

Abstract
Staphylococcus aureus is a major human opportunistic pathogen responsible for a broad spectrum of infections ranging from benign skin infection to more severe life threatening disorders (e.g. pneumonia, sepsis), particularly in intensive care patients. Scavenger receptors (SR-A and CD36) are known to be involved in S. aureus recognition by immune cells in addition to MARCO, TLR2, NOD2 and α5β1 integrin. In the present study, we further deciphered the contribution of SR-A and CD36 scavenger receptors in the control of infection of mice by S. aureus. Using double SR-A/CD36 knockout mice (S/C-KO) and S. aureus strain HG001, a clinically relevant non-mutagenized strain, we showed that the absence of these two scavenger receptors was protective in peritoneal infection. In contrast, the deletion of these two receptors was detrimental in pulmonary infection following intranasal instillation. For pulmonary infection, susceptible mice (S/C-KO) had more colony-forming units (CFU) in their broncho-alveolar lavages fluids, associated with increased recruitment of macrophages and neutrophils. For peritoneal infection, susceptible mice (wild-type) had more CFU in their blood, but recruited less macrophages and neutrophils in the peritoneal cavity than resistant mice. Exacerbated cytokine levels were often observed in the susceptible mice in the infected compartment as well as in the plasma. The exception was the enhanced compartmentalized expression of IL-1β for the resistant mice (S/C-KO) after peritoneal infection. A similar mirrored susceptibility to S. aureus infection was also observed for MARCO and TLR2. Marco and tlr2 -/- mice were more resistant to peritoneal infection but more susceptible to pulmonary infection than wild type mice. In conclusion, our results show that innate immune receptors can play distinct and opposite roles depending on the site of infection. Their presence is protective for local pulmonary infection, whereas it becomes detrimental in the peritoneal infection.
Introduction
Staphylococcus aureus is a major human opportunistic pathogen responsible for a broad spectrum of infections ranging from food poisoning and superficial skin abscesses to more serious diseases such as pneumonia, meningitis, osteomyelitis, endocarditis, septicemia or toxic shock syndrome [1]. Systemic diseases caused by S. aureus reflect its impressive capacity to subvert the defenses of the human innate immune system [2]. The unique adaptive potential displayed by S. aureus has made it one of the major causes of nosocomial infections nowadays. It is also the most commonly found bacterium in patients admitted in intensive care units (ICU), representing 30% of infections and including 14% of methicillin-resistant strains (MRSA) [3].
Several innate immune receptors are involved in the detection of S. aureus. The membrane Toll-like receptor 2 (TLR2) is involved in the detection of lipoteichoic acid (LTA), lipoproteins and eventually peptidoglycan [4]–[8]. The cytosolic nucleotide oligomerization domain 2 (NOD2) protein permits the detection of bacterial peptidoglycan, via its minimal muramyl dipeptide moiety [9], [10]. Host cell integrin α5β1 is involved in the host response to S. aureus [11]–[13] as well as other receptors belonging to the family of scavenger receptors, such as SR-A, MARCO (class A) and CD36 (class B) [14]–[20].
The relative importance of these various innate immune receptors in the resistance to S. aureus infection remains controversial. Studies published so far are difficult to compare, because they used different strains, more or less virulent, or clinical isolates. S. aureus expresses multiple virulence factors, including exotoxins, LTA, adhesins, hemolysins, cytotoxins, staphylokinase, plasminogen activating factor and capsular polysaccharide that may modulate its pathogenicity. In addition, the various in vivo models also differ in terms of inoculation route. As an example, the absence of TLR2 had minimal impact, when S. aureus was administered in the peritoneum or subcutaneously [21], [22]. In contrast, other studies using different bacterial strains and intravenous infection found tlr2-/- mice more susceptible to S. aureus [23], [24]. Similarly for NOD2, recent studies report very different results. In one model of peritoneal infection, nod2-/- mice were highly susceptible to S. aureus [25], whereas this was not seen in a skin or a lung model of infection [26], [27]. These contradictions may be due to the use of S. aureus strains of different virulence, different bacterial loads, and different route of infection.
In this study, we focused on the role of SR-A and CD36 in host resistance to S. aureus infection, because few data are available on knockout mice for these receptors in vivo with this pathogen. Both scavenger receptors are expressed by peritoneal [28] and alveolar macrophages [29]. They often share similar activities [29]–[30] and silencing of one favors the upregulation of the other [31]. These observations suggest that there might be some redundancy with these two receptors, therefore we used double KO mice.
Most of the studies showing their role in the detection of S. aureus were performed in vitro and/or with blocking antibodies. In vivo infection (cutaneous or systemic) was only performed for CD36 [17], [23]. Because S. aureus is present for 68% of the reported cases in the lungs and 22% in the abdomen of infected ICU patients [3], we chose to compare pulmonary versus peritoneal infection. In the light of the heterogeneity of the strains used so far to study S. aureus-host interaction, we decided to use a well-characterized strain (HG001) [32], derived from the NCTC 8325 strain, for which the complete genome sequence is available [33].
After infection, we did a survey in terms of mortality, bacterial load locally and in the blood, of neutrophil and macrophage recruitment, induction of cytokines and chemokines and histology. Our data show that the outcome of wild type (C57BL/6J) and SR-A/CD36 deficient mice (S/C-KO) depends on the site of infection, with mirrored results after intra-nasal or intra-peritoneal infection. SR-A and CD36 expression is protective in pulmonary, but deleterious in peritoneal infection.
Materials and Methods
Mice
The studies were performed with 8 to 12 week-old male C57BL/6(J) (B6J) purchased from Janvier (Le Genest-St.-Isle, France), and knockout mice bread in our animal facilities and all in a C57BL/6 background. The single knockout sr-a-/- and cd36-/- mice were derived from double knockout sr-a/cd36-/- mice kindly provided by Dr Valérie Quesniaux (CNRS, Orléans, France). Animals were housed in the Institut Pasteur animal facilities under specific pathogen-free conditions. All mice stayed at least 1 week in the same room before experimentation. Protocols, performed in compliance with the NIH Animal Welfare Insurance #A5476-01 issued on 02/07/2007, and the French and European Union guidelines and regulations on handling, care and protection of Laboratory Animals (http://ec.europa.eu/environment/chemicals/lab_animals/home_en.htm). Protocols were approved by the veterinary staff of Institut Pasteur animal care and Ile-de-France Paris 1 ethical committee (authorization n°: 2011-0002).
Bacteria, bacterial growth and infection
S. aureus strain HG001 [32] a rsbU + variant of strain NCTC 8325, a genetically tractable, clinically relevant non-mutagenized strain was used. HG001 is repaired for the rsbU allele, which restores the activity of the alternative sigma factor Sigma B. This strain has never been subjected to mutagenesis or UV treatment. It is methicillin sensitive and contains three resident prophages, including the one carrying the staphylokinase gene. The strain makes efficient biofilms and is highly virulent, with a pathogenicity reportedly similar to that of the USA300 strain. Bacteria were isolated on trypto-caseine soja agar medium (TSA) (BD Biosciences, Franklin Lakes, NJ, USA). One colony was pre-cultured overnight in trypto-caseine soja broth (TSB - BD Biosciences). The next day, bacteria were cultured in TSB at 37°C until they reached the exponential growth phase. Bacteria were then centrifuged (10 minat 4,000 g and 4°C), washed once with saline (Fresenius Kabi, Bad Homburg, Germany) and put in suspension at the required concentration. For intranasal infection, mice were anesthetized with an intraperitoneal injection of 100 µl of PBS containing 0.2% of xylazine (Rompun; Bayer; Leverkusen, Germany) and 1% of ketamine (Imalgen 1000; Merial; Lyon, France). They were then instilled intranasally with 20 µl of the bacterial suspension (containing 109 bacteria) at day 0. After instillation, mice were sustained in a vertical position during 30 sec to allow the bacteria to go down the broncho-alveolar tree. For the peritoneal model of infection, mice were injected i.p. with 5×107 CFU/g of body weight.
Broncho-alveolar lavages (BAL) and peritoneal lavages (PL) fluids and plasma
Mice were sacrificed post-infection with pentobarbital (CEVA santé animale; Libourne, France) diluted 1∶5 with saline. Lungs were flushed with 500 µl of saline to obtain BAL fluids. Cells were isolated from the peritoneal cavity by washing with ice-cold RPMI 1640 (Glutamax; Lonza, Basel, Switzerland). Blood was taken by intra-cardiac puncture with a 1 ml syringe containing 100 µl of heparin (100 U.I/ml) (Sanofi-Synthelabo, Le Plessis Robinson, France). Blood, BAL and PL fluids were plated at various dilutions on TSA Petri dishes for colony forming unit (CFU) counting, the remaining volume was centrifuged at 300 g for 10 min at 4°C. The leukocyte pellet from BAL and PL fluids was analyzed by flow cytometry. Supernatants and plasma were filtered (0.22 µm) using 160 Spin X columns (CoStar, Corning Lifesciences, Lowell, MA, USA) and kept at −20°C for cytokine dosage.
Cell counting and flow cytometry
Leukocytes in BAL and PL fluids were resuspended in 180 µl MACS buffer (PBS containing 0.5% FCS and 2 mM EDTA) with 20 µl mouse Fc block reagent (Myltenyi Biotec, Auburn, CA), incubated for 10 min at 4°C and divided in two. One half was incubated for 30 min on ice in MACS buffer with an anti-F4/80 antibody coupled to pacific blue (Biolegend, San Diego, CA) and an anti-Gr1 antibody coupled to PE (Miltenyi Biotec). The other half was incubated with a rat IgG2b-pacific blue (Biolegend) and a rat IgG2b-PE (AbD Serotec, Düsseldorf, Germany) as isotype controls. Cells were then washed and resuspended in 300 µL of MACS buffer. Data were acquired on 30 µl of the cell suspension using a MACSQuant flow cytometer that allows absolute cell counting. Neutrophil and macrophage counts were determined by gating on Gr1-positive/F4/80-negative or F4/80-positive cells, respectively, using the MACSQuantify software and taking into account the total volume of the samples.
Histological analysis
Lungs from mice infected intranasally were removed and immediately fixed in 10% neutral-buffered formalin. After 48 h of fixation, lung samples were embedded in paraffin; 4 micrometer sections were cut and stained with hematoxylin and eosin, or Gram to highlight tissue lesions or to detect bacteria, respectively. A double blind histological analysis was performed.
ELISA
IL-1β, IL-6, IL-10, TNFα and KC concentrations in BAL, PL fluids and plasma were determined by ELISA as specified by the manufacturer (DuoSet, R&D systems, Minneapolis, MN).
Statistical analysis
Data were expressed as individual values and median. Statistical analysis was performed with the Graph Pad Prism software using Mann-Whitney signed rank test. The mortality was analyzed using the Kaplan-Meier test. A P value below 0.05 was considered to be significant.
Results
The susceptibility to S. aureus depends on the site of infection
In the light of the heterogeneity of the bacterial strains used so far in the literature to study S. aureus-host interaction, we decided to perform our study with a well-characterized strain (HG001) [32].
The mortality of wild type C57BL/6 (B6J) and sr-a/cd36 double KO mice (S/C-KO) was followed after intra-peritoneal (i.p.) or intranasal (i.n.) infection. As shown in Figure 1, B6J mice were resistant to i.n. infection, whereas the knockout mice were highly sensitive. Interestingly, the results were completely mirrored when the infection was i.p.; in this case B6J mice were highly susceptible to infection as 80% of them succumbed to it, whereas knockout mice were resistant. Using single knockout sr-A -/- and cd36 -/- mice, we observed very similar results (Figure S1). Even if the absence of one receptor was sufficient to observe a different phenotype in terms of mortality, we preferred to investigate double knockout animals to have both types of scavenger receptors absent and avoid that one of them substitute the absence of the other [31], preventing us to have a distinct phenotype. Consequently, the rest of the study was performed with double knockout mice. Interestingly, the deletion of two other innate immune receptors involved in S. aureus recognition, namely TLR2 and MARCO ended to a similar observation. Indeed, the deletion of MARCO or TLR2 was protective in peritoneal infection while in contrast their absence was detrimental after pulmonary infection with S. aureus (figure S2).
Figure 1. Survival curves.
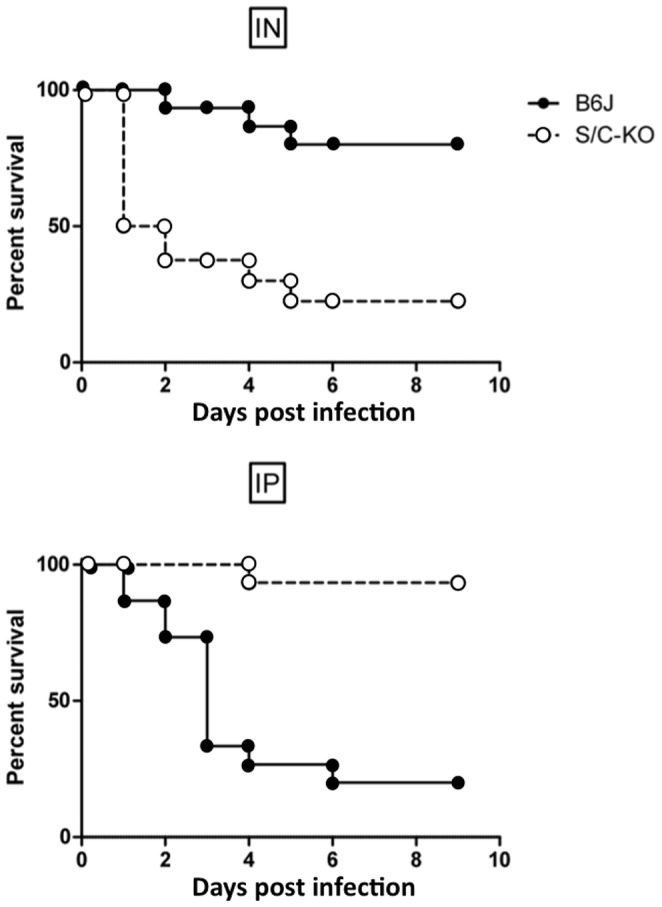
Survival curves of C57BL/6 (B6J) and double SR-A/CD36 knockout (S/C-KO) mice after a pulmonary infection following an intranasal inoculation (IN) of 109 CFU of S. aureus (upper panel) or a peritoneal injection (IP) of 5×107 CFU of S. aureus/g of mice (lower panel). The results have been acquired with n = 14 B6/J and n = 14 S/C-KO for lung infection, and n = 15 B6/J and n = 15 S/C-KO for peritoneal infection.
Bacterial load and phagocytes counts in the two models of infection are different
To better characterize the difference observed for mortality between the strains depending on the route of infection, we analyzed the colony-forming units (CFU) locally and in the blood of infected animals. For the intranasal infection model, the susceptible mice (S/C-KO) had more CFU in the bronchoalveolar lavage (BAL) fluids soon after infection (1.5 hours) (Figure 2). Furthermore, for i.n. route the bacteria rarely spread in the blood: circulating colonies were recovered only in few mice suggesting that the infection remained local in contrast to the i.p. infection, which became rapidly systemic with higher amounts of CFU in the blood of wild type (susceptible) mice. In contrast no difference was seen within the peritoneal cavity.
Figure 2. Bacterial counts in broncho-alveolar lavage (BAL) fluids (after intranasal inoculation of 109 CFU of S. aureus), in peritoneal lavage (PL) fluids (after peritoneal injection of 5×107 CFU of S. aureus/g of mice, and in blood for both types of infection.

Samples from C57BL/6 (B6J) and double SR-A/CD36 knockout (S/C-KO) were harvested 1.5 h and 3 h after infection. (R) and (S) stand for resistant and susceptible mice depending on the route of infection. All data acquired after 1.5 h are the mean of n = 5 mice, data acquired after 3 h are the mean of 10, 10, 7 and 5 mice in the peritoneal cavity, the broncho-alveolar lavages, and in the blood (after intra-nasal infection and intraperitoneal infection), respectively. * P<0.05; ** P<0.01 between wild type and deficient mice.
There was no difference between B6J and S/C-KO mice in terms of number of resident macrophages in the peritoneal cavity or the broncho-alveolar space before infection (data not shown). Significantly more macrophages and neutrophils were found at 1.5 hours for resistant mice (S/C-KO) after i.p. infection within the peritoneal cavity (Figure 3). This difference was not significant anymore at 3 hours, even if a trend was observed for more phagocytes in the case of resistant mice. Surprisingly, after i.n. infection, an early increase in the number of macrophages and neutrophils was also found for S/C-KO mice, within the broncho-alveolar lavages despite their higher susceptibility to S. aureus in this case.
Figure 3. Macrophage and neutrophil counts in broncho-alveolar lavage (BAL) fluids (after intranasal inoculation of 109 CFU of S. aureus), and in peritoneal lavage (PL) fluids (after peritoneal injection of 5×107 CFU of S. aureus/g of mice.

Samples from C57BL/6 (B6J) and double SR-A/CD36 knockout (S/C-KO) were harvested 1.5 h and 3 h after infection. (R) and (S) stand for resistant and susceptible mice depending on the route of infection. All data acquired after 1.5 h are the mean of n = 5 mice, and all data acquired after 3 h are the mean of 10 mice. ** P<0.01 between wild type and deficient mice.
Lung lesions evaluated by histology
Histology was performed to see if the differences seen in terms of mortality or bacterial load were linked to a difference in terms of lesions. Histological analysis revealed similar lesions, in nature and severity, in control and S/C-KO mice for the lungs after 12 hours post i.n. infection, i.e. a multifocal to coalescing bronchopneumonia, each focus measuring from 100 to 800 µm in diameter, and characterized by infiltrates of neutrophils (a high proportion of these neutrophils being fragmented and karyorrhectic) and macrophages, mostly centered around bronchi and bronchioles but also extending to alveoli. A high density of Gram-positive bacteria was observed in both groups. These results suggest that in the early phase of the infectious process the deficiency in SR-A and CD36 does not significantly modify the lesional inflammatory response (data not shown).
Cytokine production for resistant and susceptible mice in i.p. versus i.n infection
Next we measured the presence of cytokines and chemokines (IL-1β, IL-6, IL-10, and KC) in the lavage fluids of the infected compartment and in the plasma. After i.p. infection (Figure 4), locally very few differences were seen between susceptible and resistant mice. Only IL-1ß was locally present at higher concentrations for resistant mice (S/C-KO), at both time points. Interestingly, in the same time the levels of the anti-inflammatory cytokine IL-10 were significantly lower. For the other cytokines found in peritoneal lavages, comparable levels were detected in both groups (including TNFα, data not shown). In the plasma, differences were also noted. Less IL-1β and KC were found at 3 hours for the resistant mice (S/C-KO), as well as less IL-10 both at 1.5 and 3 hours post-infection. No difference was seen between susceptible and resistant mice for plasmatic TNFα (data not shown). For the i.n. infection, cytokine levels were very low locally at 1.5 hours but increased levels of IL-6, KC and IL-1β were found at 3 hours for susceptible mice (S/C-KO) (Figure 5). IL-10 was detected at 3 hours, but without significant difference between the two groups. Similarly, TNFα was present in BAL fluids but no difference was seen between susceptible and resistant mice (data not shown). The same situation was observed in the plasma, where more IL-1β was found at the early time point. Of note are the high early levels of IL-10 in both sensitive and resistant mice. At 3 hours, IL-6, KC and IL-10 were significantly increased for susceptible mice (S/C-KO). The levels of IL-1β in the plasma were comparable to those of the i.p. infection, but those of KC and IL-6 were much lower, probably because as seen in Figure 2 few CFU spread in the bloodstream in this model. TNFα levels were very low in the plasma and most of the time below the detection limit (data not shown).
Figure 4. Cytokine levels (IL-6, KC, IL-10 and IL-1β) in peritoneal lavage (PL) fluids and in plasma after peritoneal injection of 5×107 CFU of S. aureus/g of mice.

Samples from C57BL/6 (B6J) and double SR-A/CD36 knockout (S/C-KO) were harvested 1.5 h and 3 h after infection. (R) and (S) stand for resistant and susceptible mice. All data acquired after 1.5 h are the mean of n = 5 mice, data acquired after 3 h are the mean of 10 and 5 mice in the peritoneal cavity and in the blood compartment, respectively. * P<0.05.
Figure 5. Cytokine levels (IL-6, KC, IL-10 and IL-1β) in broncho-alveolar lavage (BAL) fluids and in plasma after intranasal inoculation of 109 CFU of S. aureus.

Samples from C57BL/6 (B6J) and double SR-A/CD36 knockout (S/C-KO) were harvested 1.5 h and 3 h after infection. (R) and (S) stand for resistant and sensitive mice. All data acquired after 1.5 h are the mean of n = 5 mice, and all data acquired after 3 h are the mean of 10 mice. * P<0.05 ; ** P<0.01 ; *** P<0.001 between wild type and deficient mice.
Discussion
The aim of this study was to revisit the resistance of mice to S. aureus infection, evaluating the impact of the route of infection and the role of scavenger receptors SR-A and CD36 in the resistance or susceptibility. Our objective was to reconcile some contradictory data found in the literature about the susceptibility of mice deficient for innate immune receptors. This contradiction may be due to the use of more or less virulent S. aureus strains, including clinical isolates, not fully characterized for the expression of virulence factors. To overcome this difficulty, we used a well-characterized strain of S. aureus (HG001) [32], which expresses virulence factors known to be important for S. aureus pathogenicity. The second source of dissonance in the literature may be due to the site of infection. We thus chose to compare two compartments: the peritoneal cavity and the lungs, which are the most frequent sites of infection with S. aureus for patients admitted in ICU. These two models give also the advantage to compare a systemic (i.p.) to a more localized infection (i.n.), as assessed by the very low spreading of CFU in the blood in the latest model.
Comparing the S/C-KO mice with their WT counterpart (B6J) we found interestingly that mice susceptible to i.p. infection were resistant to i.n. infection and vice versa. S/C-KO mice were resistant to i.p. infection. They had less CFU spreading in the blood and higher numbers of macrophages and neutrophils were found locally in the early time. This observation is in agreement with the fact that both cell types contribute to the anti-infectious process against S. aureus thanks to their phagocytic activity [34]–[38]. The involvement of CD36 [17], [18] and SR-A [39], [40] in S. aureus phagocytosis has been clearly established. The reduced susceptibility of S/C-KO mice to i.p. infection may be due to the reduced spreading of the bacteria into the bloodstream as compared to WT animals. It results that the systemic inflammatory response, illustrated by cytokines, is reduced in S/C-KO mice as compared to B6J mice. It is well known that high levels of pro-inflammatory cytokines in the blood create a systemic inflammation that may lead to organ dysfunction and death [41]. Regarding the intra-peritoneal infection, the striking difference is the higher level of both local and systemic IL-10 in the susceptible mice (B6J) and the higher level of IL-1β in the peritoneal cavity of resistant mice (S/C-KO). Indeed, on one hand IL-10 is known to be an immunosuppressive cytokine than can alter the immune defense [42]–[44], and on the other hand IL-1β and the inflammasome required for its maturation both contribute to the protection against S. aureus infection [45], [46].
Interestingly, the same S/C-KO mouse strain was highly susceptible to i.n. infection by S. aureus. They had more CFU in the lungs, but paradoxically they had also more macrophages and neutrophils. After intra-nasal delivery of S. aureus and pulmonary infection, susceptible S/C-KO mice displayed also higher levels of cytokines, both locally and systemically. This observation is reminiscent of the detrimental role of cytokines of which excessive amounts can lead to shock and multiple organ failure. For example, higher plasma levels of cytokines were found in the susceptible mouse strain (A/J) as compared to a more resistant one (C57BL/6) after systemic infection with S. aureus [35]. Furthermore, a protective therapeutic approach against lethal S. aureus peritoneal infection in mice was associated with a reduced level of inflammatory cytokines [47]. In our model, the absence of SR-A and CD36 induces a higher recruitment of phagocytes and a stronger inflammatory response in the lungs, which probably leads to the dysfunction of this organ and death. For ICU patients, lungs are most of the time the first organ showing failure. The phenomenon observed for S/C-KO mice is very fast, as most of the animals die within 24 hours, before creating visible lesions in the lungs.
Most importantly, our data demonstrate that depending upon the compartment, similar receptors can play contrasting role. In the case of S. aureus infection, such beneficial or detrimental role has already been attributed to plasminogen [48] and to gamma-interferon [49]. In the later case, the report demonstrated that the mechanisms of host defense are greatly influenced by their tissue environment since this cytokine had a positive effect to control systemic sepsis but a negative role for S. aureus-associated arthritis. Similarly, IL-10 was protective in Francisella tularensis pulmonary infection but deleterious after cutaneous inoculation [50]. In addition, the cells are greatly influenced by their local microenvironment. For example, macrophages are extremely different from one compartment to another and their capacity to respond to bacteria or bacterial products differs from one site to another as shown for intestinal, peritoneal, alveolar, spleen, microglial macrophages and monocytes [12], [51]–[53].
However, we failed to find any significant differences in terms of cytokine production when comparing the in vitro capacity of peritoneal macrophages of wild type mice and S/C-KO mice to produce IL-1β, IL-6, TNF and KC in response to LPS and heat-killed S. aureus RN1HG (data not shown). A similar observation was made for alveolar macrophages, and for the phagocytic activity of both types of macrophages from wild type and S/C-KO mice (data not shown).
Regarding cell surface receptors involved in the recognition of pathogen-associated molecular patterns, their role may vary depending on the tissue. This has been shown for TLR2 of which the presence within the kidney influences S. aureus clearance, whereas its expression in lungs, liver or spleen does not affect the bacterial clearance [54]. A similar observation in a peritonitis model was reported for TLR4 of which the expression on myeloid cells was protective while its expression on hepatocytes was deleterious [55].
In conclusion, our report demonstrates that similar cell surface innate immune receptors (CD36, SR-A, MARCO and TLR2) play different roles against S. aureus infection, depending on the site of infection. It enlightens the concept of compartmentalization of the innate immune response of which mechanisms can be completely different depending on the tissue/organ where it occurs [56]. A careful analysis, keeping in mind this concept of compartmentalization, should avoid dogmatic statements about the role of the different actors involved in anti-infectious processes of the host.
Supporting Information
Mortality of single knockout mice after infection with S. aureus is similar to that of double deficient mice. (A) Mortality was followed after intranasal (IN) or (B) intra-peritoneal (IP) infection. WT C57BL/6J (black) and cd36-/- (blue) and sr-a-/- (red) were compared. The results were acquired with n = 11 and n = 7 C57BL/6J, n = 8 and n = 5 cd36 -/-, and n = 8 and n = 6 sr-a -/- mice, for peritoneal and pulmonary infections, respectively.
(TIF)
Survival curves of C57BL/6J, marco -/- and tlr2 -/- mice after either a pulmonary infection following an intranasal (i.n.) inoculation of 109 CFU of S. aureus (left figures) or a peritoneal injection (i.p.) of 5×107 CFU of S. aureus /g of mice (right figures). The results were acquired with n = 10 and n = 5 C57BL/6J, n = 8 and n = 5 marco -/-, and n = 10 and n = 17 C57BL/6J n = 9 and n = 10 tlr2 -/-, for peritoneal and pulmonary infections, respectively.
(TIF)
Acknowledgments
The authors are grateful to Dr Geneviève Milon for precious advice and permanent support, to Dr Hiroshi Suzuki for the authorization to use sr-a knockout mice, to Dr Maria Febbraio for the authorization to use cd36 knockout mice, Dr Dawn Bowdish for the authorization to use Marco knockout mice, and to Dr Valérie Quesniaux for the kind gift of sr-a/cd36 double knockout and Marco knockout mice. The authors thank Drs Tarek Msadek, Sarah Dubrac, and Olivier Poupel (Unité de Recherche Biologie des Bactéries pathogènes à Gram-positif, Inst. Pasteur) for helpful discussion. This manuscript is dedicated to our distinguished and bright colleague, Dr. Minou Adib-Conquy, who supervised this work and who passed away in November, 2013.
Funding Statement
This study and Charlène Blanchet were supported by Institut Pasteur, Programme Transversal de Recherche (grant and fellowship PTR336). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Lowy FD (1998) Staphylococcus aureus infections. N Engl J Med 339: 520–532. [DOI] [PubMed] [Google Scholar]
- 2. Nizet V (2007) Understanding how leading bacterial pathogens subvert innate immunity to reveal novel therapeutic targets. J Allergy Clin Immunol 120: 13–22. [DOI] [PubMed] [Google Scholar]
- 3. Vincent JL, Sakr Y, Sprung CL, Ranieri VM, Reinhart K, et al. (2006) Sepsis in European intensive care units: results of the SOAP study. Crit Care Med 34: 344–353. [DOI] [PubMed] [Google Scholar]
- 4. Lien E, Sellati TJ, Yoshimura A, Flo TH, Rawadi G, et al. (1999) Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J Biol Chem 274: 33419–33425. [DOI] [PubMed] [Google Scholar]
- 5. Bubeck Wardenburg J, Williams WA, Missiakas D (2006) Host defenses against Staphylococcus aureus infection require recognition of bacterial lipoproteins. Proc Natl Acad Sci U S A 103: 13831–13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schmaler M, Jann NJ, Ferracin F, Landolt LZ, Biswas L, et al. (2009) Lipoproteins in Staphylococcus aureus mediate inflammation by TLR2 and iron-dependent growth in vivo. J Immunol 182: 7110–7118. [DOI] [PubMed] [Google Scholar]
- 7. Travassos LH, Girardin SE, Philpott DJ, Blanot D, Nahori MA, et al. (2004) Toll-like receptor 2-dependent bacterial sensing does not occur via peptidoglycan recognition. EMBO Rep 5: 1000–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dziarski R, Gupta D (2005) Staphylococcus aureus peptidoglycan is a toll-like receptor 2 activator: a reevaluation. Infect Immun 73: 5212–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, et al. (2003) Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 278: 8869–8872. [DOI] [PubMed] [Google Scholar]
- 10. Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, et al. (2003) Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J Biol Chem 278: 5509–5512. [DOI] [PubMed] [Google Scholar]
- 11. Sinha B, Francois PP, Nusse O, Foti M, Hartford OM, et al. (1999) Fibronectin-binding protein acts as Staphylococcus aureus invasin via fibronectin bridging to integrin alpha5beta1. Cell Microbiol 1: 101–117. [DOI] [PubMed] [Google Scholar]
- 12. Kapetanovic R, Parlato M, Fitting C, Quesniaux V, Cavaillon JM, et al. (2011) Mechanisms of TNF induction by heat-killed Staphylococcus aureus differ upon the origin of mononuclear phagocytes. Am J Physiol Cell Physiol 300: C850–859. [DOI] [PubMed] [Google Scholar]
- 13. Liang X, Ji Y (2007) Involvement of alpha5beta1-integrin and TNF-alpha in Staphylococcus aureus alpha-toxin-induced death of epithelial cells. Cell Microbiol 9: 1809–1821. [DOI] [PubMed] [Google Scholar]
- 14. van der Laan LJ, Dopp EA, Haworth R, Pikkarainen T, Kangas M, et al. (1999) Regulation and functional involvement of macrophage scavenger receptor MARCO in clearance of bacteria in vivo. J Immunol 162: 939–947. [PubMed] [Google Scholar]
- 15. Arredouani MS, Palecanda A, Koziel H, Huang YC, Imrich A, et al. (2005) MARCO is the major binding receptor for unopsonized particles and bacteria on human alveolar macrophages. J Immunol 175: 6058–6064. [DOI] [PubMed] [Google Scholar]
- 16. Peiser L, Gough PJ, Kodama T, Gordon S (2000) Macrophage class A scavenger receptor-mediated phagocytosis of Escherichia coli: role of cell heterogeneity, microbial strain, and culture conditions in vitro. Infect Immun 68: 1953–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stuart LM, Deng J, Silver JM, Takahashi K, Tseng AA, et al. (2005) Response to Staphylococcus aureus requires CD36-mediated phagocytosis triggered by the COOH-terminal cytoplasmic domain. J Cell Biol 170: 477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baranova IN, Kurlander R, Bocharov AV, Vishnyakova TG, Chen Z, et al. (2008) Role of human CD36 in bacterial recognition, phagocytosis, and pathogen-induced JNK-mediated signaling. J Immunol 181: 7147–7156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sever-Chroneos Z, Krupa A, Davis J, Hasan M, Yang C, et al. (2011) Surfactant protein A (SP-A)-mediated clearance of Staphylococcus aureus involves binding of SP-A to the staphylococcal adhesin eap and the macrophage receptors SP-A receptor 210 and scavenger receptor class A. J Biol Chem 286: 4854–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bocharov AV, Baranova IN, Vishnyakova TG, Remaley AT, Csako G, et al. (2004) Targeting of scavenger receptor class B type I by synthetic amphipathic alpha-helical-containing peptides blocks lipopolysaccharide (LPS) uptake and LPS-induced pro-inflammatory cytokine responses in THP-1 monocyte cells. J Biol Chem 279: 36072–36082. [DOI] [PubMed] [Google Scholar]
- 21. Mullaly SC, Kubes P (2006) The role of TLR2 in vivo following challenge with Staphylococcus aureus and prototypic ligands. J Immunol 177: 8154–8163. [DOI] [PubMed] [Google Scholar]
- 22. Miller LS, O'Connell RM, Gutierrez MA, Pietras EM, Shahangian A, et al. (2006) MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. Immunity 24: 79–91. [DOI] [PubMed] [Google Scholar]
- 23. Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, et al. (2005) CD36 is a sensor of diacylglycerides. Nature 433: 523–527. [DOI] [PubMed] [Google Scholar]
- 24. Takeuchi O, Hoshino K, Akira S (2000) Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol 165: 5392–5396. [DOI] [PubMed] [Google Scholar]
- 25. Deshmukh HS, Hamburger JB, Ahn SH, McCafferty DG, Yang SR, et al. (2009) The Critical Role of NOD2 in Regulating the Immune Response to Staphylococcus aureus. Infect Immun 77: 1376–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hruz P, Zinkernagel AS, Jenikova G, Botwin GJ, Hugot JP, et al. (2009) NOD2 contributes to cutaneous defense against Staphylococcus aureus through {alpha}-toxin-dependent innate immune activation. Proc Natl Acad Sci U S A 106: 12873–12878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kapetanovic R, Jouvion G, Fitting C, Parlato M, Blanchet C, et al. (2010) Contribution of NOD2 to lung inflammation during Staphylococcus aureus-induced pneumonia. Microbes Infect 12: 759–767. [DOI] [PubMed] [Google Scholar]
- 28. Guest CB, Hartman ME, O'Connor JC, Chakour KS, Sovari AA (2007) Phagocytosis of cholesteryl ester is amplified in diabetic mouse macrophages and is largely mediated by CD36 and SR-A. PLoS One 2: e511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Józefowski S, Kobzik L (2004) Scavenger receptor A mediates H2O2 production and suppression of IL-12 release in murine macrophages. J Leukoc Biol 76: 1066–74. [DOI] [PubMed] [Google Scholar]
- 30. Bieghs V, Verheyen F, van Gorp PJ, Hendrikx T, Wouters K, et al. (2012) Internalization of modified lipids by CD36 and SR-A leads to hepatic inflammation and lysosomal cholesterol storage in Kupffer cells. PLoS One 7: e34378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mäkinen PI, Lappalainen JP, Heinonen SE, Leppänen P, Lähteenvuo MT, et al. (2010) Silencing of either SR-A or CD36 reduces atherosclerosis in hyperlipidaemic mice and reveals reciprocal upregulation of these receptors. Cardiovasc Res 88: 530–8. [DOI] [PubMed] [Google Scholar]
- 32. Herbert S, Ziebandt AK, Ohlsen K, Schafer T, Hecker M, et al. (2010) Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect Immun 78: 2877–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gillaspy AF, Worrell V, Orvis J, Roe BA, Dyer DW, et al. (2006) The Staphylococcus aureus NCTC 8325 Genome. In: Fischetti VA, Novick RP, Ferretti JJ, Portnoy DA, Rood JI, editors. Gram-Positive Pathogens. 2nd ed. Washington, DC: ASM Press. pp. 381–412. [Google Scholar]
- 34. Verdrengh M, Tarkowski A (2000) Role of macrophages in Staphylococcus aureus-induced arthritis and sepsis. Arthritis Rheum 43: 2276–2282. [DOI] [PubMed] [Google Scholar]
- 35. von Köckritz-Blickwede M, Rohde M, Oehmcke S, Miller L, Cheung A, et al. (2008) Immunological mechanisms underlying the genetic predisposition to severe Staphylococcus aureus infection in the mouse model. Am J Pathol 173: 1657–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schwartz J, Leidal K, Femling J, Weiss J, Nauseef W (2009) Neutrophil bleaching of GFP-expressing staphylococci: probing the intraphagosomal fate of individual bacteria. J Immunol 183: 2632–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ip W, Sokolovska A, Charriere G, Boyer L, Dejardin S, et al. (2010) Phagocytosis and phagosome acidification are required for pathogen processing and MyD88-dependent responses to Staphylococcus aureus. J Immunol 184: 7071–7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Martin F, Parker D, Harfenist B, Soong G, Prince A (2011) Participation of CD11c(+) leukocytes in methicillin-resistant Staphylococcus aureus clearance from the lung. Infect Immun 79: 1898–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ono K, Nishitani C, Mitsuzawa H, Shimizu T, Sano H, et al. (2006) Mannose-binding lectin augments the uptake of lipid A, Staphylococcus aureus, and Escherichia coli by Kupffer cells through increased cell surface expression of scavenger receptor A. J Immunol 177: 5517–5523. [DOI] [PubMed] [Google Scholar]
- 40. Amiel E, Alonso A, Uematsu S, Akira S, Poynter ME, et al. (2009) Pivotal Advance: Toll-like receptor regulation of scavenger receptor-A-mediated phagocytosis. J Leukoc Biol 85: 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Adib-Conquy M, Cavaillon JM (2007) Stress molecules in sepsis and systemic inflammatory response syndrome. FEBS Lett 581: 3723–3733. [DOI] [PubMed] [Google Scholar]
- 42. Fernandes D, Baldwin C (1995) Interleukin-10 downregulates protective immunity to Brucella abortus. Infect Immun 63: 1130–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang X, HayGlass K, Brunham R (1996) Genetically determined differences in IL-10 and IFN-gamma responses correlate with clearance of Chlamydia trachomatis mouse pneumonitis infection. J Immunol 156: 4338–4344. [PubMed] [Google Scholar]
- 44. Wang M, Jeng K, Ping L (1999) Exogenous cytokine modulation or neutralization of interleukin-10 enhance survival in lipopolysaccharide-hyporesponsive C3H/HeJ mice with Klebsiella infection. Immunology 98: 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hultgren O, Svensson L, Tarkowski A (2002) Critical role of signaling through IL-1 receptor for development of arthritis and sepsis during Staphylococcus aureus infection. J Immunol 168: 5207–5212. [DOI] [PubMed] [Google Scholar]
- 46. Miller L, Pietras E, Uricchio L, Hirano K, Rao S, et al. (2007) Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J Immunol 179: 6933–6942. [DOI] [PubMed] [Google Scholar]
- 47. Ahn J, Song J, Yun Y, Jeong G, Choi I (2006) Protection of Staphylococcus aureus-infected septic mice by suppression of early acute inflammation and enhanced antimicrobial activity by ginsan. FEMS Immunol Med Microbiol 46: 187–197. [DOI] [PubMed] [Google Scholar]
- 48. Guo Y, Li J, Hagström E, Ny T (2011) Beneficial and detrimental effects of plasmin(ogen) during infection and sepsis in mice. PLoS One 6: e24774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao Y, Nilsson I, Tarkowski A (1998) The dual role of interferon-gamma in experimental Staphylococcus aureus septicaemia versus arthritis. Immunology 93: 80–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Metzger DW, Salmon SL, Kirimanjeswara G (2013) Differing Effects of Interleukin-10 on Cutaneous and Pulmonary Francisella tularensis Live Vaccine Strain Infection. Infect Immun 81: 2022–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Smythies LE, Sellers M, Clements RH, Mosteller-Barnum M, Meng G, et al. (2005) Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest 115: 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Philippart F, Fitting C, Cavaillon JM (2012) Lung microenvironment contributes to the resistance of alveolar macrophages to develop tolerance to endotoxin. Crit Care Med 40: 2987–2996. [DOI] [PubMed] [Google Scholar]
- 53. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, et al. (2012) Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 13: 1118–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gillrie M, Zbytnuik L, McAvoy E, Kapadia R, Lee K, et al. (2010) Divergent roles of Toll-like receptor 2 in response to lipoteichoic acid and Staphylococcus aureus in vivo. Eur J Immunol 40: 1639–50. [DOI] [PubMed] [Google Scholar]
- 55. Deng M, Scott MJ, Loughran P, Gibson G, Sodhi C, et al. (2013) Lipopolysaccharide clearance, bacterial clearance, and systemic inflammatory responses are regulated by cell type-specific functions of TLR4 during sepsis. J Immunol 190: 5152–5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cavaillon J-M, Annane D (2006) Compartmentalization of the inflammatory response in sepsis and SIRS. J Endotoxin Res 12: 151–170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mortality of single knockout mice after infection with S. aureus is similar to that of double deficient mice. (A) Mortality was followed after intranasal (IN) or (B) intra-peritoneal (IP) infection. WT C57BL/6J (black) and cd36-/- (blue) and sr-a-/- (red) were compared. The results were acquired with n = 11 and n = 7 C57BL/6J, n = 8 and n = 5 cd36 -/-, and n = 8 and n = 6 sr-a -/- mice, for peritoneal and pulmonary infections, respectively.
(TIF)
Survival curves of C57BL/6J, marco -/- and tlr2 -/- mice after either a pulmonary infection following an intranasal (i.n.) inoculation of 109 CFU of S. aureus (left figures) or a peritoneal injection (i.p.) of 5×107 CFU of S. aureus /g of mice (right figures). The results were acquired with n = 10 and n = 5 C57BL/6J, n = 8 and n = 5 marco -/-, and n = 10 and n = 17 C57BL/6J n = 9 and n = 10 tlr2 -/-, for peritoneal and pulmonary infections, respectively.
(TIF)