

Abstract
Background
D-2,3-butanediol has many industrial applications such as chiral reagents, solvents, anti-freeze agents, and low freezing point fuels. Traditional D-2,3-butanediol producing microorganisms, such as Klebsiella pneumonia and K. xoytoca, are pathogenic and not capable of producing D-2,3-butanediol at high optical purity. Bacillus licheniformis is a potential 2,3-butanediol producer but the wild type strain (WX-02) produces a mix of D- and meso-type isomers. BudC in B. licheniformis is annotated as 2,3-butanediol dehydrogenase or acetoin reductase, but no pervious experiment was performed to verify this hypothesis.
Results
We developed a genetically modified strain of B. licheniformis (WX-02 ΔbudC) as a D-2,3-butanediol producer with high optimal purity. A marker-less gene deletion protocol based on a temperature sensitive knock-out plasmid T2-Ori was used to knock out the budC gene in B. licheniformis WX-02. The budC knock-out strain successfully abolished meso-2,3-butanediol production with enhanced D-2,3-butanediol production. No meso-BDH activity was detectable in cells of this strain. On the other hand, the complementary strain restored the characteristics of wild strain, and produced meso-2,3-butanediol and possessed meso-BDH activity. All of these data suggested that budC encoded the major meso-BDH catalyzing the reversible reaction from acetoin to meso-2,3-butanediol in B. licheniformis. The budC knock-out strain produced D-2,3-butanediol isomer only with a high yield of 30.76 g/L and a productivity of 1.28 g/L-h.
Conclusions
We confirmed the hypothesis that budC gene is responsible to reversibly transfer acetoin to meso-2,3-butanediol in B. licheniformis. A mutant strain of B. licheniformis with depleted budC gene was successfully developed and produced high level of the D-2,3-butanediol with high optimal purity.
Keywords: Bacillus licheniformis; D-2,3-butanediol; BudC gene; meso-2,3-butanediol dehydrogenase
Background
D-2,3-butanediol as one of the promising bulk chemicals has extensive applications in cosmetics, foods, transport fuels, medicines, and polymers industries [1]. In general, 2,3-butanediol exists in three stereoisomeric forms: D-2,3-butanediol, L-2,3-butanediol and meso-2,3-butanediol [2]. All these isomers are valuable chemicals that provide chiral groups in drugs [3]. D-2,3-butanediol is also used as an antifreeze agent because of its low freezing point (-60°C) [1]. The production of 2,3-butanediol with high optical purities is therefore highly desirable [4,5].
Although many microorganisms are capable of synthesizing 2,3-butanediol, the production processes are hindered by various limitations. For example, traditional 2,3-butanediol producing microorganisms, such as Klebsiella pneumonia and K. xoytoca, are pathogenic [2,6] and produce a mixture of meso- and L-isomers with low yield and productivity [2,7]. Non-pathogenic species such as Paenibacillus polymyxa can produce D-2,3-butanediol with a high (up to 98%) enantioselective purity; however, the cell density and the overall D-2,3-butanediol productivity is low as the cells need to be grown in micro-aerobic conditions [1,3]. The growth of P. polymyxa also needs yeast extract and tryptone, which increases the medium cost and the production recovery cost [8]. Bacillus licheniformis, which is a generally-regarded-as-safe (GRAS) organism, is also capable of producing 2,3-butanediol at the industrial level [6,9,10]; however, the wild-type B. licheniformis produces a mix of D- and meso-2,3-butanediol isomers [6].
The metabolic pathway from pyruvate to 2,3-butanediol has been well studied in B. subtilis. As shown in Figure 1, pyruvate is converted to α-acetolactate, and consequently to acetoin. At high dissolved-oxygen and glucose-rich conditions, acetoin can be further converted into 2,3-butanediol by the enzyme called acetoin reductase (AR). The same protein can catalyze the reverse reaction from 2,3-butanediol to acetoin as well, when dissolved oxygen is limited and glucose is depleted. In this case, however, the enzyme is called 2,3-butanediol dehydrogenase (BDH) [1,11]. The meso-AR/BDH is encoded by the bdhA gene in B. subtilis, therefore, modification of the bdhA gene may be an efficient way to increase the optical purity of D-2,3-butanediol while avoiding the formation of meso-2,3-butanediol by B. subtilis[1].
Figure 1.

Metabolic pathways to acetoin and 2,3-butanediol optical isomers in B. licheniformis. AR, acetoin reductase (forward reaction); BDH, 2,3-butanediol dehydrogenase (reverse reaction); NADH, nicotinamide adenine dinucleotide.
Research has been attempted to produce 2,3-butanediol with high optical purity using genetically engineered microorganisms. For example, Nielsen et al. [12] introduced the acetoin and meso-2,3-butanediol biosynthesis pathway in Escherichium coli by co-expression of 2,3-butanediol dehydrogenase originally derived from yeast, resulting in 1.12 g/L meso-2,3-butanediol [12]. Li et al. [13] also transferred the gene encoding 2,3-butanediol dehydrogenase from Enterobacter cloacae to E. coli for expressing L-2,3-butanediol from diacetyl with concentrations of 16.1 g/L and 26.8 g/L of L-2,3-butanediol produced in batch and fed-batch fermentation, respectively. Although production of high optical purity of 2,3-butanediol isomers has been achieved in engineered E. coli, the product yield was usually very low, mainly due to the weak overflow of the metabolic pathway in E. coli cells.
Compared to E. coli, Bacillus species such as B. licheniformis, B. subtilis, and B. amyloliquefaciens have a strong overflow metabolic pathway from glucose. Therefore, modification of the metabolic pathway of Bacillus species provides a promising way for producing pure 2,3-butanediol isomer with high product titer. In our previous work, we have isolated a strain of B. licheniformis (termed WX-02), which showed a rapid growth and capability of producing γ-poly-glutamic acid (γ-PGA) accompanied with 2,3-butanediol and acetoin [14]. Similar to B. subtilis and other B. licheniformis strains, however, B. licheniformis WX-02 produces a mixture of D-2,3-butanediol and meso-2,3-butanediol [11,14,15]. Furthermore, the genome of B. licheniformis WX-02 was sequenced and the data submitted [GenBank: AHIF01000000], but a gene similar to the bdhA gene in B. subtilis was not found in B. licheniformis WX-02. The gene budC (gene ID: 3100198) in B. licheniformis WX-02 genome is annotated as AR; this gene (budC) is the same as that of B. licheniformis ATCC 14580 (DSM 13) [16], although it has little similarity (1.67% identity aligned by UniProt (http://www.uniprot.org/?tab=align)) to bdhA in B. subtilis. The cell extract of B. licheniformis also shows AR (BDH) activity, with acetoin, D-2,3-butanediol and meso-2,3-butanediol also being identified. All these results indicate the existence of the gene encoding AR (BDH) in B. licheniformis[17,18]. Recent research by Li et al. [10] also shows that the recombinant E. coli containing the BDH and glycerol dehydrogenase (GDH) encoding gene from B. licheniformis exhibited meso-BDH and D-BDH activity in vitro[10]. The objective of this work was to investigate the specific function of budC in the metabolism of acetoin and 2,3-butanediol in B. licheniformis WX-02, followed with developing a strategy of knocking out the budC gene so the production of the sole D-2,3-butanediol isomer can be achieved.
Results
Establishment of the budC gene knock-out strain and complementary strain
Figure 2 shows the plasmids for deletion of the budC gene and for construction of the complementary strain. These two plasmids were used for transformation of B. licheniformis WX-02 for making the budC deletion mutant and its complementary strain, respectively. The positive strains were then verified by PCR and sequencing.
Figure 2.
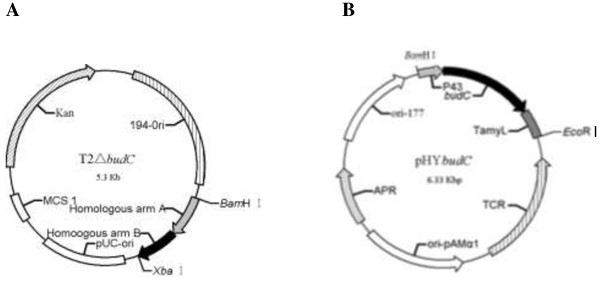
Construction of recombinant vectors. (A) Recombinant vector T2ΔbudC for budC knock-out. The vector contained a temperature-sensitive replicon from B. subtilis (194-Ori), Kanamycin-resistant gene (Kan), and the homologous arm A and B for homologous recombination. (B) Recombinant vector of pHYbudC for expression of budC in B. licheniformis. The vector contained budC expression cassette including P43 promoter, the budC gene and the α-amylase gene (amyL) transcription terminator of B. licheniformis WX-02.
The PCR results for verification of budC knock-out are shown in Figure 3A. The PCR product amplified from the genomic DNA of budC knock-out strain was about 1,500 bp; while a DNA fragment of about 2200 bp containing the budC gene and its up- and down- stream sequences was amplified from the genomic DNA of wild strain WX-02 by using primers of ΔbudC-F and ΔbudC-R as negative control. The PCR product from budC knock-out strain was purified and sequenced, and no other mutation than the budC deletion was found (data not shown), suggesting the successful construction of the budC deficient strain (WX-02 ΔbudC). The complementary strain of WX-02 ΔbudC (terms as WX-02 ΔbudC/pHYbudC), was also verified by PCR. As shown in Figure 3B, a DNA fragment of about 1500 bp was amplified from the recombinant plasmid of transformant with a matched size to the fusion fragment of P43-budC-TamyL, and then further verified by DNA sequencing (data not shown).
Figure 3.
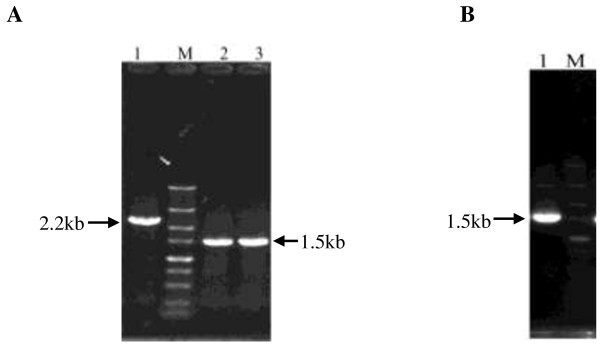
PCR verification of recombinant strains. (A) PCR verification of budC knock-out strain. M, DL5000 marker (5,000, 3,000, 2,000, 1,500, 1,000, 750, 500, 250 and 100 bp, up to bottom); lane 1, negative control (PCR product from the genomic DNA of B. licheniformis WX-02); lanes 2 and 3, fragment amplified from the genomic DNA of B. licheniformis WX-02ΔbudC. (B) PCR verification of complementary strain of B. licheniformis WX-02 ΔbudC/pHYbudC. M, DL5000 marker; lane 1, PCR product of fusion fragment of P43-budC-TamyL from recombinant strain B. licheniformis WX-02 ΔbudC/pHYbudC.
Effect of budC knock-out on meso-AR/meso and BDH activities
As described in Figure 1, the enzyme catalyzing the conversion between acetoin and 2,3-butanediol exhibits two activities depending on the culture conditions: AR activity for reduction of acetoin to 2,3-butanediol and BDH activity for dehydrogenation of 2,3-butanediol to acetoin. To investigate the effect of budC knock-out on AR and BDH activities, strains WX-02, WX-02 ΔbudC and WX-02 ΔbudC/pHYbudC were cultured for 12, 24 and 36 h; the specific activities of meso-BDH, AR, and D-BDH in the cell extracts were analyzed. As shown in Figure 4A, no meso-BDH activity was detected in the cell extracts of WX-02 ΔbudC throughout the culture; whereas both WX-02 and WX-02 ΔbudC/pHYbudC exhibited a high meso-BDH activity. As for the AR activity, WX-02 ΔbudC exhibited a very weak AR activity as compared to the other two strains (WX-02 and WX-02 ΔbudC/pHYbudC) (Figure 4B). Figure 4C shows that D-BDH activity of WX-02 ΔbudC was comparable to that of WX-02 and WX-02 ΔbudC/pHYbudC (Figure 4C). Collectively, the above results indicate that the deletion of the budC gene had a significant effect on meso-BDH, but not on D-BDH activity, indicating that the budC gene encodes meso-BDH but not D-BDH. It should also be noted that WX-02 ΔbudC/pHYbudC restored both meso-AR and meso-BDH activities compared to the budC gene knock-out strain; these two enzyme activities in WX-02 ΔbudC/pHYbudC were higher than those in WX-02 (Figure 4A and B). The reason may be due to the multicopy of the budC gene controlled by a strong promoter of P43 in WX-02 ΔbudC/pHYbudC strain as compared to the wild-type strain [19].
Figure 4.

Activity of 2,3-butanediol dehydrogenas (BDH) and acetoin reductase (AR) in different B. licheniformis. (A)meso-BDH activity; (B) AR activity; (C)D-BDH activity. The data are expressed as mean ± standard error of three replicates.
Effects of budC deletion on 2,3-butanediol configurations
Among the three stereoisomers of 2,3-butanediol, D- and L-types are racemic and can only be separated in a chiral column; they can be easily separated from the meso-type by ordinary non-chiral gas chromatograph (GC) capillary columns [4]. In this study, therefore, D-2,3-butanediol and meso-2,3-butanediol produced by B. licheniformis[2,17] were separated by ordinary non-chiral GC.
Strains WX-02, WX-02 ΔbudC and WX-02 ΔbudC/pHYbudC were respectively cultured for 24 h and then 2,3-butanediol in broth was determined. As shown in Figure 5, D-2,3-butanediol and meso-2,3-butanediol were well-separated. WX-02 ΔbudC produced D-2,3-butanediol but no meso-2,3-butanediol, whereas WX-02 and WX-02 ΔbudC/pHYbudC generated both D-2,3-butanediol and meso-2,3-butanediol (Figure 5). The result clearly shows that the synthesis of meso-2,3-butanediol in budC knocked-out strain (WX-02 ΔbudC) was successfully deleted, whereas the complementation of the budC knock-out strain (WX-02 ΔbudC/pHYbudC) restored the capability of meso-2,3-butanediol synthesis. The result confirmed that the budC gene was responsible for meso-2,3-butanediol production in B. licheniformis. Unexpectedly, the transformed strain (WX-02 ΔbudC/pHYbudC) had a higher meso-BDH activity, but this strain did not produce more meso-2,3-butanediol than the wild-type (WX-02). We believe that the synthesis of meso-2,3-butanediol in this transformant may be controlled by other rate-limiting factors. For example, the conversion from acetoin to meso-2,3-butanediol also needs nicotinamide adenine dinucleotide (NADH) as the electro-donor [6,18]; this NADH in the transformant may be the controlling factor, although the activity of meso-BDH is higher in the strain.
Figure 5.

Chromatograph profile of 2,3-butanediol produced from different B. licheniformis strain cultures. (A) WX-02; (B) WX-02 ΔbudC; (C) WX-02 ΔbudC/pHYbudC.
Production of 2,3-butanediol and acetoin by budC knock-out strain and wild-type strain
The production profile of 2,3-butanediol and acetoin by the wild strain and budC knock-out strain are presented in Figure 6. As shown in Figure 6A, wild-strain WX-02 produced both D- and meso-types of 2,3-butanediol throughout the culture. The concentration of these two isomers increased in the first 24 h; beyond this culture time, D-2,3-butanediol concentration decreased whereas meso-2,3-butanediol leveled off in the remainder of the culture period. For the culture of WX-02 ΔbudC strain, however, only the D-type of 2,3-butanediol was produced throughout all the culture. The D-2,3-butanediol concentration increased for the first 24 h, and decreased afterwards. Only a trace amount of meso-2,3-butanediol was detected at the end of culture (48 h) of WX-02 ΔbudC. Throughout all the culture period, the concentration of D-2,3-butanediol in the WX-02 ΔbudC culture was approximately the summation of the D- and meso-2,3-butanediol isomers in the WX-02 culture, indicating the meso-2,3-butanediol originally formed in the wild-type cells switched to additional D-2,3-butanediol in the WX-02 ΔbudC mutant. The above production profiles for D- and meso-type of 2,3-butanediol were also reported in the culture of B. subtilis and Serratia marcescens[9,20].
Figure 6.

Comparison of metabolites production, cell growth, and glucose consumption profile by wild-type (WX-02) and budC knock-out (WX-02 ΔbudC) B. licheniformis strains. (A)D- and meso-2,3-butanediol production. (B) Total 2,3-butanediol and acetoin production. (C) Cell density, medium pH, and residual glucose concentration. Data are expressed as mean ± standard errors of three replicates. BDL, butanediol.
As for the production of acetoin and total 2,3-butanediol, Figure 6B shows that for both the wild-type and budC gene knock-out strain, total 2,3-butanediol production increased rapidly in the first 24 h and gradually decreased afterwards; concurrently, the acetoin production of the two strains was low in the first 24 h, but increased rapidly from 24 to 36 h. The loss of the budC gene in the WX-02 ΔbudC strain resulted in more acetoin accumulation than wild strain after 36 h.
Figure 6C shows pH change, glucose utilization, and biomass density of the two strain cultures. The pH values were low in the first 24 h, indicating the synthesis of organic acids by the strains. This low pH level favored the synthesis of 2,3-butanediol (Figure 6B). In the later stage of culture, the slight increase in pH favored the conversion of D-2,3-butanediol to acetoin, which is evidenced by the increased concentration of acetoin in the medium (Figure 6B). The similar trend between pH and acetoin/2,3-butanediol conversion was also found in B. subtilis[21]. Figure 6B also shows that glucose for the two cultures had a similar trend; the glucose was rapidly consumed within the first 24 h, which corresponds to a rapid cell growth in the two cultures. After 24 h the glucose in the medium was almost depleted; as a result the cells of both the wild-type strain and budC-gene knock-out mutant ceased growth due to the glucose (Figure 6C). However, the wild-type cells showed a higher cell density than the mutant; the reason was probably due to the consumption of acetoin by the wild-type strain for supporting the cell growth. Indeed, it has been reported that acetoin can be a good carbon source for the culture of B. licheniformis once the major carbon source, glucose, is depleted [20,22]. This may also explain why the acetoin concentration in the mutant culture was much higher than that in the wild-type strain (Figure 6B).
Discussion
As a valuable compound, D-2,3-butanediol has been widely used as a major composition in solvents, anti-freeze agents, synthetic rubber, and plastics [2]. It can also be used as a potential fuel with a low freezing point and its heating value is comparable to that of ethanol and methanol [6]. Various efforts have been attempted for producing optical purity of D-2,3-butanediol by genetically modified microorganisms; however, the yield of D-2,3-butanediol has still been very low. For example, recombinant E. coli expressing the enzyme BDH was found to produce 6.1 g/L of D-2,3-butanediol [21], and B. licheniformis with a deleted lactate dehydrogenase gene (ldh) to produce 13.77 g/L of D-2,3-butanediol [6]. Heterologous expression of acetoin reductase of Clostridium beijerinckii in C. acetobutylicum has resulted in a range of 1.8 to 1.98 g/L D-2,3-butanediol [7].
The strain B. licheniformis, WX-02, used in this study was previously isolated for the production of γ-PGA with 2,3-butanediol and acetoin being the co-products. This strain can grow in a simple medium containing glucose, glutamic acid, and mineral salts [14]. As the strain WX-02 produces mixed stereoisomers of 2,3-butanediol, modification of its metabolic pathway for sole production of pure isomer of D-2,3-butanediol is desirable. To date, there have been several challenges to making a recombinant strain of B. licheniformis, including low transformation efficiency and a lack of information about the meso-BDH encoding gene. For example, Wang et al. [6] successfully deleted the ldh gene from genomic DNA in B. licheniformis by transforming protoplasts of the cells with a recombinant knock-out plasmid; however, the designed protoplast system was very complicated and the transformation efficiency was low [6]. In this paper, we successfully transformed B. licheniformis WX-02 with a recombinant knock-out plasmid with high efficiency. It demonstrates that T2-ori-based knock-out plasmid and the electro-transformation approach can be used for the metabolic modification of the B. licheniformis WX-02 strain for producing pure D-2,3-butanediol with high titer.
Previous reports showed that the bdhA gene encoding BDH is responsible for catalyzing acetoin to 2,3-butanediol in B. subtilis, and the insertion inactivation of bdhA completely blocks 2,3-butanediol synthesis [11]. For B. licheniformis, however, no bdhA gene was found in the genome. In the attempt for identifying the gene responsible for catalyzing acetoin to 2,3-butanediol in B. licheniformis, Wang et al. [6] reported the depletion of the ldh gene for the production of high optical purity of D-2,3-butanediol in B. licheniformis with 13.77 g/L D-2,3-butanediol being produced in optimized conditions [6]. However, our previous study in knocking out the ldh gene in B. licheniformis WX-02 to block lactate accumulation resulted in reduced acetoin and 2,3-butanediol production (unpublished data). The recent report showed that the budC gene might be the gene encoding meso-BDH in B. licheniformis according to the detectable enzyme activity of the recombinant protein of bdh (same as budC) gene by E. coli[10]. Therefore, we hypothesized that the budC gene is responsible for catalyzing acetoin to meso-2,3-butanediol in B. licheniformis.
Our previous research shows that the budC gene in B. licheniformis might be annotated as BDH for this species [16]. In this work, we confirmed the budC in B. licheniformis as the gene encoding meso-BDH for the reversible reaction from acetoin to meso-2,3-butanediol [18], based on the fact that the deletion of budC gene in B. licheniformis WX-02 completely blocked meso-2,3-butanediol production with significant enhanced production of D-2,3-butanediol (Figure 6A). However, the BudC protein sequence [NCBI: YP_006713433.1], aligned by blastP in the NCBI non-redundant protein database, showed that only one protein from B. sonorensis annotated as 2,3-BDH, AR or diacetyl reductase was similar (E-value <8e-112) to BudC protein. Moreover, the BudC protein sequence has a very low identity with the BDH found in other Bacillus species. For example, it has only 11.67%, 9.98% and 11.58% similarity to the BDHs from B. subtilis 168 (NP_388505.1), B. cereus YUF-4 (BAB60856.1) and B. amyloliquefaciens DSM 7 (YP_003919213.1), respectively. All these results indicate that unique BDH-encoding gene in B. licheniformis is different from other Bacillus genus.
Although the deletion of budC gene caused a slight decrease (about 5 to 10%) in cell growth (Figure 6C), it significantly enhanced the D-2,3-butanediol production (Figure 6C) (30.76 g/L) compared to both the wild strain in this work and the genetically modified strain with the deletion of the ldh gene (13.77 g/L) [6]. Finally, it should be noted that even when budC was deleted from the B. licheniformis WX-02 genome, there was still a small amount of meso-2,3-butanediol found at the end of fermentation period (Figure 6A). This may be due to the existence of other genes encoding minor meso-BDHs, or acetylacetoin reductase catalyzing acetoin to meso-2,3-butanediol [18]. Indeed, low concentration of glucose and high concentration of acetoin, as found in this work (Figure 6A and C), can induce acetylacetoin synthase to transform acetoin to meso-2,3-butanediol through the 2,3-butanediol cycle [18].
Conclusions
In summary, this report revealed the specific function of budC for the transformation between acetoin and 2,3-butanediol in B. licheniformis. The D-2,3-butanediol production level obtained in this work was the highest among the reported Bacillus genus. The study provides a deep understanding of acetoin and 2,3-butanediol metabolism in B. licheniformis, and a possible way for enhancing the production of pure D-2,3-butanediol isomer through genetic modification.
Materials and methods
Cell strain, plasmids, primers and growth media
Experiments were performed with the strains and plasmids listed in Table 1. The oligonucleotide primers listed in Table 2 were designed on the basis of B. licheniformis WX-02 genome sequence [GenBank: AHIF00000000] [16]. Luria-Bertani (LB) medium was prepared for culture of E. coli DH5α and also B. licheniformis[23]. Medium used for culturing B. licheniformis was a slight modification of that described in a previous report [24], consisting of (per liter) 120 g glucose, 33 g corn steep liquor, 9.00 g (NH4)2SO4, 1.00 g K2HPO4, 1.50 g MgSO4, 0.50 g NaCl, 0.12 g ZnCl2, 1 mg FeCl3, and 1 mg MnSO4. The medium was adjusted to 7.0 before autoclaving at 121°C for 15 minutes.
Table 1.
Bacterial strains and plasmids used in this study
| Strains and plasmids | Characteristics a | Source or reference |
|---|---|---|
|
E. coli strains |
|
|
| DH5α |
F– Φ80d/lacZΔM15, Δ(lacZYA-argF) U169, recA1, endA1, hsdR17 (rK–, mK+), phoA, supE44, λ–, thi-1, gyrA96, relA1 |
Laboratory stock |
|
B. licheniformis strains |
|
|
| WX-02 |
CCTCC M208065, wild type |
Laboratory stock [14] |
| WX-02 ΔbudC |
budC knock-out mutant of WX-02 |
This study |
| WX-02 ΔbudC/pHYbudC |
plasmid-based budC complementation strain of WX-02 ΔbudC by introduction of pHYbudC, Tcr |
This study |
| Plasmids |
|
|
| T2(2)-ori |
E. coli-B. licheniformis shuttle vector, oripUC/orits, temperature-sensitive, Kanr |
Laboratory stock |
| T2ΔbudC |
T2(2)-ori derivative containing homologous arms for budC knock-out |
This study |
| pHY300PLK |
E. coli-B. licheniformis shuttle vector, Apr(E. coli), Tcr(E. coli and B. licheniformis) |
TaKaRa |
| pHYbudC | pHY300PLK derivative containing budC, P43 promoter and TamyL (amyL terminator), Apr(E. coli), Tcr(E. coli and B. licheniformis) | This study |
aTcr, tetracycline resistance; Apr, ampicillin resistance; orits, temperature-sensitive Bacillus origin of replication; Kanr, Kanamycin resistance; CCTCC, China Center for Type Culture Collection.
Table 2.
Primers used in this study
| Primer name | a Sequence 5′ → 3′ |
|---|---|
| ΔbudC-A-F |
CGCGGATCCAAAGCGCATGTTTTAAAAC |
| ΔbudC-A-R |
CCGCCCTCCATATAGAATATAATTTTAAAAATAAACATCTTCTTTCTATAAGTAA |
| ΔbudC-B-F |
ACCAATTACTTATAGAAAGAAGATGTTTATTTTTAAAATTATATTCTATATGGAG |
| ΔbudC-B-R |
GCTCTAGACCTCGCACTAGTGTATTTTGAAAC |
| ΔbudC-F |
CGAACTCCATGAACTGACAGTC |
| ΔbudC-R |
TTGCTATTTCCTGTTATGACC |
| P43-budC-TamyL-1 |
CGCGGATCCTGTCGACGTGCATGCAGG |
| P43-budC-TamyL-2 |
CAATTTTTCCAGATACTTTACTCATGTGTACATTCCTCTCTTACCTATA |
| P43-budC-TamyL-3 |
TATAGGTAAGAGAGGAATGTACACATGAGTAAAGTATCTGGAAAAATTG |
| P43-budC-TamyL-4 |
CGTCCTCTCTGCTCTTCTATCTTTTAATTAAATACCATTCCGCCATC |
| P43-budC-TamyL-5 |
GATGGCGGAATGGTATTTAATTAAAAGATAGAAGAGCAGAGAGGACG |
| P43-budC-TamyL-6 |
CCGGAATTCGATCACCCGCGATACCGTC |
| ΔbudC A signal crossover-F |
CTTCACATGGACGATCCTAAT |
| ΔbudC A single crossover-R |
TGTTCCTCCGTAAACCGCTAAG |
| ΔbudC B single crossover-F |
CAACCACCCCTATTGAAAGCAT |
| ΔbudC B single crossover-R | GATACCTGTCCGCCTTTCTCC |
aRestriction sites highlighted in bold. Italics stands for the overlap region for splicing by overlapping extension PCR (SOE-PCR).
Chemicals and materials for cloning
Acetoin (98%) and 2,3-butanediol (98%) were purchased from Shanghai Jingchun Reagent (China). D-2,3-butanediol (>96%) was purchased from Tokyo Chemical Industry (Tokyo, Japan), meso-2,3-butanediol (99%) was purchased from Sigma-Aldrich (Sigma, St. Louis, MO, USA). All other chemicals were of analytical grade supplied by Sinopharm Chemical Reagent (Shanghai, China). T4 DNA ligase and DNA marker were purchased from Takara Bio (Dalian, China). TransStart FastPfu DNA Polymerase was purchased from TransGen Biotech (Beijing, China). Plasmid Miniprep Kit was obtained from Zoman Biotech (Beijing, China). Nucleotide sequences were determined by Beijing Genomics institution (Beijing, China).
Construction of plasmids
B. licheniformis WX-02 or B. subtilis 168 was cultured in LB medium overnight, and then collected for extraction of genomic DNA based on the method described previously [25]. The extracted genomic DNA was stored at -20°C prior to use. The gene budC was deleted by the double-crossover homologous recombination method with the primers listed in Table 2. First, two homologous arms (homologous to the 5′ and 3′ coding regions of the budC gene) of approximately 500 bp were amplified by PCR from the genomic DNA of B. licheniformis WX-02 by primers of ΔbudC-A-F and ΔbudC-A-R, ΔbudC-B-F and ΔbudC-B-R, respectively. These two homologous arms were ligated by splicing with overlapping extension PCR (SOE-PCR) with primers of ΔbudC-A-F and ΔbudC-B-R [6]. The DNA fragment was subcloned in vector T2(2)-ori joined by BamH I and Xba I restriction sites. T2(2)-ori was a previously constructed shuttle plasmid for construction of knock-out vector for B. licheniformis, with a temperature-sensitive replicon from B. subtilis to promote single crossover in bacterial cells [26]. The resulting plasmid was further verified by sequencing. A recombinant vector for budC knock-out was designated as T2ΔbudC (Figure 2A).
The fusion of the P43 promoter of B. subtilis 168, budC gene of WX-02, and terminator of amyL gene of B. licheniformis WX-02 were achieved by SOE-PCR with primers of P43-budC-TamyL-1 to 6 (Table 2) and templates of genomic DNA from B. licheniformis WX-02 or B. subtilis 168. Then the DNA fragment amplified by SOE-PCR was cloned into the plasmid of pHY300PLK joined by the BamH I and EcoR I restriction sites. The resulting plasmid was verified by sequencing. A recombinant vector for expression of budC in B. licheniformis WX-02 was designated as pHYbudC (Figure 2B).
Construction of the budC knock-out strain of WX-02
Competent cells of E. coli DH5α and B. licheniformis WX-02 were prepared for transformation of constructed plasmids as described previously [23,27]. E. coli DH5α was transferred with T2ΔbudC plasmid and cultured in LB medium with kanamycin (20 μg/mL). The plasmid T2ΔbudC isolated from the recombinant E. coli DH5α was used for transforming into B. licheniformis WX-02.
B. licheniformis WX-02 was electrotransformed with the recombinant T2ΔbudC plasmid according to the method described previously [27]; the transformants were selected by kanamycin resistance (20 μg/mL) followed with verification by PCR using the primers ΔbudC-A-F and ΔbudC-B-R (Table 2). The selected positive transformant was cultured in LB medium containing kanamycin (20 μg/mL) at 45°C for 8 h, and the temperature-sensitive replicon of the T2ΔbudC plasmid did not work at this temperature. Therefore, the high growth temperature promoted the first crossover in the cells. The mutants with kanamycin resistance were selected, and further verified by PCR with primers of ΔbudC A single crossover-F and ΔbudC A single crossover-R for crossover upstream, or ΔbudC B single crossover-F and ΔbudC B single crossover-R for crossover downstream. Then the selected colonies with single crossover were picked up and cultured in LB medium at 37°C for 8 hours, this process was repeated six times. After serial transfer without antibiotics, cells were plated on LB agar plates, and then replicated in kanamycin plates for selection of kanamycin-sensitive colonies. The budC knock-out strains that had looped out the kanamycin-resistant gene by the second crossover were selected. The mutant WX-02 ΔbudC was confirmed by PCR with primers of ΔbudC-F and ΔbudC-R (Table 2) and nucleotide sequencing.
Construction of the complementary strain of WX-02 ΔbudC
The complementation of B. licheniformis WX-02 ΔbudC was conducted with a budC expression plasmid. The B. licheniformis WX-02 ΔbudC was electrotransformed with pHYbudC DNA according to the method described previously [27], and the transformants were first selected by LB agar plates with 20 μg/mL tetracycline [27], followed with verification by PCR with primers of P43-budC-TamyL-1 and P43-budC-TamyL-6 (Table 2). The recombinant strain was designated as WX-02 ΔbudC/pHYbudC.
Detection of BDH and AR activities in cells
The wild strain WX-02, mutant strain WX-02 ΔbudC, and complementary strain WX-02 ΔbudC/pHYbudC were cultured for 12, 24 and 36 h. The cell extracts from these three cultures were prepared for determining the 2,3-BDH and AR activity based on the previous methods [11]. The reaction system contains 4 mmol/L NAD+ and 100 mmol/L 2,3-butanediol for the BDH assay or 0.2 mmol/L NADH and 50 mmol/L acetoin for the AR assay [11]. The cell extracts and reaction system were preheated at 37°C, the 200-μL reaction system was then added to a 96-well UV-star microplate (Greiner Bio-One, Germany) followed with addition of 5 μL cell extracts. The microplate was immediately put into a microplate reader (BioTek, USA) and reacted at 37°C for 5 minutes. Absorbance at 340 nm was measured initially and the end of the reaction. Under these conditions, one unit of BDH or AR activity was defined as 1 μmol of NADH produced or consumed by 1 mg of protein per minute. The protein concentration of cell extracts was determined by the Coomassie brilliant blue method [28].
Analysis
Cell density was determined by the optical absorbance at 600 nm (OD600). The concentration of residual glucose was measured by a biosensor equipped with a glucoseoxidase electrode (SBA-40C, China). Single colonies of the wild strain of WX-02, mutant strain of WX-02 ΔbudC, and complementary strain of WX-02 ΔbudC/pHYbudC on the LB plate were transferred into 250-mL flasks containing 50 mL LB medium and incubated at 37°C for 11 h in an orbital shaker at 180 rpm until the OD600 of the culture reached approximately 4.2. The cells were then sub-cultured for 48 h in the same conditions. The samples were collected periodically to determine the time course of cell density, residual glucose, and product (acetoin and 2,3-butanediol) concentrations using previously described methods [29].
Acetoin, D-2,3-butanediol, and meso-2,3-butanediol were extracted by ethyl acetate and then quantified using Trace GC Ultra Gas Chromatograph (Thermo, USA) equipped with a flame ionization detector and TR-WAX capillary column (30 m × 0.32 mm ID, 0.25 μm film). Nitrogen was used as the carrier gas with a flow rate of 1.0 mL/minute; the injected volume was 1 μL with a splitless injection mode. The injector temperature and the detector temperature were 215°C and 245°C, respectively. The column was maintained at 50°C for 1.5 minutes, increased at a rate of 10°C/minute to 110°C for 0.5 minutes, 5°C/minute to 150°C for 0.5 minutes, and 20°C/minute to 220°C for 1 minute. The concentration of acetoin and 2,3-butanediol was quantified using the internal standard (butanol).
Abbreviations
Amy: amylase; AR: acetoin reductase; BDH: 2,3-butanediol dehydrogenase; BDL: butanediol; bp: base pairs; GC: gas chromatograph; Ldh: lactate dehydrogenase; NADH: nicotinamide adenine dinucleotide; OD: optical density; SOE-PCR: splicing with overlapping extension PCR; γ-PGA: γ-poly-glutamic acid.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
GQ conceived of the study, performed the data analysis, and coordinated the manuscript draft and revision. YK and LL executed the experimental work and data analysis. AX and SZ executed the experimental work. ZW helped to revise and proofread the manuscript. DX helped with data analysis. SC conceived the study, and coordinated the manuscript draft and revision. All authors read and approved the final manuscript.
Contributor Information
Gaofu Qi, Email: qigaofu@mail.hzau.edu.cn.
Yanfang Kang, Email: 415534283@qq.com.
Lu Li, Email: lilu045@163.com.
Aifang Xiao, Email: 27323588@qq.com.
Shumeng Zhang, Email: garyq_2000@aliyun.com.
Zhiyou Wen, Email: wenz@iastate.edu.
Dihong Xu, Email: xudihong@mail.hzau.edu.cn.
Shouwen Chen, Email: mel212@126.com.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No.31170046 and J1103510). This work was also supported by rural areas of the national science and technology plan in the 12th five-year plan of China (No.2013AA102801-52).
References
- Gao J, Yang HH, Feng XH, Li S, Xu H. A 2,3-butanediol dehydrogenase from Paenibacillus polymyxa ZJ-9 for mainly producing R, R-2,3-butanediol: purification, characterization and cloning. J Basic Microbiol. 2012;52:1–9. doi: 10.1002/jobm.201200152. [DOI] [PubMed] [Google Scholar]
- Ji XJ, Huang H, Ouyang PK. Microbial 2,3-butanediol production: a state-of-the-art review. Biotechnol Adv. 2011;29:351–364. doi: 10.1016/j.biotechadv.2011.01.007. [DOI] [PubMed] [Google Scholar]
- Yu B, Sun J, Bommareddy RR, Song L, Zeng AP. Novel (2R,3R)-2,3-butanediol dehydrogenase from potential industrial strain Paenibacillus polymyxa ATCC 12321. Appl Environ Microbiol. 2011;77:4230–4233. doi: 10.1128/AEM.02998-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Lv C, Gao C, Qin J, Ma C, Liu Z, Liu P, Li L, Xu P. A novel whole-cell biocatalyst with NAD+ regeneration for production of chiral chemicals. PLoS One. 2010;5:e8860. doi: 10.1371/journal.pone.0008860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Qin J, Gao C, Hua D, Ma C, Lia L, Wanga Y, Xu P. Production of (2S,3 S) - 2,3 - butanediol and (3S)-acetoin from glucose using resting cells of Klebsiella pneumonia and Bacillus subtilis. Bioresour Technol. 2011;102:10741–10744. doi: 10.1016/j.biortech.2011.08.110. [DOI] [PubMed] [Google Scholar]
- Wang QZ, Chen T, Zhao X, Chamu J. Metabolic engineering of thermophilic Bacillus licheniformisfor chiral pure D-2,3-butanediol production. Biotechnol Bioeng. 2012;109:1610–1621. doi: 10.1002/bit.24427. [DOI] [PubMed] [Google Scholar]
- Siemerink MA, Kuit W, Contreras AML, Eggink G, van der Oost J, Kengen SW. D - 2, 3 - butanediol production due to heterologous expression of an acetoin reductase in Clostridium acetobutylicum. Appl Environ Microbiol. 2011;77:2582–2588. doi: 10.1128/AEM.01616-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang W, Ma Y, Zeng AP. Medium optimization and proteome analysis of (R, R) - 2,3 - butanediol production by Paenibacillus polymyxa ATCC 12321. Appl Microbiol Biot. 2013;97:585–597. doi: 10.1007/s00253-012-4331-6. [DOI] [PubMed] [Google Scholar]
- Jurchescu IM, Hamann J, Zhou X, Ortmann T, Kuenz A, Prüße U, Lang S. Enhanced 2,3-butanediol production in fed-batch cultures of free and immobilized Bacillus licheniformis DSM 8785. Appl Microbiol Biot. 2013;97:6715–6723. doi: 10.1007/s00253-013-4981-z. [DOI] [PubMed] [Google Scholar]
- Li L, Zhang L, Li K, Wang Y, Gao C, Han B, Ma C, Xu P. A newly isolated Bacillus licheniformis strain thermophilically produces 2,3-butanediol, a platform and fuel biochemical. Biotechnol Biofuels. 2013;6:123. doi: 10.1186/1754-6834-6-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson WL. The Bacillus subtilis ydjL (bdhA) gene encodes acetoin reductase/2,3 - butanediol dehydrogenase. Appl Environ Microbiol. 2008;74:6832–6838. doi: 10.1128/AEM.00881-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen DR, Yoon SH, Yuan CJ, Prather KLJ. Metabolic engineering of acetoin and meso-2,3-butanediol biosynthesis in E. coli. Biotechnol J. 2010;5:274–284. doi: 10.1002/biot.200900279. [DOI] [PubMed] [Google Scholar]
- Li L, Wang Y, Zhang L, Ma C, Wang A, Tao F, Xu P. Biocatalytic production of (2S,3S)-2,3-butanediol from diacetyl using whole cells of engineered Escherichia coli. Bioresour Technol. 2012;115:111–116. doi: 10.1016/j.biortech.2011.08.097. [DOI] [PubMed] [Google Scholar]
- Wei XT, Ji ZX, Chen SW. Isolation of halotolerant Bacillus licheniformis WX-02 and regulatory effects of sodium chloride on yield and molecular sizes of poly-γ-glutamic acid. Appl Biochem Biotechnol. 2010;160:1332–1340. doi: 10.1007/s12010-009-8681-1. [DOI] [PubMed] [Google Scholar]
- Renna MC, Najimudin N, Winik LR, Zahler SA. Regulation of the Bacillus subtilis alsS, alsD, and alsR genes involved in post-exponential-phase production of acetoin. J Bacteriol. 1993;175:3863–3875. doi: 10.1128/jb.175.12.3863-3875.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yangtse W, Zhou Y, Lei Y, Qiu Y, Wei X, Ji Z, Qi G, Yong Y, Chen L, Chen S. Genome Sequence of Bacillus licheniformis WX-02. J Bacteriol. 2012;194:3561–3562. doi: 10.1128/JB.00572-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannini PP, Mantovani M, Medici A, Pedrini P. Production of 2,3-butanediol by Bacillus stearothermophilus: fermentation and metabolic pathway. Chem Eng. 2008;14:281–286. [Google Scholar]
- Giovannini PP, Mantovani M, Grandini A, Medici A, Pedrini P. New acetoin reductases from Bacillus stearothermophilus: meso- and 2R, 3R-butanediol as fermentation products. J Mol Catal B Enzym. 2011;69:15–20. doi: 10.1016/j.molcatb.2010.12.004. [DOI] [Google Scholar]
- Zhang XZ, Cui ZL, Hong Q, Li SP. High-level expression and secretion of methyl parathion hydrolase in Bacillus subtilis WB800. Appl Environ Microbiol. 2005;71:4101–4103. doi: 10.1128/AEM.71.7.4101-4103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao B, Zhang LY, Sun J, Su G, Wei D, Chu J, Zhu J, Shen Y. Characterization and regulation of the 2,3-butanediol pathway in Serratia marcescens. Appl Microbiol Biot. 2012;93:2147–2159. doi: 10.1007/s00253-011-3608-5. [DOI] [PubMed] [Google Scholar]
- Ali NO, Bignon J, Rapoport G, Debarbouille M. Regulation of the acetoin catabolic pathway is controlled by sigma L in Bacillus subtilis. J Bacteriol. 2001;183:2497–2504. doi: 10.1128/JB.183.8.2497-2504.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanh TN, Jürgen B, Bauch M, Liebeke M, Lalk M, Ehrenreich A, Evers S, Maurer K, Antelmann H, Ernst F, Homuth G, Hecker M, Schweder T. Regulation of acetoin and 2, 3-butanediol utilization in Bacillus licheniformis. Appl Microbiol Biotechnol. 2010;87:2227–2235. doi: 10.1007/s00253-010-2681-5. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. The 3rd ed., Cold Spring Horbor laboratory [Z] NY: Cold Spring Harbor; 2001. [Google Scholar]
- Liu Y, Zhang S, Yong YC, Ji Z, Ma X, Xu Z, Chen S. Efficient production of acetoin by the newly isolated Bacillus licheniformis strain MEL09. Process Biochem. 2011;46:390–394. doi: 10.1016/j.procbio.2010.07.024. [DOI] [Google Scholar]
- Sohail M. A simple and rapid method for preparing genomic DNA from gram-positive bacteria. Mol Biotechnol. 1998;10:191–193. doi: 10.1007/BF02760866. [DOI] [PubMed] [Google Scholar]
- Guerout-Fleury AM, Shazand K, Frandsen N, Stragier P. Antibiotic-resistance cassettes for Bacillus subtilis. Gene. 1995;167:335–336. doi: 10.1016/0378-1119(95)00652-4. [DOI] [PubMed] [Google Scholar]
- Xue G, Johnson JS, Dalrymple BP. High osmolarity improves the electro-transformation efficiency of the gram-positive bacteria Bacillus subtilis and Bacillus licheniformis. J Microbiol Meth. 1999;34:183–191. doi: 10.1016/S0167-7012(98)00087-6. [DOI] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Gao J, Xu H, Li QJ, Feng XH, Li S. Optimization of medium for one-step fermentation of inulin extract from Jerusalem artichoke tubers using Paenibacillus polymyxa ZJ-9 to produce R, R-2,3-butanediol. Bioresour Technol. 2010;101:7076–7082. doi: 10.1016/j.biortech.2010.03.143. [DOI] [PubMed] [Google Scholar]