA sequencing (data not shown).
5.6. Purification of Campylobacter jejuni MOMP and generation of rabbit anti-MOMP antiserum
Campylobacter MOMP was purified to homogeneity according to a previously described method [38]. New Zealand White female rabbits were immunized subcutaneously three times at two-week intervals with 30 µg of purified NCTC11168 MOMP emulsified in Freud's complete (first immunization only) or incomplete adjuvant. Rabbit anti-MOMP antisera was used in immunoblotting experiments 1 : 5000 diluted in blocking buffer (1% BSA in PBS-T) and incubated for 2 h. After washing three times in PBS-T, membranes were incubated for 2 h with goat anti-rabbit IgG–alkaline phosphatase conjugate (Sigma), at a dilution of 1 : 2000 in blocking buffer. After washing with PBS-T, the blots were developed using BCIP/NBT-Blue liquid substrate (Sigma).
5.7. Identification of glycopeptides from purified MOMP
MOMP was purified under non-denaturing conditions and analysed by Nanoflow LC-MS/MS FT/ICR following in-gel protein digestion as previously described [34]. Briefly, the gel pieces were destained by washing three times in 25 mM NH4HCO3 in 50% CH3CN and once in 25 mM NH4HCO3 in 50% CH3OH. Gel pieces were dried in a vacuum centrifuge and incubated with digestion buffer (50 mM NH4HCO3, 10 ng µl−1 trypsin) at 37°C overnight. Peptides were extracted in 50% CH3CN/1% CH3COOH and the supernatant was evaporated to dryness in a vacuum centrifuge. Prior to MS analysis, the peptides were reconstituted in 0.2% HCOOH. After digestion, samples were subjected to LC-MS/MS analysis using a hybrid linear ion trap-FTICR mass spectrometer operated in data-dependent mode, automatically switching to MS/MS mode. MS-spectra were acquired in the FTICR, while MS/MS-spectra were acquired in the LTQ-trap. For each FTICR scan, the six most intense, double-, triple- and quadruple-charged, ions were sequentially fragmented in the linear trap by collision-induced dissociation. All tandem mass spectra were searched by Mascot (Matrix Science, London, UK) against the SwissProt database (release 54.0). Glycan structure and position were determined by the manual interpretation of the CID fragmentation spectra, using the Xcalibur software v. 2.0.7 (Thermo Scientific).
5.8. Detection of glycoproteins using lectins or anti-T antigen
Microtitre plates (NUNC Immobilizer Amino) were coated with NCTC11168 MOMP by placing 5 μg ml−1 glycoprotein in sodium carbonate buffer in each well. Untreated wells were used as negative controls. Plates were incubated at 37°C for 1 h, before being washed three times with PBS-T. Non-specific binding was blocked by adding 1% BSA/PBS to each well for 30 min at room temperature. Plates were then washed three times with PBS-T, before 3 μg ml−1 biotinylated lectins (Vector) or anti-T antigen (1 : 5000) in PBS were added and incubated for 30 min at room temperature. Following further washing with PBS-T, avidin–biotin-complex (peroxidase) was added according to the kit instructions and incubated for 30 min at room temperature following by washing three times with PBS-T. Finally, 100 µl of ABTS was applied and plates were read with an ELISA reader (Biotek EL800) at an absorbance of 405 nm.
5.9. Computational modelling
All molecular dynamic simulations were conducted by using the Amber (v. 10.0) suite of programs on the Linux/Intel PC cluster of TR-Grid maintained by TUBITAK (Scientific and Technologic Research Council of Turkey). We initiated our simulations using the following amino acid sequence:
MKLVKLSLVAALAAGAFSAANATPLEEAIKDVDVSGVLRYRYDTGNFDKNFVNNSNLNNSKQDHKYRAQVNFSAAIADNFKAFVQFDYNAADGGYGANGIKNDQKGLFVRQLYLTYTNEDVATSVIAGKQQLNLIWTDNAIDGLVGTGVKVVNNSIDGLTLAAFAVDSFMAAEQGADLLEHSNISTTSNQAPFKVDSVGNLYGAAAVGSYDLAGGQFNPQLWLAYWDQVAFFYAVDAAYSTTIFDGINWTLEGAYLGNSLDSELDDKTHANGNLFALKGSIEVNGWDASLGGLYYGDKEKASTVVIEDQGNLGSLLAGEEIFYTTGSRLNGDTGRNIFGYVTGGYTFNETVRVGADFVYGGTKTEAANHLGGGKKLEAVARVDYKYSPKLNFSAFYSYVNLDQGVNTNESADHSTVRLQLYKF.
The model was constructed based on the similar secondary structures exhibited by porins [30]. The core structure of MOMP was initiated by using the skeleton of the anion-selective porins from Co. acidovorans (1E54.pdb) and P. aeruginosa (2QTK.pdb) [55] as templates to build the beta-barrels. A combination of HyperChem (HyperChem Professional 7.51), Chimera (UCSF) [56] and the LEaP module as implemented in Amber were used to build the core and add the loops and turns. The initial structure was heated from 0 to 325 K with a restraint of 10 kcal mol−1 Å−2 on residues of beta-barrels to avoid the effect of conformational changes in loops on beta-barrels for a period of 200 ps in four steps, followed by simulations from 0 to 325 K for another period of 200 ps without any restraints in four steps. The system was further simulated at 300 K for a period of 8 ns. The ff99SB force field [57] was employed and solvation effects were incorporated using the Generalized Born model [58], as implemented [59] in Amber. A lower energy structure was chosen and this was glycosylated at the residue 268 with Gal(β1–3)-GalNAc(β1–4)-GalNAc(β1–4)-GalNAc-α-linked to the protein using xleap as implemented in Amber. The Glycam04 force field [60–62] was used to model the carbohydrate unit. The charge on the oxygen of the side chain of Thr was changed from −0.6761 to −0.4599 and the atom type of OS was assigned. The angle and dihedral parameters for dimethylether (CT-OS-CG) and dimethoxymethane (H2-CG-OS-CT) were used for the glycosylated angle and dihedral for the carbohydrate linkage. The system was minimized with 500 steps of the steepest descent minimization followed by 500 steps of conjugate gradient minimization and heated at 400 K for a period of 10 ps to avoid bad contacts with a restraint of 10 kcal mol−1 Å−2 on the protein backbone. The system was heated from 0 to 325 K for a period of 200 ps without any restraints, followed by a simulation at 300 K for a period of 3.5 ns. Root-mean-square deviation (RMSD) analysis for the complex system was carried out on the trajectories by the ptraj module of Amber (v. 10). Three-dimensional structures were displayed using Chimera (UCSF) [63], and RMSD graphics were generated by Xmgrace. Docking calculations were performed to accommodate the Lewis antigen (Leb) and H-II antigen within the cavity of the protein. Docking of the Leb was carried out using Dock v. 6.0 [64]. Docking was performed with default settings to obtain a population of possible conformations and orientations for Leb at the binding site. Spheres around the centre of the binding pocket were defined as the binding pocket for the docking runs. Dock v. 6.0 employs sphgen to produce spheres and because sphgen cannot handle more than 99 999 spheres, the residues forming loops were stripped off, and thus the calculations of spheres and grids were only performed with the beta-barrel forming the cavity. Then, the coordinates of the Leb obtained were recorded and AM1-Bcc (Austian model with Bond and charge correction) [65] atomic partial charges and atom types of general amber force field [66] were assigned for them using antechamber as implemented in Amber. Xleap was used to accommodate the Leb within the cavity of MOMP with combine command as well as to produce topology/parameter and coordinate files. The atom type of Leb was changed to those described in the Glycam04 force field [60–62]. The system was minimized, followed by MD simulation at 300 K for about 6.0 ns. The same procedure was applied to the glycosylated protein.
5.10. Auto-agglutination assay
Campylobacter jejuni strains were grown overnight in broth under microaerophilic conditions at 37°C, with shaking. Cultures were filtered to remove large aggregates (5 µm pore size filters; Millipore) and adjusted to OD600 of 1.0. One millilitre of culture was transferred to a cuvette, left at room temperature and OD600 monitored every 10–20 min for up to 6 h.
5.11. Biofilm assay
The method carried out was adapted from [67,68]. Campylobacter jejuni strains were grown overnight in MH broth under microaerophilic conditions at 37°C, with shaking. The OD600 of those cultures was measured and used to inoculate MH broth (for inhibition studies containing 10 µg ml−1 of BgAgs) to get a final OD600 of 0.02. One hundred and fifty microlitres of broth was added to wells of polystyrene 96-well plates (Nunc) with control wells containing broth alone. The plates were incubated under the same microaerophilic conditions at 37°C with shaking for 2 days. Following incubation, biofilm formation was quantified. Briefly, liquid medium was removed, and the plate washed three times with PBS-T. Then, 150 μl of 1% crystal violet was added into each well, and the plate covered and stained for 5 min. The crystal violet was removed, and the dye on the plate was resolved by adding 200 μl of 33% glacial acetic acid. Plates were then measured using an ELISA reader at 595 nm.
5.12. Caco-2 association assays
Association assays were essentially carried out as previously described [10]. Briefly, 24-well tissue culture plates were seeded with Caco-2 cells in DMEM + 5% fetal calf serum + antibiotic/antimycotic solution (Invitrogen) and incubated until fully confluent and then for an additional 3–4 days until cells were showing evidence of differentiation (formation of domes and visible thickening of the monolayer). One day prior to infection, the culture medium was changed to DMEM + 2% serum (no antibiotics). For inhibition experiments, 20 µg ml−1 purified NCTC11168 MOMP or MOMPT268T/G, in DMEM + 2% serum, was added to monolayers for 3 h at 37°C under an atmosphere of 5% CO2. After washing unbound protein three times with PBS, 108 cfu per well C. jejuni NCTC11168 or 11168MOMPT268T/G were added and monolayers incubated for 3 h at 37°C under an atmosphere of 5% CO2. Total association was assayed by washing three times in PBS, followed by homogenization of monolayers in 0.5% sodium deoxycholate in PBS. Associated C. jejuni cells were enumerated by serial dilution, and plating of aliquots onto modified CCDA agar (Oxoid). Assays were repeated three times in duplicate and statistically significant differences assessed using Student's t-test.
5.13. Chicken colonization study
Briefly, 20 six-weeks-old White Leghorn chickens derived from specific pathogen-free (SPF) parents (Lohmann's, Germany) were randomly divided into two groups (n = 10 per group) and housed in separate isolators with unlimited food and water. At day one, all birds in both groups were challenged by oral gavage with 3 × 103 cfu C. jejuni resuspended in 100 µl PBS. Necropsies were performed on all birds at 7 days post-challenge. Caeca and liver were removed aseptically to quantify colonization levels of bacteria by plating out serial dilutions of gut contents and homogenized liver onto blood agar plates containing Skirrow's antibiotics and cefoperazone (20 µg ml−1) [69]. The detection limit for colonization was 102 cfu g−1 caecal content.
Supplementary Material
Acknowledgements
We thank Dr N. Hussein (University of Duhok, Iraq) for technical assistance during the construction of plasmid pSD, and Prof. D. A. Case (Rutgers State University, NJ, USA) for providing the licence of Amber (to N.P.), TR-Grid (TUBITAK, Turkey) and DUBAP Dicle University (02-FF-20 and 05-FF-30) for the computational facilities (to N.P.). We also thank the Proteomics Core Facility at Sahlgrenska Academy, Gothenburg University, and Prof. J. Ketley (University of Leicester, UK) for providing Campylobacter strains.
Animal studies were conducted under the Animals Scientific Procedures Act (1986) and were approved by the AHVLA local ethical review committee.
Funding statement
This work was supported by the Medical Research Council (MRC), UK (grant no. G0901696).
References
- 1.Butzler JP. 2004. Campylobacter, from obscurity to celebrity. Clin. Microbiol. Infect. 10, 868–876. (doi:10.1111/j.1469-0691.2004.00983.x) [DOI] [PubMed] [Google Scholar]
- 2.Young KT, Davis LM, Dirita VJ. 2007. Campylobacter jejuni: molecular biology and pathogenesis. Nat. Rev. Microbiol. 5, 665–679. (doi:10.1038/nrmicro1718) [DOI] [PubMed] [Google Scholar]
- 3.Schaad UB. 1982. Reactive arthritis associated with Campylobacter enteritis. Pediatr. Infect. Dis. 1, 328–332. (doi:10.1097/00006454-198209000-00009) [DOI] [PubMed] [Google Scholar]
- 4.Hannu T, Mattila L, Rautelin H, Pelkonen P, Lahdenne P, Siitonen A, Leirisalo-Repo M. 2002. Campylobacter-triggered reactive arthritis: a population-based study. Rheumatology 41, 312–318. (doi:10.1093/rheumatology/41.3.312) [DOI] [PubMed] [Google Scholar]
- 5.Hughes RA, Cornblath DR. 2005. Guillain–Barre syndromé. Lancet 366, 1653–1666. (doi:10.1016/S0140-6736(05)67665-9) [DOI] [PubMed] [Google Scholar]
- 6.Lecuit M, et al. 2004. Immunoproliferative small intestinal disease associated with Campylobacter jejuni. N. Engl. J. Med. 350, 239–248. (doi:10.1056/NEJMoa031887) [DOI] [PubMed] [Google Scholar]
- 7.Schwerer B. 2002. Antibodies against gangliosides: a link between preceding infection and immunopathogenesis of Guillain–Barré syndrome. Microbes Infect. 4, 373–384. (doi:10.1016/S1286-4579(02)01550-2) [DOI] [PubMed] [Google Scholar]
- 8.Min T, Vedadi M, Watson DC, Wasney GA, Munger C, Cygler M, Matte A, Young NM. 2009. Specificity of Campylobacter jejuni adhesin PEB3 for phosphates and structural differences among its ligand complexes. Biochemistry 48, 3057–3067. (doi:10.1021/bi802195d) [DOI] [PubMed] [Google Scholar]
- 9.Muller A, León-Kempis Mdel R, Dodson E, Wilson KS, Wilkinson AJ, Kelly DJ. 2007. A bacterial virulence factor with a dual role as an adhesin and a solute-binding protein: the crystal structure at 1.5 Å resolution of the PEB1a protein from the food-borne human pathogen Campylobacter jejuni. J. Mol. Biol. 372, 160–171. (doi:10.1016/j.jmb.2007.06.041) [DOI] [PubMed] [Google Scholar]
- 10.Ashgar SS, Oldfield NJ, Wooldridge KG, Jones MA, Irving GJ, Turner DP, Ala'Aldeen DAA. 2007. CapA, an autotransporter protein of Campylobacter jejuni, mediates association with human epithelial cells and colonization of the chicken gut. J. Bacteriol. 189, 1856–1865. (doi:10.1128/JB.01427-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin S, Joe A, Lynett J, Hani EK, Sherman P, Chan VL. 2001. JlpA, a novel surface-exposed lipoprotein specific to Campylobacter jejuni, mediates adherence to host epithelial cells. Mol. Microbiol. 39, 1225–1236. (doi:10.1111/j.1365-2958.2001.02294.x) [DOI] [PubMed] [Google Scholar]
- 12.Konkel ME, Garvis SG, Tipton SL, Anderson JDE, Cieplak JW. 1997. Identification and molecular cloning of a gene encoding a fibronectin-binding protein (CadF) from Campylobacter jejuni. Mol. Microbiol. 24, 953–963. (doi:10.1046/j.1365-2958.1997.4031771.x) [DOI] [PubMed] [Google Scholar]
- 13.Schroder W, Moser I. 1997. Primary structure analysis and adhesion studies on the major outer membrane protein of Campylobacter jejuni. FEMS Microbiol. Lett. 150, 141–147. (doi:10.1016/S0378-1097(97)00111-0) [DOI] [PubMed] [Google Scholar]
- 14.Guerry P. 2007. Campylobacter flagella: not just for motility. Trends Microbiol. 15, 456–461. (doi:10.1016/j.tim.2007.09.006) [DOI] [PubMed] [Google Scholar]
- 15.Wacker M, et al. 2002. N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science 298, 1790–1793. (doi:10.1126/science.298.5599.1790) [DOI] [PubMed] [Google Scholar]
- 16.Szymanski CM, Burr DH, Guerry P. 2002. Campylobacter protein glycosylation affects host cell interactions. Infect. Immun. 70, 2242–2244. (doi:10.1128/IAI.70.4.2242-2244.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez F, Sharma R, Hinton M, Bedford MR. 2000. Diet influences the colonisation of Campylobacter jejuni and distribution of mucin carbohydrates in the chick intestinal tract. Cell. Mol. Life Sci. 57, 1793–1801. (doi:10.1007/PL00000659) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howard SL, et al. 2009. Campylobacter jejuni glycosylation island important in cell charge, legionaminic acid biosynthesis, and colonization of chickens. Infect. Immun. 77, 2544–2556. (doi:10.1128/IAI.01425-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guerry P, Ewing CP, Schirm M, Lorenzo M, Kelly J, Pattarini D, Majam G, Thibault P, Logan S. 2006. Changes in flagellin glycosylation affect Campylobacter autoagglutination and virulence. Mol. Microbiol. 60, 299–311. (doi:10.1111/j.1365-2958.2006.05100.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Young NM, et al. 2002. Structure of the N-linked glycan present on multiple glycoproteins in the Gram-negative bacterium, Campylobacter jejuni. J. Biol. Chem. 277, 42 530–42 539. (doi:10.1074/jbc.M206114200) [DOI] [PubMed] [Google Scholar]
- 21.Ruiz-Palacios GM, Cervantes LE, Ramos P, Chavez-Munguia B, Newburg DS. 2003. Campylobacter jejuni binds intestinal H(O) antigen (Fucα1, 2Galβ1, 4GlcNAc), and fucosyloligosaccharides of human milk inhibit its binding and infection. J. Biol. Chem. 278, 14 112–14 120. (doi:10.1074/jbc.M207744200) [DOI] [PubMed] [Google Scholar]
- 22.Newburg DS, Ruiz-Palacios GM, Morrow AL. 2005. Human milk glycans protect infants against enteric pathogens. Annu. Rev. Nutr. 25, 37–58. (doi:10.1146/annurev.nutr.25.050304.092553) [DOI] [PubMed] [Google Scholar]
- 23.Day CJ, Tram G, Hartley-Tassell LE, Tiralongo J, Korolik V. 2013. Assessment of glycan interactions of clinical and avian isolates of Campylobacter jejuni. BMC Microbiol. 13, 228 (doi:10.1186/1471-2180-13-228) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hakomori S. 1984. Tumor-associated carbohydrate antigens. Annu. Rev. Immunol. 2, 103–126. (doi:10.1146/annurev.iy.02.040184.000535) [DOI] [PubMed] [Google Scholar]
- 25.Lloyd KO. 1987. Blood group antigens as markers for normal differentiation and malignant change in human tissues. Am. J. Clin. Pathol. 87, 129–139. [DOI] [PubMed] [Google Scholar]
- 26.Greenwell P. 1997. Blood group antigens: molecules seeking a function? Glycoconj. J. 14, 159–173. (doi:10.1023/A:1018581503164) [DOI] [PubMed] [Google Scholar]
- 27.Borén T, Falk P, Roth KA, Larson G, Normark S. 1993. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science 262, 1892–1895. (doi:10.1126/science.8018146) [DOI] [PubMed] [Google Scholar]
- 28.Ilver D, et al. 1998. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 279, 373–377. (doi:10.1126/science.279.5349.373) [DOI] [PubMed] [Google Scholar]
- 29.Mahdavi J, et al. 2002. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 297, 573–578. (doi:10.1126/science.1069076) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Q, Meitzler JC, Huang S, Morishita T. 2000. Sequence polymorphism, predicted secondary structures, and surface-exposed conformational epitopes of Campylobacter major outer membrane protein. Infect. Immun. 68, 5679–5689. (doi:10.1128/IAI.68.10.5679-5689.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thibault P, Logan SM, Kelly JF, Brisson JR, Ewing CP, Trust TJ, Guerry P. 2001. Identification of the carbohydrate moieties and glycosylation motifs in Campylobacter jejuni flagellin. J. Biol. Chem. 276, 34 862–34 870. (doi:10.1074/jbc.M104529200) [DOI] [PubMed] [Google Scholar]
- 32.Nothaft H, Szymanski CM. 2010. Protein glycosylation in bacteria: sweeter than ever. Nat. Rev. Microbiol. 8, 765–778. (doi:10.1038/nrmicro2383) [DOI] [PubMed] [Google Scholar]
- 33.Golden NJ, Acheson DW. 2002. Identification of motility and autoagglutination Campylobacter jejuni mutants by random transposon mutagenesis. Infect. Immun. 70, 1761–1771. (doi:10.1128/IAI.70.4.1761-1771.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shevchenko A, Wilm M, Vorm O, Mann M. 1996. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal. Chem. 68, 850–858. (doi:10.1021/ac950914h) [DOI] [PubMed] [Google Scholar]
- 35.Bunn-Moreno MM, Campos-Neto A. 1981. Lectin(s) extracted from seeds of Artocarpus integrifolia (jackfruit): potent and selective stimulator(s) of distinct human T and B cell functions. J. Immunol. 127, 427–429. [PubMed] [Google Scholar]
- 36.Sastry MV, Banarjee P, Patanjali SR, Swamy MJ, Swarnalatha GV, Surolia A. 1986. Analysis of saccharide binding to Artocarpus integrifolia lectin reveals specific recognition of T-antigen (β-D-Gal(1--3)D-GalNAc). J. Biol. Chem. 261, 11 726–11 733. [PubMed] [Google Scholar]
- 37.Jeanteur D, Lakey JH, Pattus F. 1991. The bacterial porin superfamily: sequence alignment and structure prediction. Mol. Microbiol. 5, 2153–2164. (doi:10.1111/j.1365-2958.1991.tb02145.x) [DOI] [PubMed] [Google Scholar]
- 38.Bolla JM, Loret E, Zalewski M, Pages JM. 1995. Conformational analysis of the Campylobacter jejuni porin. J. Bacteriol. 177, 4266–4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhuang J, Engel A, Pages JM, Bolla JM. 1997. The Campylobacter jejuni porin trimers pack into different lattice types when reconstituted in the presence of lipid. Eur. J. Biochem. 244, 575–579. (doi:10.1111/j.1432-1033.1997.t01-1-00575.x) [DOI] [PubMed] [Google Scholar]
- 40.Amano J, Oshima M. 1999. Expression of the H Type 1 blood group antigen during enterocytic differentiation of Caco-2 cells. J. Biol. Chem. 274, 21 209–21 216. (doi:10.1074/jbc.274.30.21209) [DOI] [PubMed] [Google Scholar]
- 41.Grant CC, Konkel ME, Cieplak W, Jr, Tompkins LS. 1993. Role of flagella in adherence, internalization, and translocation of Campylobacter jejuni in nonpolarized and polarized epithelial cell cultures. Infect. Immun. 61, 1764–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nachamkin I, Yang XH, Stern NJ. 1993. Role of Campylobacter jejuni flagella as colonization factors for three-day-old chicks: analysis with flagellar mutants. Appl. Environ. Microbiol. 59, 1269–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wassenaar TM, Bleumink-Pluym NMC, van der Zeijst BAM. 1991. Inactivation of Campylobacter jejuni flagellin genes by homologous recombination demonstrates that flaA but not flaB is required for invasion. EMBO J. 10, 2055–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De E, Jullien M, Labesse G, Pages JM, Molle G, Bolla JM. 2000. MOMP (major outer membrane protein) of Campylobacter jejuni; a versatile pore-forming protein. FEBS Lett. 469, 93–97. (doi:10.1016/S0014-5793(00)01244-8) [DOI] [PubMed] [Google Scholar]
- 45.Moser I, Schroeder W, Salnikow J. 1997. Campylobacter jejuni major outer membrane protein and a 59-kDa protein are involved in binding to fibronectin and INT 407 cell membranes. FEMS Microbiol. Lett. 157, 233–238. (doi:10.1111/j.1574-6968.1997.tb12778.x) [DOI] [PubMed] [Google Scholar]
- 46.Cody AJ, Maiden MJ, Dingle KE. 2009. Genetic diversity and stability of the porA allele as a genetic marker in human Campylobacter infection. Microbiology 155, 4145–4154. (doi:10.1099/mic.0.031047-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szymanski CM, Logan SM, Linton D, Wren BW. 2003. Campylobacter: a tale of two protein glycosylation systems. Trends Microbiol. 11, 233–238. (doi:10.1016/S0966-842X(03)00079-9) [DOI] [PubMed] [Google Scholar]
- 48.Hitchen P, Brzostek J, Panico M, Butler JA, Morris HR, Dell A, Linton D. 2010. Modification of the Campylobacter jejuni flagellin glycan by the product of the Cj1295 homopolymeric-tract-containing gene. Microbiology 156, 1953–1962. (doi:10.1099/mic.0.038091-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seal BS, Hiett KL, Kuntz RL, Woolsey R, Schegg KM, Ard M, Stintzi A. 2007. Proteomic analyses of a robust versus a poor chicken gastrointestinal colonizing isolate of Campylobacter jejuni. J. Proteome Res. 6, 4582–4591. (doi:10.1021/pr070356a) [DOI] [PubMed] [Google Scholar]
- 50.Layton SL, Morgan MJ, Cole K, Kwon YM, Donoghue DJ, Hargis BM, Pumford NR. 2011. Evaluation of Salmonella-vectored Campylobacter peptide epitopes for reduction of Campylobacter jejuni in broiler chickens. Clin. Vaccine Immunol. 18, 49–454. (doi:10.1128/CVI.00379-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Langermann S, et al. 1997. Prevention of mucosal Escherichia coli infection by FimH-adhesin-based systemic vaccination. Science 276, 607–611. (doi:10.1126/science.276.5312.607) [DOI] [PubMed] [Google Scholar]
- 52.Houpt E, Barroso L, Lockhart L, Wright R, Cramer C, Lyerly D, Petri WA. 2004. Prevention of intestinal amebiasis by vaccination with the Entamoeba histolytica Gal/GalNac lectin. Vaccine 22, 611–617. (doi:10.1016/j.vaccine.2003.09.003) [DOI] [PubMed] [Google Scholar]
- 53.Huang S, Sahin O, Zhang Q. 2007. Infection-induced antibodies against the major outer membrane protein of Campylobacter jejuni mainly recognize conformational epitopes. FEMS Microbiol. Lett. 272, 137–143. (doi:10.1111/j.1574-6968.2007.00752.x) [DOI] [PubMed] [Google Scholar]
- 54.van Vliet AH, Wooldridge KG, Ketley JM. 1998. Iron-responsive gene regulation in a Campylobacter jejuni fur mutant. J. Bacteriol. 180, 5291–5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Biswas S, Mohammad MM, Movileanu L, van den Berg B. 2008. Crystal structure of the outer membrane protein OpdK from Pseudomonas aeruginosa. Structure 16, 1027–1035. (doi:10.1016/j.str.2008.04.009) [DOI] [PubMed] [Google Scholar]
- 56.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. (doi:10.1002/jcc.20084) [DOI] [PubMed] [Google Scholar]
- 57.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. 2006. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725. (doi:10.1002/prot.21123) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Still WC, Tempczyk A, Hawley RC, Hendrickson T. 1990. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 112, 6127–6129. (doi:10.1021/ja00172a038) [Google Scholar]
- 59.Tsui V, Case DA. 2000. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers 56, 275–291. (doi:10.1002/1097-0282(2000)56:4<275::AID-BIP10024>3.0.CO;2-E) [DOI] [PubMed] [Google Scholar]
- 60.Basma M, Sundara S, Calgan D, Vernali T, Woods RJ. 2001. Solvated ensemble averaging in the calculation of partial atomic charges. J. Comput. Chem. 22, 1125–1137. (doi:10.1002/jcc.1072) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kirschner KN, Woods RJ. 2001. Solvent interactions determine carbohydrate conformation. Proc. Natl Acad. Sci. USA 98, 10 541–10 545. (doi:10.1073/pnas.191362798) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kirschner KN, Woods RJ. 2001. Quantum mechanical study of the nonbonded forces in water–methanol complexes. J. Phys. Chem. A 105, 4150–4155. (doi:10.1021/jp004413y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gouda H, Kuntz ID, Case DA, Kollman PA. 2003. Free energy calculations for theophylline binding to an RNA aptamer: comparison of MM-PBSA and thermodynamic integration methods. Biopolymers 68, 16–34. (doi:10.1002/bip.10270) [DOI] [PubMed] [Google Scholar]
- 64.Lee MR, Duan Y, Kollman PA. 2000. Use of MM-PB/SA in estimating the free energies of proteins: application to native, intermediates, and unfolded villin headpiece. Proteins 39, 309–316. (doi:10.1002/(SICI)1097-0134(20000601)39:4<309::AID-PROT40>3.0.CO;2-S) [PubMed] [Google Scholar]
- 65.Srinivasan J, Cheatham TE, Cieplak P, Kollman PA, Case DA. 1998. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate–DNA helices. J. Am. Chem. Soc. 120, 9401–9409. (doi:10.1021/ja981844+) [Google Scholar]
- 66.Kollman PA, et al. 2000. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897. (doi:10.1021/ar000033j) [DOI] [PubMed] [Google Scholar]
- 67.O'Toole GA, Kolter R. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol. 30, 295–304. (doi:10.1046/j.1365-2958.1998.01062.x) [DOI] [PubMed] [Google Scholar]
- 68.Stepanovic S, Vukovic D, Dakic I, Savic B, Svabic-Vlahovic M. 2000. A modified microtiter-plate test for quantification of staphylococcal biofilm formation. J. Microbiol. Methods 40, 175–179. (doi:10.1016/S0167-7012(00)00122-6) [DOI] [PubMed] [Google Scholar]
- 69.Wassenaar TM, van der Zeijst BA, Ayling R, Newell DG. 1993. Colonization of chicks by motility mutants of Campylobacter jejuni demonstrates the importance of flagellin A expression. J. Gen. Microbiol. 139, 1171–1175. (doi:10.1099/00221287-139-6-1171) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.