

Abstract
Background
Duchenne muscular dystrophy (DMD) is a severe and progressive muscle-wasting disorder caused by mutations in the dystrophin gene that result in the absence of the membrane-stabilising protein dystrophin. Dystrophic muscle fibres are susceptible to injury and degeneration, and impaired muscle regeneration is associated with fibrotic deposition that limits the efficacy of potential pharmacological, cell- and gene-based therapies. Novel treatments that can prevent or attenuate fibrosis have important clinical merit for DMD and related neuromuscular diseases. We investigated the therapeutic potential for tranilast, an orally bioavailable anti-allergic agent, to prevent fibrosis in skeletal muscles of mdx dystrophic mice.
Results
Three-week-old C57Bl/10 and mdx mice received tranilast (~300 mg/kg) in their food for 9 weeks, after which fibrosis was assessed through histological analyses, and functional properties of tibialis anterior muscles were assessed in situ and diaphragm muscle strips in vitro. Tranilast administration did not significantly alter the mass of any muscles in control or mdx mice, but it decreased fibrosis in the severely affected diaphragm muscle by 31% compared with untreated mdx mice (P < 0.05). A similar trend of decreased fibrosis was observed in the tibialis anterior muscles of mdx mice (P = 0.10). These reductions in fibrotic deposition were not associated with improvements in maximum force-producing capacity, but we did observe small but significant improvements in the resistance to fatigue in both the diaphragm and TA muscles of mdx mice treated with tranilast.
Conclusion
Together these findings demonstrate that administration of potent antifibrotic compounds such as tranilast could help preserve skeletal muscle structure, which could ultimately increase the efficacy of pharmacological, cell and gene replacement/correction therapies for muscular dystrophy and related disorders.
Background
Duchenne muscular dystrophy (DMD) is a severe, X-linked genetic muscle-wasting disorder characterised by progressive muscle weakness that culminates in respiratory failure and premature death. The disease affects approximately 1:3,500 live male births worldwide, and affected boys are usually wheelchair bound by their early teens and experience a severely reduced quality of life. DMD is caused by mutations in the dystrophin (dmd) gene resulting in very low levels or a complete absence of the dystrophin protein, a key structural element of muscle fibres that renders them highly susceptible to damage. As a consequence, dystrophic muscles are characterised by inflammation and ongoing cycles of degeneration and regeneration. This environment limits muscle regenerative capacity and there is concomitant replacement of formerly functional muscle fibres with adipose and fibrotic material (reviewed in [1]).
The mdx mouse is the most commonly used animal model of DMD. It arose from a natural mutation in a colony of C57BL/10 mice in which a premature stop codon was introduced into the dmd gene, resulting in a complete loss of the dystrophin protein [2,3]. While this model is a good genocopy of the human disease, the pathology of the mdx mouse does not mimic that observed in DMD. Mdx mice exhibit a mild pathology with a slightly reduced lifespan (18–24 months) and functional decline of hindlimb muscles becoming evident at approximately 18 months of age [4], which is most likely a result of modifier genes in the C57BL/10 strain that can alter the effect of dystrophin loss on skeletal muscle pathology. Unlike the hindlimb muscles, the diaphragm of mdx mice undergoes progressive deterioration of muscle structure and function and is therefore a preferred muscle when examining the efficacy of potential therapeutic agents of clinical relevance [5-7].
As DMD is caused by mutations in a single gene, one of the most promising therapies is through gene replacement. However, while gene replacement or correction studies are likely to provide an eventual cure for DMD, several barriers need to be overcome including the presence of fibrosis within dystrophic skeletal muscles (reviewed in [8]). Fibrosis not only creates a physical barrier, but also replaces the muscle fibres that can be targeted, limiting the efficacy of cell- and gene-based therapies. Attenuating fibrotic infiltration may be needed to optimise gene, cell and pharmacological therapies.
Various agents with antifibrotic properties have been trialled to reduce fibrosis deposition in skeletal muscle. Suramin, a TGF-β inhibitor, and interleukin-15 (IL-15) have been shown to reduce muscle fibrosis but can have side effects when administered systemically [9-11]. Another compound with antifibrotic properties is tranilast, an orally bioavailable antiallergic agent that has been approved for use in the human population in Japan and South Korea since 1982 for the treatment of bronchial asthma, atopic dermatitis and allergic rhinitis [12-17]. Since that time, the effectiveness of tranilast as a therapeutic agent for a range of fibrotic disorders and its mechanism of action have been studied extensively both in vitro and in vivo (reviewed in [18]). In 1992, Suzawa and colleagues [19] demonstrated that tranilast suppressed release of profibrotic cytokines from monocyte-macrophages in vitro[19], highlighting tranilast’s antifibrotic properties. Tranilast has subsequently been demonstrated to reduce tuberointerstitial and heart fibrosis in diabetic rat models and to block TGF-β-induced fibrosis in vitro and in vivo[20-24]. In addition, tranilast administration was found to be efficacious in reducing muscle fibrosis in the Bio14.6 hamster model of limb-girdle muscular dystrophy and reducing serum creatine kinase levels in mdx dystrophic mice, effects they suggest may be a result of tranilast-mediated inhibition of the Ca2+-permeable growth factor-regulated channel (TRPV2 or GRC) [18,25]. In this study, we report that short-term administration of tranilast in mdx mice decreases fibrosis in skeletal muscle and improves the resistance to muscle fatigue. Together these findings demonstrate that tranilast has therapeutic potential to combat fibrosis in muscle diseases such as DMD.
Results
Tranilast does not alter skeletal muscle mass or strength
At the end of the 9-week treatment period, the tibialis anterior (TA), soleus (SOL), extensor digitorum longus (EDL), plantaris (PLANT), gastrocnemius (GAST), quadriceps (QUAD) and heart muscles from both non-treated and treated mdx mice were significantly larger than those from non-treated and treated control mice (Figure 1A). These differences cannot be attributed to differences in food intake as daily intake was not different between strains or treatment groups (data not shown) and averaged 3.3 g/mouse/day. Administration of tranilast did not significantly alter the mass of any of the tested skeletal muscles in control or mdx mice (Figure 1A). Consequently, 9-week treatment with tranilast did not affect whole-body strength (Figure 1B) or mobility (Figure 1C), as assessed by grip strength and rotarod performance.
Figure 1.
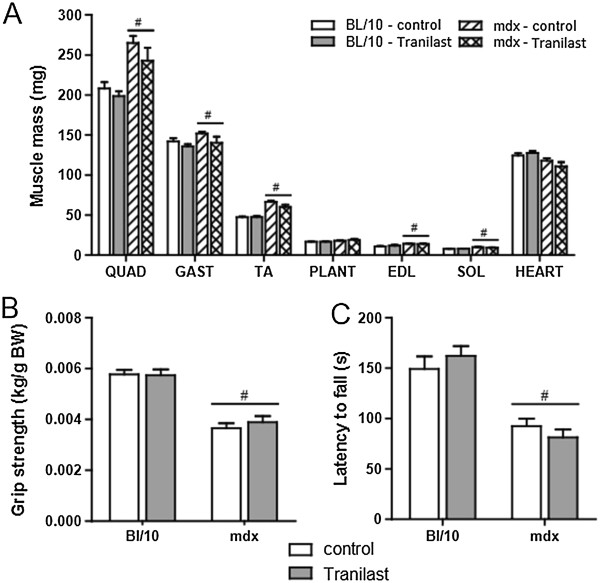
Tranilast effects on muscle mass and whole body strength of mdx mice. Three-week-old male C57Bl/10 control mice or mdx mice received either vehicle or tranilast treatment for 9 weeks. (A) Individual muscle masses at the end of the treatment period. (B) Grip strength was measured 3 days prior to endpoint. (C) Whole body function and coordination was assessed by rotarod performance 2 days prior to endpoint. QUAD: quadriceps; GAST: gastrocnemius; TA: tibialis anterior; PLANT: plantaris; EDL: extensor digitorum longus; SOL: soleus. #P < 0.05 group main effect, mdx vs. control.
Fibrotic deposition is decreased in muscles of tranilast-treated dystrophic mice
The TA and diaphragm muscles of mdx mice contained three- and nine-fold more fibrosis, respectively, compared with control mice (Figure 2). Tranilast administration to young mdx mice for 9 weeks resulted in a significant three-fold (31%) decrease in fibrosis in the diaphragm compared with untreated mdx mice (Figure 2A-C, P < 0.05). A similar trend (30% decrease, P = 0.10) was observed in the TA muscles of mdx mice. The level of fibrosis in the TA muscles and diaphragm of control animals was naturally very low and unchanged with tranilast administration (Figure 2C). Analysis of gene expression revealed that Collagen 1a1, 3a1, 5a1 and TGFβ genes were all more highly expressed in the diaphragm of mdx compared to C57BL/10 mice, but tranilast treatment had no effect on the expression levels of these genes (Table 1). Tranilast treatment resulted in a change in fibre type distribution in the TA muscles of mdx mice with an increased proportion of type IIa fibres with a concomitant decrease in type IIb/x fibres compared with muscles from untreated mdx mice (Table 2). No significant differences were observed between tranilast-treated and control mdx mice in fibre cross-sectional area or oxidative enzyme capacity in either the TA or diaphragm muscles (Table 2).
Figure 2.

Tranilast effects on fibrosis in the diaphragm and TA of mdx mice. At endpoint, the diaphragm muscle of vehicle-treated C57BL/10 (A), vehicle-treated mdx (B), tranilast-treated C57BL/10 (C) or tranilast-treated mdx (D) mice was excised and collagen infiltration visualised in cross-sections after Van Gieson’s staining. (E) Fibrotic accumulation in Van Gieson’s stained sections from diaphragm and tibialis anterior muscles of untreated and treated mice. #P < 0.05 group main effect, mdx vs. control. *P < 0.05 mdx treated vs. mdx control; n.s, not significant.
Table 1.
Tranilast administration does not cause a long-term change in fibrotic gene expression in the diaphragm
| CON + VEH | CON + TRA | mdx- VEH | mdx-TRA | |
|---|---|---|---|---|
|
Col1a1 |
1.00±0.09 |
0.83±0.13 |
3.42±0.42* |
3.37±0.33* |
|
Col2a1 |
1.00±0.28 |
1.23±0.47 |
0.79±0.20 |
0.92±0.34 |
|
Col3a1 |
1.00±0.10 |
0.89±0.13 |
3.72±0.26* |
3.27±0.34* |
|
Col4a1 |
1.00±0.06 |
0.87±0.11 |
1.31±0.14 |
1.35±0.13 |
|
Col5a1 |
1.00±0.09 |
0.91±0.08 |
3.46±0.24* |
3.40±0.36* |
|
COl6a1 |
1.00±0.06 |
0.87±0.11 |
1.31±0.14 |
1.35±0.13 |
|
Fn1 |
1.00±0.14 |
1.34±0.07 |
1.00±0.14 |
1.01±0.18 |
| TGFβ1 | 1.00±0.11 | 0.86±0.07 | 1.48±0.04* | 1.45±0.12* |
At the conclusion of the treatment period RNA was extracted and real-time PCR analysis was used to examine the expression level of genes known to be associated with fibrosis. *P < 0.05 mdx vs. control.
Table 2.
Tranilast administration causes a shift in fibre type proportions in the TA muscles of mdx mice
| CON + VEH | CON + TRA | mdx + VEH | mdx + TRA | |
|---|---|---|---|---|
|
Tibialis anterior | ||||
| Fibre CSA (μm
2
) |
1,811.4 ± 54.9 |
1,859.7 ± 74.3 |
1,890.2 ± 54.0 |
1,932.2 ± 113.6 |
| SDH intensity (o.d.) |
1,645.4 ± 57.4 |
1,622.3 ± 36.6 |
1,497.2 ± 74.1† |
1,464.7 ± 48.9† |
| % Type IIa fibres |
11.0 ± 3.1 |
9.1 ± 1.6 |
4.9 ± 0.6 |
10.8 ± 0.5* |
|
Diaphragm | ||||
| Fibre CSA (μm
2
) |
1,010.6 ± 69.3 |
949.8 ± 69.8 |
764.9 ± 39.9† |
874.6 ± 65.3† |
| SDH intensity (o.d.) |
2,004.0 ± 63.9 |
1,725.6 ± 212.3 |
1,897.3 ± 46.2† |
1,793.1 ± 124.1† |
| % Type IIa fibres | 63.8 ± 1.6 | 67.8 ± 3.9 | 71.5 ± 2.6† | 78.6 ± 3.3† |
At the conclusion of the treatment period the fibre cross-sectional area (CSA), fibre type and oxidative enzyme activity were determined by histological evaluations of cross-sections stained with H&E and reacted for succinate dehydrogenase (SDH) activity. †P < 0.05, group main effect; *P < 0.05, tranilast-treated vs. vehicle-treated mdx mice.
Tranilast administration improves resistance to muscle fatigue in dystrophic mice
Dystrophic mdx mice exhibited a ~40% reduction in diaphragm- and TA-specific force compared with control (Figure 3A/B). Nine-week treatment with tranilast did not improve whole-body strength or mobility and did not improve maximum force-producing capacity in the TA or diaphragm muscles of control or mdx mice (i.e., for TA muscle: C57BL/10 control 1,825 ± 35 mN vs. C57Bl/10 tranilast 1,743 ± 55 mN; mdx control 1,456 ± 88 mN vs. mdx tranilast 1,177 ± 139 mN). However, force production during a 4-min fatiguing stimulation protocol was improved in both the diaphragm and TA muscles of tranilast-treated mdx mice (Figure 3B, C).
Figure 3.

Tranilast effects on fatigue of diaphragm muscle strips and tibialis anterior muscles of mdx mice. Analysis of the frequency-force relationship showed significant reductions in specific force in the mdx TA and diaphragm compared to controls, which was not altered with administration of tranilast (A/B). At the conclusion of treatment, resistance to fatigue was determined from intermittent stimulation of (C) diaphragm muscle strips evaluated in vitro or (D) TA muscles evaluated in situ, from vehicle or tranilast-treated mdx mice. Values are expressed as relative specific force (force per cross-sectional area) during the 4-min stimulation protocol. #P < 0.05 group main effect, mdx vs. control. *P < 0.05 treatment main effect.
Tranilast impairs glucose tolerance in control and dystrophic mice
To check whether tranilast administration altered glucose handling in control and dystrophic mice we also performed a glucose tolerance test. Dystrophic mdx mice exhibited impaired glucose tolerance as evidenced by a 100% higher glucose response following a single intraperitoneal injection of glucose (1 g/kg) (expressed as area under the curve, Figure 4, p < 0.05). While basal blood glucose levels were not affected by tranilast administration, ~20% increased peak blood glucose levels were observed in treated control and mdx mice compared with untreated mice during the GTT (Figure 4). Furthermore, the blood glucose response was ~70% higher in tranilast-treated control and mdx mice compared with untreated mice (Figure 4).
Figure 4.
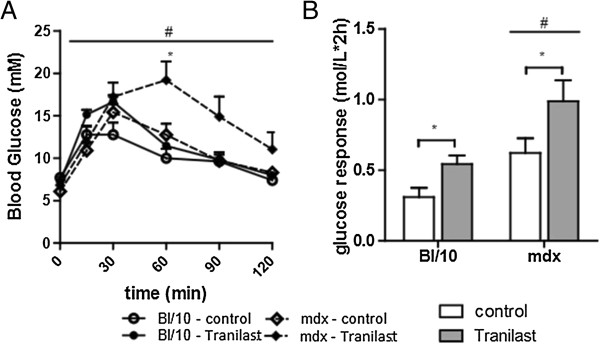
Tranilast treatment effects on glucose tolerance in mdx mice. Glucose tolerance tests were performed on vehicle and tranilast-treated control and mdx mice during the final week of treatment. Changes in blood glucose over time during the glucose tolerance test (A) and calculated glucose response (B; area under the curve). #P < 0.05 group main effect, mdx vs. control. *P < 0.05 treatment main effect.
Discussion
The identification of pharmacological agents that can prevent, reduce and/or resolve fibrotic deposition has great potential for enhancing therapies for DMD and other muscle-wasting disorders. While gene and cell therapies will eventually provide the cure for the single-gene muscle-wasting disorders, the efficacy of these approaches is likely to be hampered by the presence of significant fibrosis within affected skeletal muscles. Here we have demonstrated that one agent, tranilast, successfully reduces fibrotic deposition in skeletal muscles of mdx dystrophic mice.
Tranilast has been administered to sarcoglycan-deficient Bio14.6 hamsters, a rodent model of limb-girdle muscular dystrophy (LGMD). Treatment of 30-day-old hamsters for 120 days significantly decreased fibrosis in skeletal muscle and reduced serum creatine kinase levels and the number of centrally nucleated muscle fibres, indicating reduced muscle fibre breakdown and regeneration [25]. That study also observed a reduction in serum creatine kinase levels after a 30-day treatment in 30-day-old mdx mice [25]. We have subsequently demonstrated that oral administration of tranilast (~300 mg/kg) to young mice for 9 weeks significantly reduced fibrotic accumulation by ~30% in the diaphragm muscles of mdx mice (Figure 2A, B, C). We observed a similar trend towards a decrease in fibrosis accumulation in the TA muscles of treated mdx mice but this was not statistically significant (P = 0.10). This is most likely due to the low levels of fibrosis in the TA muscles compared with those in the diaphragm of mdx mice. The observed decrease in the diaphragm, which is the most severely affected of the muscles in the mdx mouse, indicates that tranilast was able to reduce fibrotic accumulation. We did not observe a change in the maximum force production of the TA muscle or diaphragm muscle strips, which is not surprising since a 30% reduction in fibrosis in these muscles of mdx mice would only increase the available functional tissue in the TA and diaphragm by 1.5% and 5%, respectively. We did not observe an improvement in whole-body strength or mobility of the treated mice or in maximum force of diaphragm muscle strips. Other compounds with specific muscle effects and antifibrotic properties such as IL-15 and sildenafil have been shown to improve the muscle force-producing capacity [26,27]. However, we did observe an improvement in the fatigability of both the diaphragm and TA muscles in treated mdx mice, indicating a potential improvement in muscle function. This improved fatigue resistance was not due to any changes in the muscle oxidative capacity, as there were no differences in muscle fibre oxidative capacity (SDH) between control and tranilast-treated mice (Table 1).
Although tranilast administration successfully decreased fibrotic tissue infiltration in dystrophic skeletal muscle, one concern was that it also resulted in impaired glucose tolerance in both dystrophic and control mice. Tranilast has been shown to inhibit insulin secretion in rats [28], so long-term treatment may not be possible using this specific compound. However, the effective human dose of tranilast for fibrotic pathology has been shown to be 5 mg/kg, and subsequent safety information regarding the use of tranilast in humans at this dose has not indicated toxicity issues associated with effects on insulin secretion [18]. It remains to be determined whether this dose, which is significantly lower than the dose used in the present study, would be sufficient to alter fibrosis in human skeletal muscle. Therefore it is possible that newer generation drugs may be required for therapeutic application. To this end, more targeted drugs based on the structure of tranilast are being developed that can attenuate interstitial fibrosis in the hearts of diabetic rats without causing hyperglycaemia [29].
Conclusion
Interventions to minimise fibrosis are important not just for skeletal muscle diseases but also to enhance functional recovery after serious muscle injuries. Although tranilast decreased fibrosis in dystrophic skeletal muscles, functional benefits were limited to modest improvements in fatigue resistance with impaired glucose tolerance also being a limiting factor. These issues need to be overcome in order to improve the therapeutic relevance and efficacy. Together these findings demonstrate that administration of potent antifibrotic compounds such as tranilast and newer drugs could help preserve skeletal muscle structure to ultimately increase the efficacy of pharmacological, cell and gene replacement/correction therapies for muscular dystrophy and related disorders.
Methods
Animals
Three-week-old male C57BL/10 (n = 32) and C57BL/10ScSn mdx (mdx) (n = 32) mice were obtained from the Animal Resources Centre (ARC), WA, Australia. All experimental protocols were approved by the Animal Ethics Committee of The University of Melbourne and conducted in accordance with the Australian code of practice for the care and use of animals for scientific purposes as stipulated by the National Health and Medical Research Council (Australia).
Mice were allocated into one of four groups: (1) control group treated with vehicle (CON + VEH, n = 16); (2) control group treated with tranilast (CON + TRA, n = 16); (3) mdx group treated with vehicle (mdx + VEH, n = 16); (4) mdx group treated with tranilast (mdx + TRA, n = 16).
Tranilast administration
Three-week-old control (C57BL/10) and mdx mice received tranilast (kindly provided by Dr. Spencer Williams, Department of Chemistry, The University of Melbourne) for a period of 9 weeks. Mice received standard laboratory chow (Speciality Feeds, Glen Forrest, WA, Australia) with or without the addition of 2.7 g tranilast/kg. Food was made available ad libitum based on the assumption that mice would consume approximately 4 g of feed per day resulting in a dose of 400 mg/kg/day of tranilast. This dose has been shown previously to reduce collagen infiltration in heart [20] and kidney [30] of diabetic rats. Food intake was monitored throughout the treatment period [20] and as mice consumed an average 3.3 g/day, daily tranilast intake averaged 293 mg/day.
Measurement of whole body strength
Whole body strength, whole body mobility and coordination were assessed in control C57BL/10, treated C57BL/10, control mdx and treated mdx mice at 2 (mobility) or 3 (body strength) days prior to endpoint by means of a grip strength meter (Columbus Instruments, Columbus, OH) and rotarod performance test (Rotamex-5, Columbus Instruments) as described previously [31].
Glucose tolerance test
Glucose tolerance tests were performed on control C57BL/10, treated C57BL/10, control mdx and treated mdx mice 5 days prior to endpoint. Following an overnight fast, a basal blood sample was taken from the tail vein (23 G needle) and analysed for glucose concentration using a glucometer. Mice then received an intraperitoneal injection of glucose solution (1 g/kg body mass). At 15, 30, 60, 90 and 120 min after the injection of the glucose solution, a blood sample was collected from the tail vein (23 G needle) and analysed for glucose concentration.
Assessment of contractile properties of skeletal muscle and tissue collection
At the conclusion of the treatment period, mice were anesthetised with sodium pentobarbitone (Nembutal; 60 mg/kg; Sigma-Aldrich) via i.p. injection. The methods for assessment of the contractile properties of the mouse tibialis anterior (TA) muscle in situ have been described in detail previously [32]. At the conclusion of the contractile measurements in situ, the TA muscle was carefully excised, blotted on filter paper and weighed on an analytical balance, followed by freezing in thawing isopentane for later histological examination. Soleus (SOL), extensor digitorum longus (EDL), plantaris (PLAN), gastrocnemius (GAST) and quadriceps (QUAD) muscles were excised, blotted on filter paper, trimmed of their tendons and adhering tissue, weighed and frozen in liquid nitrogen.
The entire diaphragm and rib cage were then surgically excised and costal diaphragm muscle strips composed of longitudinally arranged full-length muscle fibres were isolated and prepared for functional assessment in vitro, as described in detail elsewhere [32]. Upon completion of the functional analyses, the diaphragm muscle strip was trimmed of tendon and any adhering non-muscle tissue, blotted once on filter paper and weighed on an analytical balance. The muscle strips were then frozen in thawing isopentane for later histological examination. Mice were killed as a consequence of diaphragm and heart excision while deeply anesthetised.
Skeletal muscle histology and fibrosis
Serial sections (5 μm) were cut transversely through the diaphragm and the TA muscle using a refrigerated (-20°C) cryostat (CTI Cryostat; IEC, Needham Heights, MA). Sections were stained with haematoxylin and eosin (H&E) to reveal the general muscle architecture. Muscle fibre cross-sectional area, oxidative enzyme capacity and fibre type were determined as described previously [33]. Briefly, TA and diaphragm sections were reacted histochemically for succinate dehydrogenase (SDH) activity and immunoreacted with antibodies to laminin (Sigma-Aldrich) and myosin IIa, N2.261 (The University of Iowa, Department of Biology, Iowa City, IA; USA) in order to determine the oxidative capacity, CSA of individual myofibers and proportion of type IIA fibres respectively. Muscle collagen content was assessed from Van Gieson-stained cross-sections [32]. Digital images of stained sections were obtained using an upright microscope with a camera (Axio Imager D1, Carl Zeiss, Wrek, Göttingen, Germany), controlled by AxioVision AC software (AxioVision AC Rel. 4.8, Carl Zeiss Imaging Solutions, Wrek, Göttingen, Germany). Images were quantified using AxioVision 4.8 software.
Analysis of gene expression
At the conclusion of the treatment period, diaphragm muscles were excised and total RNA was extracted using a commercially available kit, according to the manufacturer’s instructions (PureLink RNA Mini Kit, Invitrogen). The RNA concentration was determined by a spectrophotometer at 260 nm and subsequently transcribed into cDNA using the Superscript VILO cDNA synthesis kit (Invitrogen). Real-time RT-PCR was performed as described previously [34] using the following forward and reverse primer sequences: Col1a1, 5′-CACCCTCAAGAGCCTGAGTC-3′and 5′-GTTCGGGCTGATGTACCAGT-3′; Col2a1, 5′-GCCAAGACCTGAAACTCTGC-3′ and 5′-GCCATAGCTGAAGTGGAAGC-3′; Col3a1, 5′-ACCAAAAGGTGATGCTGGAC-3′ and 5′-GACCTCGTGCTCCAGTTAGC-3′; Col4a1, 5′-AAAGGGAGAAAGAGGCTTGC-3′ and 5′-CCTTTGTACCGTTGCATCCT-3′; Col5a1, 5′-GGTCCCTGACACACCTCAGT-3′ and 5′-TGCTCCTCAGGAACCTCTGT-3′; Col6a1, 5′-CCCCATTGGACCTAAAGGAT-3′ and 5′-TCTCCCACTTCACCCTCATC-3′; Fn1, 5′-ACCACCCAGAACTACGATGC-3′ and 5′-GGAACGTGTCGTTCACATTG-3′; TGFβ1, 5′- TGAGTGGCTGTCTTTTGACG-3′ and 5′- TCTCTGTGGAGCTGAAGCAA-3′. Gene expression was quantified using a cycle threshold (CT) method, whereby a ΔCT was calculated by subtracting the 18S CT from the gene CT. The relative gene expression was then calculated using the expression 2-ΔCT.
Statistical analyses
Data were analysed with the GraphPad Prism software. Statistical significance was determined using a two-way analysis of variance (ANOVA; for comparison of treatment and group), with significance set at P < 0.05. A Tukey post hoc multiple comparison test was used where appropriate to determine significance between groups. For fatigue data comparing multiple time points, a two-way repeated measures ANOVA was used. Values are presented as mean ± SEM.
Abbreviations
CSA: Cross-sectional area; DMD: Duchenne muscular dystrophy; EDL: Extensor digitorum longus; GAST: Gastrocnemius; LGMD: Limb girdle muscular dystrophy; mdx: Muscular dystrophy X-linked; PLAN: Plantaris; QUAD: Quadriceps; SOL: Soleus; TA: Tibialis anterior; TGFβ: Transforming growth factor-β; TRA: Tranilast; VEH: Vehicle.
Competing interests
None of the authors have any competing interests.
Authors’ contribution
KS, SMG, DIS, RK and GSL conceived and designed the experiments. MT, KS, AC and TN performed the experiments. KS, MT, SMG, DIS, RK and GSL have analysed and interpreted the data. KS, MT, DIS, RK and GSL have drafted and revised the manuscript. All authors have given final approval of the latest version of the manuscript.
Contributor Information
Kristy Swiderski, Email: kristys@unimelb.edu.au.
Michelle Todorov, Email: m.todorov@student.unimelb.edu.au.
Stefan M Gehrig, Email: sgehrig@unimelb.edu.au.
Timur Naim, Email: tnaim@unimelb.edu.au.
Annabel Chee, Email: aymchee@unimelb.edu.au.
David I Stapleton, Email: dis@unimelb.edu.au.
René Koopman, Email: rkoopman@unimelb.edu.au.
Gordon S Lynch, Email: gsl@unimelb.edu.au.
Acknowledgements
Supported by a Research Collaboration Grant from The University of Melbourne. K.S. is supported by an Early Career Fellowship from the National Health and Medical Research Council.
References
- Mann CJ, Perdiguero E, Kharraz Y. et al. Aberrant repair and fibrosis development in skeletal muscle. Skelet Muscle. 2011;1:21. doi: 10.1186/2044-5040-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulfield G, Siller WG, Wight PA. et al. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicinski P, Geng Y, Ryder-Cook AS. et al. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- Lynch GS, Hinkle RT, Chamberlain JS. et al. Force and power output of fast and slow skeletal muscles from mdx mice 6–28 months old. J Physiol. 2001;535:591–600. doi: 10.1111/j.1469-7793.2001.00591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stedman HH, Sweeney HL, Shrager JB. et al. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352:536–539. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- Lynch GS, Rafael JA, Hinkle RT. et al. Contractile properties of diaphragm muscle segments from old mdx and old transgenic mdx mice. Am J Physiol. 1997;272:C2063–C2068. doi: 10.1152/ajpcell.1997.272.6.C2063. [DOI] [PubMed] [Google Scholar]
- Hara H, Wakayama Y, Kojima H. et al. Aquaporin 4 expression in the mdx mouse diaphragm. Acta Histochem Cytochem. 2011;44:175–182. doi: 10.1267/ahc.11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir LA, Chamberlain JS. Emerging strategies for cell and gene therapy of the muscular dystrophies. Expert Rev Mol Med. 2009;11:e18. doi: 10.1017/S1462399409001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YS, Li Y, Foster W. et al. The use of suramin, an antifibrotic agent, to improve muscle recovery after strain injury. Am J Sports Med. 2005;33:43–51. doi: 10.1177/0363546504265190. [DOI] [PubMed] [Google Scholar]
- Chan YS, Li Y, Foster W. et al. Antifibrotic effects of suramin in injured skeletal muscle after laceration. J Appl Physiol. 2003;95:771–780. doi: 10.1152/japplphysiol.00915.2002. [DOI] [PubMed] [Google Scholar]
- Pistilli EE, Alway SE. Systemic elevation of interleukin-15 in vivo promotes apoptosis in skeletal muscles of young adult and aged rats. Biochem Biophys Res Comm. 2008;373:20–24. doi: 10.1016/j.bbrc.2008.05.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia Mesa M. New approach to the mechanism of antiasthmatic action of Tranilast. Allergologia et immunopath. 1990;18:53–56. [PubMed] [Google Scholar]
- Kim SJ, Kim JW, Kim Y. et al. Effects of tranilast and pentoxifylline in a mouse model of chronic asthma using house dust mite antigen. J Asthma. 2009;2009(46):884–894. doi: 10.3109/02770900903089998. [DOI] [PubMed] [Google Scholar]
- Kondo N, Fukutomi O, Kameyama T. et al. Inhibition of proliferative responses of lymphocytes to food antigens by an anti-allergic drug, N(3′,4′-dimethoxycinnamoyl) anthranilic acid (Tranilast) in children with atopic dermatitis. Clin Exp Allergy. 1992;22:447–453. doi: 10.1111/j.1365-2222.1992.tb00146.x. [DOI] [PubMed] [Google Scholar]
- Kondo N, Fukutomi O, Shinbara M. et al. Inhibition of interferon-gamma and interleukin-2 production from lymphocytes stimulated with food antigens by an anti-allergic drug, Tranilast, in patients with food-sensitive atopic dermatitis. Biotherapy. 1994;8:19–22. doi: 10.1007/BF01878117. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Kanai K, Asano K. et al. Suppression of matrix metalloproteinase production in nasal fibroblasts by tranilast, an antiallergic agent, in vitro. Mediators Inflam. 2005;2005:150–159. doi: 10.1155/MI.2005.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Tanaka K, Kaneshige H. et al. The effects of long term Tranilast administration on bronchial hypersensitivity in asthmatics. Panminerva medica. 1989;31:88–93. [PubMed] [Google Scholar]
- Rogosnitzky M, Danks R, Kardash E. Therapeutic potential of tranilast, an anti-allergy drug, in proliferative disorders. Anticancer Res. 2012;32:2471–2478. [PubMed] [Google Scholar]
- Suzawa H, Kikuchi S, Ichikawa K. et al. Inhibitory action of tranilast, an anti-allergic drug, on the release of cytokines and PGE2 from human monocytes-macrophages. Jap J Pharmacol. 1992;60:85–90. doi: 10.1254/jjp.60.85. [DOI] [PubMed] [Google Scholar]
- Martin J, Kelly DJ, Mifsud SA. et al. Tranilast attenuates cardiac matrix deposition in experimental diabetes: role of transforming growth factor-β. Cardiovasular Res. 2005;65:694–701. doi: 10.1016/j.cardiores.2004.10.041. [DOI] [PubMed] [Google Scholar]
- Mifsud S, Kelly DJ, Qi W. et al. Intervention with tranilast attenuates renal pathology and albuminuria in advanced experimental diabetic nephropathy. Nephron Physiol. 2003;95:83–91. doi: 10.1159/000074845. [DOI] [PubMed] [Google Scholar]
- Miyajima A, Asano T, Asano T. et al. Tranilast ameliorates renal tubular damage in unilateral ureteral obstruction. J Urology. 2001;165:1714–1718. doi: 10.1016/S0022-5347(05)66400-2. [DOI] [PubMed] [Google Scholar]
- Miyazawa K, Kikuchi S, Fukuyama J. et al. Inhibition of PDGF- and TGF-beta 1-induced collagen synthesis, migration and proliferation by tranilast in vascular smooth muscle cells from spontaneously hypertensive rats. Atherosclerosis. 1995;118:213–221. doi: 10.1016/0021-9150(95)05607-6. [DOI] [PubMed] [Google Scholar]
- Yamada H, Tajima S, Nishikawa T. et al. Tranilast, a selective inhibitor of collagen synthesis in human skin fibroblasts. J Biochem. 1994;116:892–897. doi: 10.1093/oxfordjournals.jbchem.a124612. [DOI] [PubMed] [Google Scholar]
- Iwata Y, Katanosaka Y, Shijun Z. et al. Protective effects of Ca2+ handling drugs against abnormal Ca2+ homeostasis and cell damage in myopathic skeletal muscle cells. Biochem Pharmacol. 2005;70:740–751. doi: 10.1016/j.bcp.2005.05.034. [DOI] [PubMed] [Google Scholar]
- Harcourt LJ, Holmes AG, Gregorevic P. et al. Interleukin-15 administration improves diaphragm muscle pathology and function in dystrophic mdx mice. Am J Pathol. 2005;166:1131–1141. doi: 10.1016/S0002-9440(10)62333-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percival JM, Whitehead NP, Adams ME. et al. Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J Pathol. 2012;228:77–87. doi: 10.1002/path.4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi S, Ozaki N, Umeda H. et al. Tranilast inhibits glucose-induced insulin secretion from pancreatic beta-cells. Horm Metab Res. 2008;40:518–523. doi: 10.1055/s-2008-1073163. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Edgley AJ, Cox AJ. et al. FT011, a new anti-fibrotic drug, attenuates fibrosis and chronic heart failure in experimental diabetic cardiomyopathy. Eur J Heart Failure. 2012;14:549–562. doi: 10.1093/eurjhf/hfs011. [DOI] [PubMed] [Google Scholar]
- Kelly DJ, Zhang Y, Gow R. et al. Tranilast attenuates structural and functional aspects of renal injury in the remnant kidney model. J Am Soc Nephrol. 2004;15:2619–2629. doi: 10.1097/01.ASN.0000139066.77892.04. [DOI] [PubMed] [Google Scholar]
- Murphy KT, Chee A, Trieu J. et al. Importance of functional and metabolic impairments in the characterization of the C-26 murine model of cancer cachexia. Dis Model Mech. 2012;5:533–545. doi: 10.1242/dmm.008839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KT, Chee A, Gleeson BG. et al. Antibody-directed myostatin inhibition enhances muscle mass and function in tumor-bearing mice. Am J Physiol. 2011;301:R716–R726. doi: 10.1152/ajpregu.00121.2011. [DOI] [PubMed] [Google Scholar]
- Gehrig SM, Koopman R, Naim T. et al. Making fast-twitch dystrophic muscles bigger protects them from contraction injury and attenuates the dystrophic pathology. Am J Path. 2010;176:29–33. doi: 10.2353/ajpath.2010.090760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryall JG, Schertzer JD, Murphy KT. et al. Chronic β2-adrenoceptor stimulation impairs cardiac relaxation via reduced SR Ca2+-ATPase protein and activity. Am J Physiol Heart Circ Physiol. 2008;294:H2587–H2595. doi: 10.1152/ajpheart.00985.2007. [DOI] [PubMed] [Google Scholar]