

Abstract
Esophageal squamous cell carcinoma (ESCC) is one of the most aggressive forms of human cancer with poor prognosis due to late diagnosis and metastasis. Common genomic alterations in ESCC include p53 mutation, p120ctn inactivation, and overexpression of oncogenes such as cyclin D1, EGFR, and c-Met. Using esophageal epithelial cells transformed by the overexpression of EGFR and p53R175H, we find novel evidence of a functional link between p53R175H and the c-Met receptor tyrosine kinase to mediate tumor cell invasion. Increased c-Met receptor activation was observed upon p53R175H expression and enhanced further upon subsequent EGFR overexpression. We inhibited c-Met phosphorylation, resulting in diminished invasion of the genetically transformed primary esophageal epithelial cells (EPC-hTERT-EGFR-p53R175H), suggesting that the mechanism of increased invasiveness upon EGFR and p53R175H expression may be the result of increased c-Met activation. These results suggest that the use of therapeutics directed at c-Met in ESCC and other squamous cell cancers.
Keywords: p53 mutation, c-Met, esophageal cancer, tumor invasion
Introduction
Human squamous cell carcinomas are the most common type of epithelial cancers. One subtype, esophageal squamous cell carcinoma (ESCC), is an aggressive cancer with poor prognosis due to late diagnosis, local and distant metastases, and limited therapeutic options. The other subtype of esophageal cancer is esophageal adenocarcinoma, with the fastest rate of increase of any cancer in the US.1 Risk factors for ESCC include cigarette-smoking, alcohol use, and certain mineral (e.g., selenium, zinc, molybdenum) and vitamin (A, C, E, and K) deficiencies.2,3 The development of ESCC is a multistep, progressive process, and a number of genetic alterations in the tumor cells have been identified.4,5 Among the oncogenes that are activated include cyclin D1 and EGFR, and the tumor suppressor genes that are inactivated include p53, p120ctn, and p16INK4a.2 EGFR overexpression and p53 mutations are particularly common in premalignant lesions.6-8 TP53 mutations result in the stabilization and accumulation of mutant p53 protein. Mutations fall into two categories: (1) mutations that directly block DNA binding by p53 (R273H mutation-archetype) and (2) mutations that alter the global conformation of the p53 protein (R175H mutation-archetype). It has been shown to influence tumorigenesis in multiple ways, including loss of the gatekeeper functions of wild-type (WT) p53 by acting as a dominant negative directly on WT p53, and more recently appreciated, through WT p53-independent gain of function (GOF) mechanisms. In the latter context, the putative mechanisms involve abrogation of the effects of Tp63, an ortholog of Tp53, and direct DNA binding with activation of a different repertoire of genes by mutant p53. Clinically, tumors with p53 mutations carry a worse prognosis than those that are p53 null.9,10 p53 null cells can be transformed by expression of mutant p53.11 Tumors in p53R172H/+ transgenic mice tend to be of more epithelial origin compared with p53−/− or p53+/− murine tumors and metastasize more than those from p53+/− mice.11,12
The c-Met tyrosine kinase receptor activates a signaling response program termed “invasive growth” that is necessary both during normal embryonic development and adult tissue repair.13,14 It is activated upon binding its ligand, namely hepatocyte growth factor (HGF). Specifically, there is transphosphorylation of Tyr 1234 and Tyr 1235 on the intracellular domain of c-Met, induction of docking molecules, and the activation of diverse signaling pathways, such as Ras, PI3K, STAT, β-catenin, and Notch pathways.15 Perturbed activation of c-Met signaling has been shown to be important for neoplastic transformation in a wide variety of tumor types, including ESCC, largely through c-Met amplification or overexpression, although rare receptor mutations have been detected.16,17
There is limited evidence linking p53 mutation and c-Met signaling in cancer. Sarcomas arising in Li–Fraumeni patients have increased c-Met protein levels.18 WT p53 was shown recently to regulate c-Met expression and influence cell migration and invasion in normal and transformed ovarian epithelium.19 We have reported previously, using a model that recapitulates early genetic alternations in ESCC, that expression of mutant p53R175H and EGFR could transform immortalized human primary esophageal epithelial keratinocytes (EPC-hTERT), thereby dramatically increasing their migratory and invasive capabilities.5 In this study, we report a previously unidentified direct connection between p53R175H and increased c-Met receptor activity that is linked directly to tumor invasion.
Results
Mutant p53R175H expression leads to c-Met activation in a HGF-ligand-independent manner
We have shown previously that c-Met phosphorylation was increased in EPC-hTERT-cells retrovirally infected to express p53R175H and its activation was further increased upon co-expression with EGFR.20 This specific pattern of activation was not shared with other selected receptor tyrosine kinases as IGF1Rβ and EGFR phosphorylation (data not shown). In a certain context, EGF and HGF cooperate to promote cell proliferation, scatter and invasion in mouse mammary cells21. Certainly, EGFR inhibition has led to the evaluation of targeting c-Met for EGFR inhibitor resistant tumors.22,23 Yet, inhibition of EGFR tyrosine kinase activity did not affect c-Met activation (Fig. 1A). That being said, our results are not inconsistent with reports of the lack of crosstalk between EGFR and c-Met in mouse adult liver oval cells.24 In our experimental conditions, a c-Met inhibitor decreased c-Met phosphorylation, but interestingly, c-Met phosphorylation was not affected by an HGF blocking antibody (previously optimized for blocking efficiency20) in EPC-hTERT-p53R175H ± EGFR cells (Fig. 1B and C). This suggests that HGF was not responsible for inducing c-Met activation under these experimental conditions. Further support of this conclusion emerges from the finding that HGF was undetectable in cell culture media (keratinocyte serum free medium, KSFM) or in conditioned media collected from EPC2-hTERT-EGFR-p53R175H cells (data not shown). c-Met phosphorylation was reduced when EPC2-hTERT-EGFR-p53R175H cells were cultured in keratinocyte basal medium (KBM) or in KSFM without the added bovine pituitary extract (BPE) (Fig. 1D), but was constitutively phosphorylated in KSFM with BPE ± additional EGF (normal culture conditions). It is conceivable there is a factor(s) in BPE that influence Met phosphorylation. In order to determine if the p53R175H-mediated c-Met phosphorylation was the result of the potential expression and autocrine secretion of an unidentified ligand capable of activating c-Met, cells with low/absent c-Met phosphorylation (EPC2-hTERT-EGFR and EPC2-hTERT, respectively) were treated with conditioned media collected from EPC2-hTERT-EGFR-p53R175H cells (Fig. 1E). No increase in c-Met phosphorylation was observed in either EPC2-hTERT-EGFR or EPC2-hTERT cells, thereby leading us to conclude that c-Met activation upon p53R175H expression is ligand-independent, either HGF or some other ligand.

Figure 1. p53R175H expression leads to c-Met activation in an HGF-independent manner. (A) Inhibition with AG11478 EGFR tyrosine kinase inhibitor (1uM) for 120 min. (B and C) Inhibition with anti-HGF antibody or the PHA665752 phospho-Met inhibitor. (D) Activation of phospho-Met in EPC2-hTERT-EGFR-p53R175H can be attenuated by using keratinocyte basal medium (KBM) or using keratinocyte serum free medium (KSFM) without BPE (bovine pituitary extract) and EGFR supplements. (E) Treatment of cells with condition media from EPC2-hTERT-EGFR-p53R175H cells does not activate phospho-Met in EPC2-hTERT-neo-puro or EPC2-hTERT-EGFR-puro.
Phospho-Met expression is specific to the p53R175H mutation
A survey of primary esophageal keratinocytes expressing additional p53 mutations (R273H, V143A, or R248W) described previously5,25 revealed that the activation of the c-Met receptor upon p53 mutation was exclusive to R175H (Fig. 2A). Esophageal keratinocytes isolated from p53 null and WT mice with overexpression of the human p53R175H mutant (previously described)26 did not have similar patterns of c-Met activation (Fig. 2B), which might point to species difference in the origin of the esophageal keratinocytes or the potential combinatorial effects of EGFR and p53R175H.
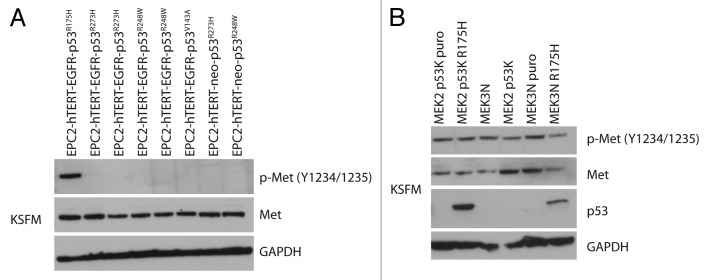
Figure 2. Phospho-Met expression is specific to the p53R175H. (A) Panel of four different p53 mutations R175H, R273H, R248W, and V143A were analyzed by western blotting for phospho-Met expression. Only the cells expressing the p53R175H mutant have increased phospho-Met expression. (B) Mouse esophageal keratinocytes (MEK) isolated from p53 null mice (MEK3N p53K puro, MEK2N p53K) and wild-type p53 mice (MEK3N, MEK3N puro) were transfected with human p53R175H, in both the p53 null (MEK2 p53K p53R175H) and wt p53 (MEK3N p53R175H). The p53R175H did not have the same effect on activation of phospho-Met as in human keratinocytes.
Restoration of WT p53 function reverses p53R175H mutant activation of phospho-Met
In order to determine if the increased c-Met phosphorylation was linked directly to p53 mutation, three pharmacologic activators of WT p53 signaling were used. 5-iminodaunorubicin has been identified for its cytotoxicity independent of the cellular p53 status and does so by activating p53 family member p73 and downstream p21.27 CP-31398 and PRIMA-1 have been shown to bind to mutant p53 and alter its conformation so as to restore proper DNA binding.28,29 Treatment with either 5-ID or CP-31398 compound resulted in a loss of c-Met phosphorylation in EPC-hTERT-EGFR-p53R175H cells with a concomitant upregulation of p21 (Fig. 3A and B). PRIMA-1 treatment did not show an effect on phospho-Met or activation of p21 at 1 µM or 10 µM concentrations; treatments at higher doses proved to be toxic to the cells (Fig. 3B). Both 5-ID and CP-31398 compounds inhibited invasion of EPC-hTERT-EGFR-p53R175H cells as well as proliferation (Fig. 3C–E). These experiments give credence to the premise that the increased tumor cell invasion apparent with p53R175H mutation is mediated in part by phosho-Met.

Figure 3. Restoration of WT p53 function reverses p53R175H mutant activation of phospho-Met. (A and B) EPC2-hTERT-EGFR-p53R175H cells treated for 24hrs with 5-iminodaunorubicin (5-ID) in a range from 0.1–10 μM (A), CP-31398 (50 ng/mL–5 μg/mL) (B) show decrease in phospho-Met levels by western blotting; treatment with PRIMA1 (1–10 μM) (B) had no effect in the levels of phopho-Met. (C) Treatment with 5-ID (range 0.1–10 μM) and CP-31398 (range 0.01–10 μg/mL) inhibited cells growth of EPC2-hTERT-EGFR-p53R175H in WST-1 assay (C). Error bars represent ± SEM, and the Student t test was used to determine significance (*, P ≤ 0.05). (D) Treatment of EPC2-hTERT-EGFR-p53R175H with 5-ID (1 uM and 5 uM) and CP-31398 (2 ug/mL and 5 ug/mL) caused a decrease in boyden-chamber invasion assay. (C–D) Error bars represent ± SEM, and the Student t test was used to determine significance (*, P ≤ 0.05). (E). H&E staining of organotypic 3D cultures show 5-ID (3 μM) and CP-31398 (5 μg/mL) treatment can reduce invasion, magnification 10×.
Discussion
ESCC is typically inoperable and fatal when detected after tumor cells have metastasized. We have studied the genetic influences on esophageal tumor cell invasion, one of the initial critical steps of tumor dissemination and metastasis. We report a novel connection between mutant p53R175H expression and activation of the c-Met receptor tyrosine kinase that is HGF ligand independent and potentially independent of other ligands. The specific nature of the DNA contact inhibition by p53R175 and how it activates c-Met tyrosine kinase activity is interesting. Mutant p53 has been demonstrated to result in enhanced integrin and epidermal growth factor receptor (EGFR) trafficking, which depends upon Rab-coupling protein (RCP) and results in constitutive activation of EGFR/integrin signaling.30 To that end, mutant p53 may enhance c-Met signaling to promote cell scattering and invasion through both TAp63-dependent and -independent mechanisms;30,31 however, this is dependent upon HGF ligand binding the c-Met receptor, a scenario not present in our results, suggesting that our specific interrelationship between mutant p53 and c-Met is mediated through another mechanism. Another possibility is that mutant p53 may have a direct transcriptional effect upon c-Met by virtue of its GOF properties.32 Recently, the miR34 family of microRNA was identified as a p53 transcriptional product and c-Met was confirmed to be a target of the miR34 family.33-35 We did find that mir34a is decreased in the p53R175H cells (data not shown). However, we do not find that overexpressing mir34a had an effect on either c-Met expression levels or phospho-Met levels (data not shown).
ESCC is a difficult cancer to treat. Currently, neo-adjuvant or adjuvant therapy involves chemotherapy and radiation. Biologics have gained some attention, although EGFR inhibition is not standard. Since p53R175H is present in a subset of ESCC,36 it would be intriguing to consider Met inhibition therapy in this context in future pre-clinical study.
Materials and Methods
Cell culture
Primary human esophageal keratinocytes, designated EPC2-hTERT, and their derivatives are described previously.37 The following mediums where used keratinocyte basal medium (KBM) from Lonza or using keratinocyte serum free medium (KSFM) (Invitrogen) without BPE (bovine pituitary extract) and EGFR supplements. Stable transduction of primary esophageal cells with retroviral vectors was described previously.26,37,38 Briefly, pFB-neo retroviral vectors (Stratagene) containing the entire coding sequence for the human EGFR (pFB-neo-WT-hEGFR), pBabe-puro-p53R175H or pBabe-puro-p53R273H or pBabe-puro-R248W or pBabe-puro-V143A were transfected into Phoenix-Ampho packaging cells (gift of Dr. Garry Nolan, Stanford University) using LipofectAMINE 2000 reagent (Invitrogen), according to the manufacturer’s instructions. Culture supernatants from individual Phoenix-Ampho cells were used to infect EPC2-hTERT cells. Cells were passaged 48 h after infection and selected with G418 (300 μg/mL), puromycin (0.5 μg/mL), for a period of 7 d, resulting in generation of control EPC2-hTERT-EGFR-p53R175H, EPC2-hTERT-EGFR-p53R273H, EPC2-hTERT-EGFR-p53R248W, EPC2-hTERT-EGFR-p53V143A, EPC2-hTERT-neo-p53R273H and EPC2-hTERT-neo-p53R248W cells. Independent infections and selections were performed to generate two additional cell lines of each genotype. Mouse esophageal keratinocytes (MEK) isolated from p53 null mice (MEK3N p53K puro, MEK2N p53K) and wild-type p53 mice (MEK3N, MEK3N puro) previously decribed,38 were transfected with human p53R175H, in both the p53 null (MEK2 p53K p53R175H) and wt p53 (MEK3N p53R175H). In both context the p53R175H mutant did not have the same effect on activation of phospho-Met as in the human keratinocytes. The following reagents were used in cell culture studies: AG1478 (Sigma) EGFR tyrosine kinase inhibitor (1 μM) for 120min; PHA665752 phospho-Met inhibitor (Tocris Bioscience); 5-iminodaunorubicin (5-ID) (gift of Dr Wafik El-Deiry) range from 0.1–10 μM; CP-3139829 (50 ng/mL–5 μg/mL) (Tocris Biosciences) and PRIMA128 (1–10 μM) (Cayman Chemical).
Conditioned media treatment
Condition media of EPC2-hTERT-EGFR-p53R175H cells grown in 2D cultures in KSFM medium, was collected after 48 h, collected medium was spun down for 5 min at 1000 rpm to remove any cellular debris. EPC2-hTERT-neo-puro or EPC2-hTERT-EGFR-puro cells (0.5 × 106 per plate) where plated 24 h before treatment. Condition medium was added after 24 h of growth, cells where harvested 24 h after for protein lysis previously described.
Western blot analysis
Cells were harvested in lysis buffer (50 mM Tris·HCl [pH 8], 150 mM NaCl, 1% Nonidet P-40, 1% Triton X-100, 2 mM sodium orthovanadate, 10 mM sodium fluoride, 5 mM sodium pyrophosphate, protease inhibitor tablet [Roche]). Thirty micrograms of protein were run on a 4–12% SDS/PAGE Bis-Tris gel (Invitrogen) and transferred to a poly (vinylidene difluoride) membrane (Immobilon-P; Millipore). Membranes were blocked in 5% nonfat milk (Bio-Rad Life Science) in PBS-T [1× PBS without Ca2+ and Mg2+ (Invitrogen) and 0.1% Tween 20] for 1 h at room temperature. Membranes were then probed with primary antibody diluted in 5% milk in PBS-T overnight at 4 °C, washed with PBS-T, and incubated with anti-mouse or anti-rabbit secondary antibodies (1:5000 in PBS-T) for 1 h at room temperature and washed in PBS-T. The signal was visualized using an enhanced chemiluminescence solution (ECL Plus; GE Healthcare Life Sciences) and exposed to Blue Lite Autorad film (ISC-BioExpress).
Antibodies
Antibodies were obtained from the following sources: EGFR(Ab-12) (1:1000) (NeoMarkers/Thermo Fisher Scientific), phospho-EGFR (Tyr1068) (1:500) and phospho-Met (Tyr1234/1235) (1:500) Cell Signaling Technology, Met antibody (C-28) (1:1000) Santa Cruz Biotechnology, p53 (Ab-6) (1:1000) Calbiochem, GAPDH (1:15000) Chemicon/Millipore, Cell Signaling and p21 (WAF1 Ab-1) (1:1000) Oncogene. Anti-mouse and anti-rabbit horseradish peroxidase (HRP)-conjugated antibodies were from GE Healthcare Life Sciences. Mouse IgG1 control antibody was from R&D Systems. 2B8 mouse anti-human HGF IgG1 generated from hybridoma (AVEO Pharmaceuticals) and mouse IgG1 control antibody from R&D Systems, where used in HGF blocking antibody experiment, as previously described.20
Cell proliferation WST-1 assay
Five thousand cells were plated in 100 μl of KSFM in a 96-well plate for 72 h before reading with WST-1 reagent (Roche). After 48 h of growth, we added 5-ID and CP-31398 reagents at described concentrations. After 24 h 10 ul of WST-1 reagent and incubated at 37 °C for 2 h. Following incubation plate was read using ELISA plate reader to measure absorbency. All experiments were performed in triplicate on three independent days.
Boyden chamber invasion assay
For invasion assays, insert plates (8-μm pore size, 24-well insert) coated with growth factor reduced Matrigel matrix were used (BD Biosciences). Inserts were placed in a 24-well plate containing DMEM + 10% serum to stimulate cell invasion. 1 × 105 cells in serum-free medium were placed in each insert (plus the 5-ID or CP-31398). Twenty hours later, the cells remaining inside the insert were removed with a cotton swab and the invading cells on the insert bottom were labeled with 4 μg/mL Calcein AM dye (Invitrogen) in Hanks’s balanced salt solution (HBSS) (Invitrogen) for 30 min at 37 °C. The labeled cells were then read on a Biotek FLX800 multidetection microplate reader (BioTek) at 485 nm excitation and 528 nm detection. All experiments were performed in triplicate on three independent days.
Organotypic culture
H&E staining of organotypic 3D cultures show 5-ID and CP-31398 treatment can reduce invasion. The employment of EPC cells and their derivative cell lines in organotypic culture was done as described previously.39 The cultures were harvested and fixed for 1 h or overnight in 10% buffered formalin phosphate (Fisher) before being paraffin-embedded and sectioned (Leica RM2155 microtome; Leica Microsystems) for hematoxylin and eosin (H&E) staining.
Disclosure of Potential Conflicts of Interest
The authors have no conflicts of interest to disclose.
Acknowledgments
The authors would like to thank the members of the Rustgi lab for technical support and helpful discussions. This work was supported by NIH/NCI P01 CA098101, NIH/NCI U01 CA 143056, NIH/NIDDK P30 DK050306 Center for Molecular Studies in Digestive and Liver Diseases (and Molecular Pathology and Imaging, Molecular Biology/Gene Expression, Cell Culture Core Facilities), American Cancer Society Grant RP-10-033-01-CCE (AKR) NIH T32 NIH/NRSA F32 DK082149 (KDG), NIH T32 CA09140 (MEV), and NIH T32 CA115299 (GSW).
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/25406
References
- 1.Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349:2241–52. doi: 10.1056/NEJMra035010. [DOI] [PubMed] [Google Scholar]
- 2.Okano J, Snyder L, Rustgi AK. Genetic alterations in esophageal cancer. Methods Mol Biol. 2003;222:131–45. doi: 10.1385/1-59259-328-3:131. [DOI] [PubMed] [Google Scholar]
- 3.Rustgi AK. Models of esophageal carcinogenesis. Semin Oncol. 2006;33(Suppl 11):S57–8. doi: 10.1053/j.seminoncol.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 4.Lehrbach DM, Nita ME, Cecconello I. Molecular aspects of esophageal squamous cell carcinoma carcinogenesis. Arq Gastroenterol. 2003;40:256–61. doi: 10.1590/S0004-28032003000400011. [DOI] [PubMed] [Google Scholar]
- 5.Okawa T, Michaylira CZ, Kalabis J, Stairs DB, Nakagawa H, Andl CD, et al. The functional interplay between EGFR overexpression, hTERT activation, and p53 mutation in esophageal epithelial cells with activation of stromal fibroblasts induces tumor development, invasion, and differentiation. Genes Dev. 2007;21:2788–803. doi: 10.1101/gad.1544507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Itakura Y, Sasano H, Shiga C, Furukawa Y, Shiga K, Mori S, et al. Epidermal growth factor receptor overexpression in esophageal carcinoma. An immunohistochemical study correlated with clinicopathologic findings and DNA amplification. Cancer. 1994;74:795–804. doi: 10.1002/1097-0142(19940801)74:3<795::AID-CNCR2820740303>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 7.Parenti AR, Rugge M, Frizzera E, Ruol A, Noventa F, Ancona E, et al. p53 overexpression in the multistep process of esophageal carcinogenesis. Am J Surg Pathol. 1995;19:1418–22. doi: 10.1097/00000478-199512000-00008. [DOI] [PubMed] [Google Scholar]
- 8.Volant A, Nousbaum JB, Giroux MA, Roué-Quintin I, Metges JP, Férec C, et al. p53 protein accumulation in oesophageal squamous cell carcinomas and precancerous lesions. J Clin Pathol. 1995;48:531–4. doi: 10.1136/jcp.48.6.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alsner J, Sørensen SB, Overgaard J. TP53 mutation is related to poor prognosis after radiotherapy, but not surgery, in squamous cell carcinoma of the head and neck. Radiother Oncol. 2001;59:179–85. doi: 10.1016/S0167-8140(01)00301-2. [DOI] [PubMed] [Google Scholar]
- 10.Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med. 2007;357:2552–61. doi: 10.1056/NEJMoa073770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–60. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 12.Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–72. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Comoglio PM, Trusolino L. Invasive growth: from development to metastasis. J Clin Invest. 2002;109:857–62. doi: 10.1172/JCI15392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–16. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
- 15.Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834–48. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- 16.Knudsen BS, Vande Woude G. Showering c-MET-dependent cancers with drugs. Curr Opin Genet Dev. 2008;18:87–96. doi: 10.1016/j.gde.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 17.Joffre C, Barrow R, Ménard L, Calleja V, Hart IR, Kermorgant S. A direct role for Met endocytosis in tumorigenesis. Nat Cell Biol. 2011;13:827–37. doi: 10.1038/ncb2257. [DOI] [PubMed] [Google Scholar]
- 18.Rong S, Donehower LA, Hansen MF, Strong L, Tainsky M, Jeffers M, et al. Met proto-oncogene product is overexpressed in tumors of p53-deficient mice and tumors of Li-Fraumeni patients. Cancer Res. 1995;55:1963–70. [PubMed] [Google Scholar]
- 19.Hwang CI, Matoso A, Corney DC, Flesken-Nikitin A, Körner S, Wang W, et al. Wild-type p53 controls cell motility and invasion by dual regulation of MET expression. Proc Natl Acad Sci U S A. 2011;108:14240–5. doi: 10.1073/pnas.1017536108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grugan KD, Miller CG, Yao Y, Michaylira CZ, Ohashi S, Klein-Szanto AJ, et al. Fibroblast-secreted hepatocyte growth factor plays a functional role in esophageal squamous cell carcinoma invasion. Proc Natl Acad Sci U S A. 2010;107:11026–31. doi: 10.1073/pnas.0914295107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Accornero P, Miretti S, Cucuzza LS, Martignani E, Baratta M. Epidermal growth factor and hepatocyte growth factor cooperate to enhance cell proliferation, scatter, and invasion in murine mammary epithelial cells. J Mol Endocrinol. 2010;44:115–25. doi: 10.1677/JME-09-0035. [DOI] [PubMed] [Google Scholar]
- 22.Karamouzis MV, Konstantinopoulos PA, Papavassiliou AG. Targeting MET as a strategy to overcome crosstalk-related resistance to EGFR inhibitors. Lancet Oncol. 2009;10:709–17. doi: 10.1016/S1470-2045(09)70137-8. [DOI] [PubMed] [Google Scholar]
- 23.Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J Biol Chem. 2000;275:8806–11. doi: 10.1074/jbc.275.12.8806. [DOI] [PubMed] [Google Scholar]
- 24.Martínez-Palacián A, del Castillo G, Herrera B, Fernández M, Roncero C, Fabregat I, et al. EGFR is dispensable for c-Met-mediated proliferation and survival activities in mouse adult liver oval cells. Cell Signal. 2012;24:505–13. doi: 10.1016/j.cellsig.2011.09.031. [DOI] [PubMed] [Google Scholar]
- 25.Michaylira CZ, Wong GS, Miller CG, Gutierrez CM, Nakagawa H, Hammond R, et al. Periostin, a cell adhesion molecule, facilitates invasion in the tumor microenvironment and annotates a novel tumor-invasive signature in esophageal cancer. Cancer Res. 2010;70:5281–92. doi: 10.1158/0008-5472.CAN-10-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andl CD, Mizushima T, Nakagawa H, Oyama K, Harada H, Chruma K, et al. Epidermal growth factor receptor mediates increased cell proliferation, migration, and aggregation in esophageal keratinocytes in vitro and in vivo. J Biol Chem. 2003;278:1824–30. doi: 10.1074/jbc.M209148200. [DOI] [PubMed] [Google Scholar]
- 27.Wang W, Kim SH, El-Deiry WS. Small-molecule modulators of p53 family signaling and antitumor effects in p53-deficient human colon tumor xenografts. Proc Natl Acad Sci U S A. 2006;103:11003–8. doi: 10.1073/pnas.0604507103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lambert JM, Gorzov P, Veprintsev DB, Söderqvist M, Segerbäck D, Bergman J, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009;15:376–88. doi: 10.1016/j.ccr.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 29.Wischhusen J, Naumann U, Ohgaki H, Rastinejad F, Weller M. CP-31398, a novel p53-stabilizing agent, induces p53-dependent and p53-independent glioma cell death. Oncogene. 2003;22:8233–45. doi: 10.1038/sj.onc.1207198. [DOI] [PubMed] [Google Scholar]
- 30.Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139:1327–41. doi: 10.1016/j.cell.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 31.Muller PA, Trinidad AG, Timpson P, Morton JP, Zanivan S, van den Berghe PV, et al. Mutant p53 enhances MET trafficking and signalling to drive cell scattering and invasion. Oncogene. 2013;32:1252–65. doi: 10.1038/onc.2012.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu X, Liu DP, Xu Y. The gain of function of p53 cancer mutant in promoting mammary tumorigenesis. Oncogene. 2012 doi: 10.1038/onc.2012.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Migliore C, Petrelli A, Ghiso E, Corso S, Capparuccia L, Eramo A, et al. MicroRNAs impair MET-mediated invasive growth. Cancer Res. 2008;68:10128–36. doi: 10.1158/0008-5472.CAN-08-2148. [DOI] [PubMed] [Google Scholar]
- 34.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Siemens H, Neumann J, Jackstadt R, Mansmann U, Horst D, Kirchner T, et al. Detection of miR-34a promoter methylation in combination with elevated expression of c-Met and β-catenin predicts distant metastasis of colon cancer. Clin Cancer Res. 2013;19:710–20. doi: 10.1158/1078-0432.CCR-12-1703. [DOI] [PubMed] [Google Scholar]
- 36.Soussi T, Asselain B, Hamroun D, Kato S, Ishioka C, Claustres M, et al. Meta-analysis of the p53 mutation database for mutant p53 biological activity reveals a methodologic bias in mutation detection. Clin Cancer Res. 2006;12:62–9. doi: 10.1158/1078-0432.CCR-05-0413. [DOI] [PubMed] [Google Scholar]
- 37.Harada H, Nakagawa H, Oyama K, Takaoka M, Andl CD, Jacobmeier B, et al. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Mol Cancer Res. 2003;1:729–38. [PubMed] [Google Scholar]
- 38.Takaoka M, Harada H, Deramaudt TB, Oyama K, Andl CD, Johnstone CN, et al. Ha-Ras(G12V) induces senescence in primary and immortalized human esophageal keratinocytes with p53 dysfunction. Oncogene. 2004;23:6760–8. doi: 10.1038/sj.onc.1207923. [DOI] [PubMed] [Google Scholar]
- 39.Kalabis J, Wong GS, Vega ME, Natsuizaka M, Robertson ES, Herlyn M, et al. Isolation and characterization of mouse and human esophageal epithelial cells in 3D organotypic culture. Nat Protoc. 2012;7:235–46. doi: 10.1038/nprot.2011.437. [DOI] [PMC free article] [PubMed] [Google Scholar]