

Abstract
Aim
Drug toxicities, compliance and penetrance into viral reservoirs have diminished the efficacy of long-term antiretroviral therapy (ART) for treatment of HIV infection. Cell-targeted nanoformulated ART was developed to improve disease outcomes. However, rapid noninvasive determination of drug biodistribution is unrealized. To this end, small magnetite ART (SMART) nanoparticles can provide assessments of ART biodistribution by MRI.
Materials & methods
Poly(lactic-co-glycolic acid), 1,2-distearoyl-sn-glycero-3-phosphocholine- and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(methoxy-PEG 2000)-encased particles were synthesized with atazanavir (ATV) and magnetite. Uptake and retention of ATV and magnetite administered at 3:1 ratios (weight/weight) were determined in human monocyte-derived macrophages and mice.
Results
SMART particles were taken up and retained in macrophages. In mice, following parenteral SMART injection, magnetite and drug biodistribution paralleled one another with MRI signal intensity greatest in the liver and spleen at 24 h. Significantly, ATV and magnetite levels correlated.
Conclusion
SMART can permit rapid assessment of drug tissue concentrations in viral reservoirs.
Keywords: HIV, macrophage, monocyte, nanomedicine, nanoparticles, small magnetite antiretroviral therapy
Eradication of HIV-1 in infected human hosts will necessitate antiretroviral drug delivery to viral sanctuaries with the secondary elimination of latent or restricted infections [1]. The former can be facilitated through targeted nanoparticle drug delivery, but to achieve its full potential would require improved virus target tissue drug bioavailability. One major hurdle towards achieving this goal is the measurement of anti-retroviral therapeutics (ART) distribution in viral reservoirs outside of conventional plasma drug levels [2]. We hypothesized that one way to achieve this goal is by synthesizing combinations of ultra-small magnetite particles and ART in a single nanoparticle, then using the particle as a probe for drug tissue distribution measurements. If successful, such small magnetite ART (SMART) could permit rapid pharmacokinetic and pharmacodynamic evaluations of ART in virus target tissues, such as the lymph nodes and brain. In this manner, drug biodistribution would be readily quantitated by a conventional MRI scan. Such an approach would also provide the potential for delivering packaged medicines to sites of limited viral growth and serve, in part, to eliminate residual virus in an infected human host. The approach is realistic as it has previously been used for targeted cancer drug delivery utilizing T2- or T2*-quantification by MRI [3–6].
To perform proof-of-concept studies we placed magnetite, also referred to as superpara-magnetic iron oxide particles, into lipid-coated polylactic-co-glycolic acid (PLGA) nanoparticles with a commonly used antiretroviral protease inhibitor, atazanavir (ATV). By combining PLGA and magnetite, organic/inorganic hybrid composite biomaterials allowed combined diagnostics, or drug distribution assessments, with therapeutic ART delivery (theranostics) through a single MRI scan [7]. SMART nanoparticles were screened for optimal uptake and retention in human monocyte-derived macrophages (MDMs). This facilitated studies investigating the dynamics of in vivo drug tissue distribution. Collectively, the work provides the groundwork for the implementation of SMART systems for noninvasive drug pharmacokinetics for the inevitable goal of viral elimination.
Materials & methods
Material preparation & characterization
PLGA was purchased from Sigma Chemical Co. (MO, USA); 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) and 1,2-distearoyl-sn -glycero-3-phosphoethanolamine-N -(methoxy-PEG 2000) (mPEG-DSPE) were from Corden Pharma International (Plankstadt, Germany); ATV sulfate was from Longshem Co. (Shanghai, China); organic solvents were from Acros Organics (NJ, USA). PLGA, DSPC and mPEG-DSPE encased the SMART particle containing ATV (free base) and magnetite. The magnetite particles were synthesized as follows: 6 mmol tris(acetylacetonato) iron(III) was mixed with 30 mmol 1,2-hexadecanediol, 18 mmol oleic acid, 18 mmol olylamine and 60 ml benzyl ether in a three-neck round-bottomed flask equipped with condenser, magnetic stirrer, thermograph and heating mantle, and stirred under nitrogen. The mixture was slowly heated to 110°C and maintained at that temperature for 1 h, then slowly heated to 200°C. Reflux was maintained after it reached 200°C for 2 h, and the mixture was then slowly heated to 298°C and kept at reflux for another 1.5 h. After cooling to room temperature, a dark homogeneous colloidal suspension was obtained. The suspension was precipitated in ethanol with a magnetic field. The black precipitate was dissolved in hexane with the presence of oleic acid and oleylamine and the solution was centrifuged at 3800 × g for 10 min to remove any undispersed residue. The black solution was reprecipitated in ethanol and centrifuged at 10,000 × g for 30 min. Solid products were obtained by drying the precipitate under vacuum, which generated the final dry particles.
SMART composition & characterization
Preparation of the drug-loaded DSPC/mPEG-DSPE shell and PLGA core particle was as follows. First, a weighed amount of PLGA, ATV and magnetite were dissolved in chloroform (oil phase) with a weight ratio of magnetite to ATV of 1:3. Second, the aqueous phase was prepared by hydration of DSPC and mPEG-DSPE films. The oil phase was added to the DSPC and mPEG-DSPE aqueous solution dropwise, with constant stirring and then sonicated for 60 s followed by a 20 s break on an ice bath. This procedure was repeated for three cycles. Chloroform was then removed by stirring overnight. Third, the particle suspension was centrifuged at 500 × g for 5 min. The supernatant fluids were collected to remove the aggregated nanoparticles. A high-speed 50,000 × g centrifugation for 20 min was used to collect the nanoparticles. After washing twice with phosphate-buffered saline (PBS), the nanoparticles were resuspended. SMART size and size distribution were measured by dynamic light scattering (Malvern Zetasizer Nano Series Nano-ZS, Malvern Instruments, MA, USA) and then diluted in ultrapure water related to mass concentrations and dispersions. Fourth, the surface charge of the SMART particles was determined by ZPlus, a ζ-potential analyzer (Malvern Zetasizer Nano Series Nano-ZS, Malvern Instruments, MA, USA). The pH value and concentration of the particles dispersion were fixed before measurements of ζ-potentials. Finally, the shape and surface morphology of the SMART particles were investigated by transmission electron microscopy (TEM), performed as previously described [8].
SMART particle stability & release of drug in isotonic solution
SMART particles were dispersed in PBS and placed into a 10 kDa dialysis tube in 2 l of PBS under stirring at 37°C. At 30 min and 1, 2, 3, 4, 6, 8 and 10 days, 100 μl of the suspension was collected. The supernatant was dissolved in 900 μl tetrahydrofuran/methanol (volume ratio 1:8) mixture. The amount of ATV and magnetite was measured by HPLC and inductively coupled plasma mass spectrometry (ICP-MS), respectively [8,9].
SMART uptake & retention by MDM
Human monocytes were obtained by leukapheresis, from HIV-1 and hepatitis B seronegative donors, then purified by counter-current centrifugal elutriation [10]. Monocytes were cultured in six-well plates at a density of 1 × 106 cells/ml in Dulbecco’s minimum essential medium containing 10% heat-inactivated pooled human serum, 1% glutamine, 50 μg/ml gentamicin, 10 μg/ml ciprofloxacin and 1000 U/ml recombinant human macrophage colony-stimulating factor [11]. After 7 days of differentiation, MDMs were treated with 100 μM SMART particles, (based upon ATV content). Uptake of SMART particles was assessed without medium change for 8 h. Adherent MDMs were collected by scraping into PBS, at 1, 2, 4 and 8 h after treatment. Cells were pelleted by centrifugation at 1000 × g for 8 min at 4°C. Cell pellets were briefly sonicated in 200 μl of methanol/acetonitrile (1:1) and centrifuged at 20,000 × g for 10 min at 4°C. To determine cell retention of SMART particles, MDMs were exposed to 100 μM SMART particles for 8 h, washed three times with PBS, and fresh media without particles was added. MDMs were cultured for an additional 15 days with half medium exchanges every other day. On days 1, 5, 10 and 15 after SMART treatment, MDMs were collected as described for cell uptake. Cell extracts were stored at −80°C until HPLC and ICP-MS analysis [8].
Prussian blue staining of MDM-retained SMART particles
MDMs were treated with 100 μM SMART particles for 24 h. Adherent MDMs were washed three times with PBS. Cells were fixed with 2% formalin/2.5% glutaraldehyde in PBS for 10min then washed twice with PBS. Fixed macrophages were treated with 5% potassium ferro-cyanide/5% hydrochloric acid (1:1) for 10 min at room temperature. Following solution aspiration the cells were washed twice with PBS. Stained cells were examined by light microscopy.
MRI phantoms & relaxivity measures
MDMs were seeded onto 12-well plates at 1 × 106 cells/ml. After the cells reached 80% confluence, the medium was changed to a medium containing 100 μM SMART particles (based on ATV content). After 24 h the treatment medium was removed and the cells were washed three times with 1 ml PBS. Cells were collected and suspended at different cell concentrations (0–5 × 106 cells/ml) in 1% agar gel. T2-relaxtivity was measured by MRI. Magnetite content in the cells was quantitated by ICP-MS.
SMART biodistribution
Biodistribution of SMART particles was determined in male Balb/cJ mice (Jackson Labs, ME, USA). SMART particles (30 mg/kg ATV) were injected via a jugular vein cannula in a total volume of 100 μl for each mouse. The mice were scanned by MRI 2 h before injection and then continuously at 0.25, 1, 2 and 4 h or at 24 h after SMART administration. Tissues were collected following the final MRI scan. Tissue drug levels were quantitated by ultra-performance liquid chromatography tandem mass spectrometry [12] and magnetite levels were determined by ICP-MS as previously described [9].
MRI acquisition
MRI was acquired using a 7T/16cm Bruker Pharmascan TM MRI/magnetic resonance spectroscopy scanner (Ettlingen, Germany) and a commercial mouse body resonator. SMART detection by MRI was carried out using T2 mapping for quantitation and T2*-weighted high-resolution imaging for detection of biodistribution throughout the body. The sequence used for T2 mapping was a Carr Purcell Meiboom Gill, phase-cycled, multislice, multiecho sequence. Forty one 0.5-mm-thick contiguous interleaved coronal images were acquired with 256 × 192 acquisition matrix and 40 mm field of view. Starting with an echo time (TE) of 10 ms, 12 echoes were acquired with 10 ms echo spacing and a repetition time of 4680 ms with signal averages of 3. The total acquisition time for this sequence was 30 min. T2*-weighted MRI was acquired using a 3D spoiled gradient recalled echo based fast low angle shot (FLASH) sequence with the following parameters: TE/repetition time/pulse angle = 3 ms/10 ms/15°, field of vision = 50 mm × 40 mm × 30 mm, acquisition matrix = 256 × 196 × 128, number of signal averages = 6. The total scan time for this sequence was 25 min.
MRI analyses
T2 maps were reconstructed using custom programs written in interactive data language (Exelis Visual Information Solutions, VA, USA). Preinjection and 24 h postinjection maps were constructed using the even-echo images from the CPMG, phase-cycled imaging data set. Mean tissue T2 was determined using region of interest (ROI) analyses before and after SMART injection for the 24 h results. Magnetite concentration was then determined from the change in tissue relaxivity:
| (Equation 1) |
using the SMART particle relaxivity per milligram magnetite (r2 [s−1 ml mg−1]) determined from the slope of magnetite concentration versus R2 in phantom studies.
Acute (0–4 h) data were acquired with in-magnet jugular vein injection, allowing sequential T2 mapping to be acquired with a T2*-weighted FLASH image acquired at the end of a 4-h period. The natural coregistration of these data allowed development of magnetite concentration maps based on relaxivity changes using custom programs written in interactive data language for the acute scanning session. The ROI analyses were performed using ImageJ software (NIH, MD, USA) [101].
Immunohistochemical identification of SMART uptake by cells
To determine cell localization of SMART, spleen and liver were collected after the final MRI scan and fixed in 10% neutral buffered formalin. Tissues were paraffin embedded and sectioned at 5 μm. To identify macrophages, sections were incubated with the antibody to ionized calcium-binding adaptor molecule 1 (Iba1, Wako Chemicals USA, Inc., VA, USA). The polymer-based HRP-conjugated antimouse and antirabbit EnVision™ (Dako, CA, USA) were used as secondary detection reagents and color developed with 3,3′-diaminobenzidine. All paraffin-embedded sections were stained with Prussian blue to identify magnetite content. Slides were imaged using a Nuance EX camera fixed onto a Nikon Eclipse E800 using Nuance software (Cambridge Research & Instrumentation, MA, USA).
Results
SMART development & in vitro evaluation
The schematic structure of SMART is represented in Figure 1a. This is composed of a hydrophobic PLGA/ATV/magnetite core and an amphiphilic DSPC and mPEG-DSPE lipid shell. DSPC and mPEG-DSPE increased SMART stability and facilitated increased systemic formulation circulation times. Both ATV and magnetite are distributed homogeneously within the core of the particle. SMART was made using a single oil-in-water emulsion with lipid surfactants. After sonication, amphiphilic lipids self-assembled into a monolayer surrounding PLGA/ATV/magnetite-containing oil droplets, achieved through hydrophobic interactions. Evaporation of chloroform under continuous magnetic stirring allowed for the formation of a lipid-coated solid PLGA/ATV/magnetite core. SMART was then purified by ultracentrifugation before further characterization. Our dynamic light scattering results indicated that the average size of the particles was 268 nm with a polydispersity of 0.2. The narrow size distribution is linked to the DSPC, which serves to stabilize the polymeric SMART in the aqueous phase. The ζ-potential of the particles is −45.2 mV, which provides the particles stability when suspended in aqueous media. Although DSPC is neutral when used as a particle coat, it exhibits nonzero mobilities in an external electric field. This may result in a higher negative charge since some anions bind to the neutral lipids making the surface more negatively charged. TEM was employed to obtain the image that best reflects SMART particle morphology (Figure 1B). This illustrated that the particles were spherical in shape with narrow size distributions. A representative particle is shown by TEM, showing the ultra small iron oxide contained within the particle’s core.
Figure 1. Development of small magnetite antiretroviral therapy nanoparticles.
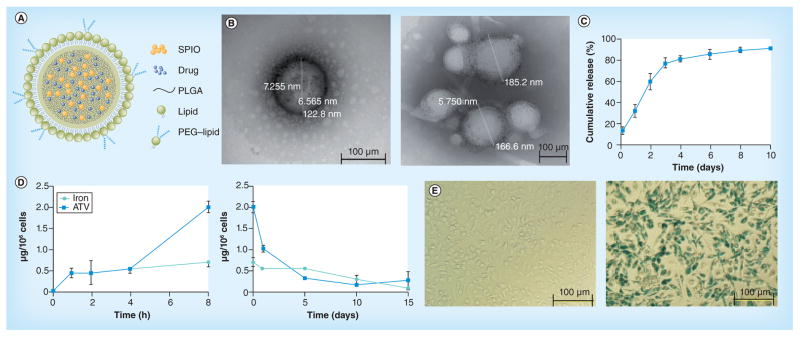
(A) The schematic structure of lipid-coated PLGA small magnetite antiretroviral therapy (SMART). ATV and SPIO are well distributed within the PLGA matrix to form the core of SMART. The PLGA core is coated with lipid monolayer to form the shell of SMART. (B) Representative transmission electron micrograph of a single SMART particle. (C) Cumulative release of ATV in isotonic solution. (D) Time course of uptake (upper panel) and retention (lower panel) of SMART in monocyte-derived macrophages (MDMs). MDMs were treated with 100 μM SMART (based on ATV content) for 1-, 2-, 4- and 8-h uptake studies. For cell retention, MDMs were treated with 100 μM SMART for 8 h, cell culture media was changed and cells were cultured for an additional 15 days. The cell lysates at indicated times were analyzed by HPLC and inductively coupled plasma mass spectrometry for ATV and magnetite quantification, respectively. Data represent the mean ± standard error of the mean (n = 3), for each time point. (E) Prussian blue staining of MDMs. MDMs were treated with phosphate-buffered saline (negative control, upper panel) or 100 μM SMART (lower panel) for 24 h, and then fixed with 2% formalin/2.5% glutaraldehyde and stained with 5% potassium ferrocyanide/5% hydrochloric acid (1:1).
ATV: Atazanavir; SPIO: Superparamagnetic iron oxide; PLGA: Polylactic-co-glycolic acid.
The data contained in Figure 1C demonstrate the stability of SMART, in that ATV is slowly released from the particle. Moreover, there was no significant burst release in the first 4 h and sustained ATV concentrations were seen over several days. The cumulative ATV release reached 80% on day 4 and 94% of ATV was released by day 10. After SMART particle characterizations were completed, the in vitro kinetics of MDM uptake and retention were determined. Our previous studies of nanoART uptake in MDMs showed that >95% of total uptake occurs by 8 h for ATV nanoART [8,13–15]. Up to 2 μg of ATV/106 cells was recorded in MDMs at 8 h with magnetite uptake reflective of particle composition (Figure 1D). The majority of the cells took up the magnetite as observed through Prussian blue staining (Figure 1E). Moreover, such staining demonstrated that magnetite-containing particles were readily incorporated in macrophages by 8 h. The controlled and sustained release profile of ATV facilitates the application of the SMART particles for the delivery of antiretroviral drugs.
Measurement of SMART particle relaxivity
Concentration-dependent relaxivity (r2 [s−1 ml mg−1]) causing increased absolute relaxivity (R2[s−1]) in tissue as a function of concentration (expressed as mg/ml magnetite) of SMART particles was determined using phantoms consisting of both free SMART particles (Figure 2a) and SMART particles taken up by MDMs (Figure 2B). The magnetite concentrations in mg/ml contained by SMART in 1% agar gels were plotted against R2 as measured by MRI. The relationship between R2 and magnetite concentration of SMART in phantoms was linear within the range of the measured magnetite concentrations. The concentration-dependent relaxivity of SMART was found to be r2 = 6200.2 s−1 ml mg−1 in MDM and r2 = 7052.1 s−1 ml mg−1 in PBS. The r2 of SMART in MDMs was used for noninvasive in vivo quantitation of magnetite concentration due to SMART influx using MRI.
Figure 2. Measuring the concentration-dependent relaxivity of small magnetite antiretroviral therapy.
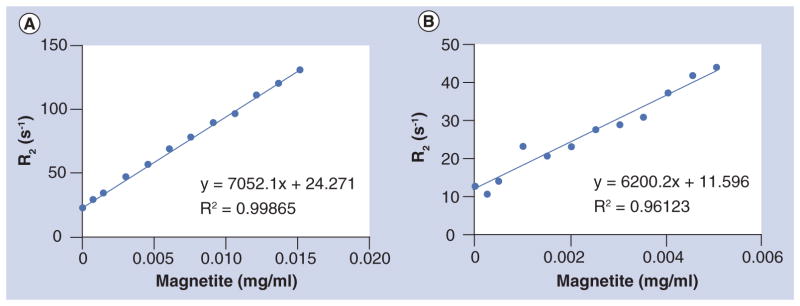
(A) In phosphate-buffered saline and (B) monocyte-derived macrophages (MDMs). MDMs were incubated with 100 μM small magnetite antiretroviral therapy (based on ATV content) for 24 h. Collected MDMs and small magnetite antiretroviral therapy were suspended in 1% agar gel. T2 was measured by MRI and magnetite content by inductively coupled mass spectrometry.
R2: Relaxivity; R2: Linear regression coefficient squared.
Real-time SMART biodistribution & pharmacokinetics
Magnetite labeling allows MRI to be used to quantify the distribution of SMART particles over time in live animals. This can be seen in Figure 3. Figure 3a shows examples of magnetite concentration (from magnetite in SMART) constructed from MRI T2 maps measured before and every 30 min for 4 h continuously after SMART injection. ROI analyses of these data from six animals are shown in Figure 3B. It can be appreciated from the images that a significant amount of the SMART is still within the vasculature, largely leading to the intensity in the kidney, as the kidney shows very little uptake by 24 h. This reflects the measured concentration in the kidney reducing over the first 4 h, while in the liver and spleen, which are organs where SMART accumulates, the mean signal is relatively constant or increases as the particles redistribute from the blood to the tissue. Significant accumulation of SMART was found in liver and spleen at 4 h, as can be appreciated in Figure 4. Figure 4 displays two of the 0.128-mm thick, T2*-weighted, high-resolution, 3D FLASH images of the same mouse before and 4 h after injection of SMART. Presence of magnetite in tissue causes a reduction of T2* to the point of complete signal loss at a TE of 3 ms in the liver, spleen and some abdominal regions. This method is not quantitative; however, it does allow ready identification of the presence of magnetite throughout the body, which can be used to guide quantitative ROI analyses using T2 maps.
Figure 3. MRI assessments of the tissue drug biodistribution and pharmocokinetics by small magnetite antiretroviral therapy particles.

After pre-MRI scan, mice were injected with small magnetite antiretroviral therapy (SMART) through a jugular vein cannula, and then scanned by MRI at continuously at 30 min intervals up to 4 h after SMART administration. Mean tissue SMART content was determined as detailed in the ‘Materials & methods’ section. Immediately after the final scan, mice were euthanized and tissues were collected for atazanavir quantification by ultra-performance liquid chromatography tandem mass spectrometry. (A) MRI-based images of magnetite concentration in kidney, spleen and liver from 0.5 to 4 h following SMART administration. (B) Mean ± scanning electron microscope (n = 6) of magnetite levels in kidney, spleen and liver over 4 h following SMART administration.
Figure 4. 3D gradient recalled echo images of the same mouse before and 4 h after injection of small magnetite antiretroviral therapy.
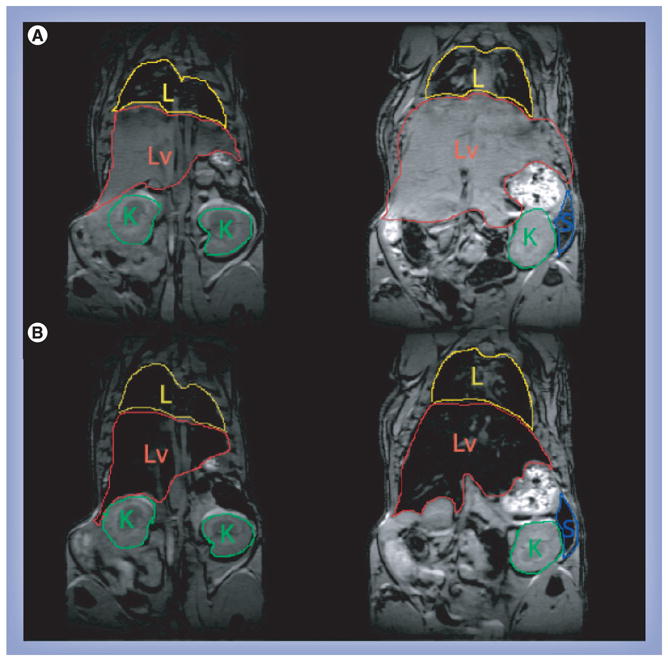
(A) Images before injection and (B) 4 h after injection. The signal from the liver is completely eliminated due to the accumulation of magnetite loaded small magnetite antiretroviral therapy.
K: Kidney; L: Lung; Lv: Liver; S: Spleen.
Correlation between magnetite & drug tissue content
Figure 5 shows the relationship between magnetite concentration and ATV concentration in the liver, spleen and kidney in four animals 24 h after injection. It can be appreciated that there is a significant positive correlation (Pearson correlation: r = 0.786; p = 0.0008). These results demonstrate the capability of MRI to be used for monitoring nanoART distribution.
Figure 5. Correlation of small magnetite antiretroviral therapy particle-associated magnetite and atazanavir in tissues 24 h after administration.<.

br>The magnetite concentration was quantified from the change in T2 weighted relaxivity (Equation 1) and the per milligram magnetite relaxivity determined as the slope of magnetite concentration versus R2 in small magnetite antiretroviral therapy particles phantom studies. ATV concentrations were quantified by ultra-performance liquid chromatography tandem mass spectrometry following the final 24 h MRI scan.
ATV: Atazanavir; R2: Linear regression coefficient squared.
Identification of magnetite–ART relationships in systemic tissues
We reasoned that cellular biodistribution of SMART was concordant with our prior results with nanoART [16]. To prove this theory, we next studied the relationships between SMART particle biodistribution and macrophages in mice following parenteral SMART injections. Animals were sacrificed 4 h after injection and tissues collected. Dual Iba-1 (for macrophages) and Prussian blue staining (for magnetite) were performed and evaluated by brightfield microscopic imaging. Prussian blue staining was almost exclusively in tissue cells identified as macrophages. As shown in Figure 6, Iba-1+ macrophages were readily seen in both the liver and spleen in parallel to the distribution of Prussian blue. The dual staining pictures showed that the SMART particles were retained in tissue macrophages. Erythrocyte hemoglobin and its breakdown products are a substantial source of iron. These are degraded after the lifespan of the erythrocyte [17]. This occurs predominantly in the spleen with iron transferred back into the blood. The highly vascularized spleen contains easily seen red blood cell breakdown products, including iron, that are phagocytized by macrophages [18]. Thus, the total iron content in the spleen is readily visualized by Prussian blue staining. Iron content is substantially increased after SMART treatments (Figure 6g & H). The iron levels in the spleen also changed from 228 μg/g before treatment to 356 μg/g after treatment, measured by ICP-MS. Endogenous iron levels from circulating red blood cells also explains the dark backgrounds seen on MRI scans in the spleen that are darker after SMART particle injections.
Figure 6. Immunohistology of Iba-1 staining and Prussian blue staining in liver and spleen.
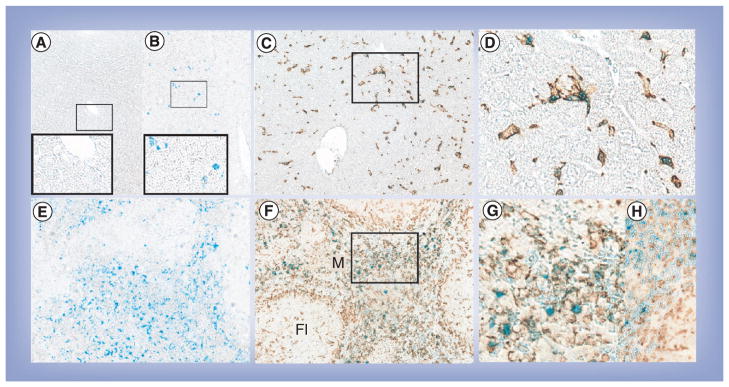
(A) Liver from control mice with Prussian blue (magnification: 200×; inset 400×). (B) Liver from small magnetite antiretroviral therapy (SMART)-treated mice with Prussian blue (magnification: 200×; inset 400×). (C) Liver from SMART-treated mice with Prussian blue and Iba-1 (magnification: 200×). (D) Enlargement from indicated section in (C). (E) Spleen from SMART-treated mice with Prussian blue (magnification: 200×). (F) Spleen from SMART-treated mice with Prussian blue and Iba-1 (magnification: 200×). (G) Enlargement from indicated section in (F). (H) Spleen from control mice with Prussian blue and Iba-1 (magnification: 400×). Livers and spleens were fixed with 10% formalin, paraffin embedded and sectioned for immunohistological analysis after the final MRI scan. Macrophages were identified by Iba1 stains (brown) and magnetite identified by Prussian blue.
Fl: Lymphoid follicle; M: Marginal zone.
Discussion
Our laboratories are known for the development of cell-based carriage and delivery of antiretroviral drugs to sites of active HIV-1 replication [8,10,13,15,19–21]. This platform can facilitate slow release of drug for time periods measured in weeks or perhaps months. This system, coined the ‘Trojan horse’ macrophage drug delivery scheme, takes full advantage of the macrophages’ substantive endosomal storage capacity, its phagocytic and secretory functions, and its high degree of mobility to facilitate drug delivery [22]. As the monocyte macrophage is a cell target for viral growth, the added benefit rests in the abilities to bring ART to subcellular sites of viral assembly [23]. Such a system, when used as a parenteral injection scheme, has previously been shown to hold significant gains over conventional native oral drug therapeutic regimens [19,21].
The principal goal in developing this polymer system rests in the ability to utilize MRI scans to rapidly assess cell and tissue drug biodistribution. The idea is that the polymer-encased dual magnetite and drug particle would permit a clear determination of drug levels in virus target tissues in a very short time interval (hours). As plasma drug levels remain the gold standard for pharmacokinetic testing, this technology clearly opens up new opportunities to develop platforms that would accelerate the elimination of, or some day the cure, viral infections. Notably, there is a considerable focus among HIV/AIDS researchers towards the development of reliable methods to bring drugs to reservoir sites with the explicit goal of eliminating the virus. Targeted drug delivery, as well as gene delivery, when combined with suitable imaging techniques could facilitate this goal by providing a ‘go/no go’ for treatment success [8]. Although this is the first time such ‘theranostics’ have been applied for HIV diagnosis and therapies, similar systems have been developed in recent years for cancer treatments [24]. Here, the application is for early diagnostics. The unique payloads of nanomaterials include fluorescent semiconductor nanocrystals (quantum dots), as well as magnetic nanoparticles, as developed in this report. All provide properties that can facilitate in vivo imaging with the help of MRI tests, as well as fluorescence-based approaches [25]. Overall, interest in this idea will likely continue to grow through the development of carrier nanoparticles designed to target specific tissue and effect local chemo-, radio- and gene-directed antiretroviral or immune modulatory therapies [26].
Liposomes and polymer nanoparticles are the two major types of nanoparticles that have been developed and evaluated for diagnostic and therapeutic purposes. Liposomes composed of natural lipids are attractive drug delivery systems because of their high biocompatibility, low immunogenicity, long systemic circulation and favorable pharmacokinetic profile. Specific targeted delivery can easily be achieved by conjugating a targeting ligand to the lipid molecule [27–29]. Liposomal formulations including Doxil® and DaunoXome® are approved by the US FDA for clinical applications [27,29,30]. However, the possible intrinsic low drug-loading capacity, fast release profiles of hydrophobic drugs and physical instability of liposomes limit their clinical applications of different drugs [31]. Polymeric nanoparticles composed of synthetic PLGA are another widely developed and studied drug delivery platform because of their high stability, relatively high drug-loading capacity for all types of drugs, biodegradability, low toxicity and controlled/sustained drug release profiles. Depending on particle composition, the drug release profiles of PLGA nanoparticles can be modulated to be within days, weeks or even months [32–34]. However, the biocompatibility/immunogenicity of nanoparticles composed of synthetic polymers, including PLGA, is not as high as liposomes. Without further chemical modification, PLGA nanoparticles are rapidly removed from the circulation by the mononuclear phagocyte system, resulting in short systemic circulation [31]. In summary, both liposomes and PLGA nanoparticles are not independently structurally robust platforms. Thus, lipid-coated polymer nano_particles, formed by combining synthetic polymers and natural lipids, have been developed as robust drug delivery platforms to combine the advantages and avoid the disadvantages of liposomes and polymer nanoparticles [35,36].
The visualization of cellular function in living organisms is not a new modality [10,37,38]. Optical x-ray, nuclear magnetic resonance, MRI and ultrasound allows 3D whole-body scans at high spatial resolution and are adept at morphological and functional evaluations. The data obtained can be enhanced by magnetite and improved image resolution. By immobilizing a specific target molecule on the surface of a magnetic particle, the molecule inherits its magnetic property. Magnetic tissue targeting using multifunctional carrier particles, such as lipid-coated polymer nanoparticles, can also facilitate effective treatments by enabling site-directed therapeutic outcomes. To this end, we selected DSPC and mPEG-DSPE as the shell and PLGA as the core of SMART system. DSPC is used to increase the biocompatibility of SMART, and mPEG-DSPE is used to build a sterically repulsive shield around SMART that reduces opsonization, prevents interactions with the mononuclear phagocyte system, allows the particles to escape renal exclusion, and increases systemic circulation. To the best of our knowledge, this is the first attempt to develop lipid-coated PLGA nanoparticles for HIV therapeutics. Our current work sought to use lipid-coated PLGA SMART to encase magnetite and ART to facilitate MDM uptake of the drug and its subsequent slow release. The long-term goals are straightforward. One is to use the synthesized SMART to facilitate drug screening for specifically targeting ligands or sugars. The second is to determine the distribution of nanoART in viral reservoirs for the ultimate eradication of HIV. In this proof-of-concept study, the jugular vein was used for the intravenous injections of SMART and ensures uniform drug and magnetite distribution, minimizes standard deviation within groups and provides rapid particle distribution. Our in vivo MRI results clearly demonstrated that the concentrations of magnetite and ATV in mouse tissues, such as liver, spleen and kidney, correlate with one another at 24 h after injection. The results support the idea that SMART technology facilitates the noninvasive evaluation of drug pharmacokinetics and biodistribution in virus target tissues by MRI. Our immunohistochemical analysis further demonstrates that SMART particles are taken up by liver and spleen macrophages. These cells serve as the reservoir of SMART particles, as well as the major targets of HIV. The advantages of using SMART rests as a cassette system for testing nanoparticles that direct nanoformulated ART to tissue viral reservoirs.
Conventional ART and our prior works with nontargeted formulations demonstrate limited entry of ART into lymph nodes and brain virus reservoirs [21]. To reduce residual HIV from these reservoirs, efforts have been made to specifically deliver ART into lymph nodes, gut and nervous system tissues. As HIV mainly infects CD4+ T cells and macrophages, various ligands targeted to these cells have been selected for the development of targeted drug delivery systems [39]. Recent works from our laboratories showed that folic acid-conjugated particles can significantly increase ART distribution to lymph nodes and HIV-infected brains, but not in the healthy brains [40]. Future work will focus on the development and evaluation of HIV reservoir-targeted nanoformulations. For this purpose, targeted SMART will firstly be developed for short-term noninvasive drug pharmacokinetics and biodistribution evaluation, and ligand screening. Our results in this report imply that SMART technology for MRI has the potential to provide rapid assessments and screening of targeted nanoformulations for the development of next-generation cell and tissue ligand-decorated particles. In summary, this initial study lays the groundwork for what will quickly follow and ultimately, we believe, lead to an effective means to combat and potentially eliminate HIV disease.
Conclusion
SMART is defined as lipid-coated PLGA nanoparticles that encase magnetite and ART. These particles permit rapid MRI determinations of antiretroviral drug levels in virus target tissues. SMART is taken up by MDMs and distributed to tissues. We propose that SMART can facilitate noninvasive evaluation of drug pharmacokinetics and biodistribution. Importantly, this advance can facilitate the testing of ligand-directed drug delivery systems designed to improve clinical outcomes for HIV/AIDS.
Future perspective
SMART facilitates the noninvasive evaluation of drug tissue biodistribution. It is designed to assess ART penetrance into viral reservoirs and provide rapid assessments of the next-generation cell and tissue ligand decorated particles. This would facilitate the development of improved nanoART designed to improve clinical outcomes and, ultimately, affect the eradication of HIV infection in its human host. This can be achieved in the future using SMART as a probe for drug screening by testing specific targeting ligands for drug biodistribution with the long-term goal of using theranostic techniques with the aim of eliminating reservoirs of residual viral infection.
Executive summary.
Small magnetite antiretroviral therapy nanoparticle development
Small magnetite antiretroviral therapy (SMART) nanoparticles were developed as probes for antiretroviral drug biodistribution.
1,2-distearoyl-sn-glycero-3-phospho-choline and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(methoxy-PEG 2000) increase biocompatibility of SMART particles.
Polylactic-co-glycolic acid SMART encases magnetite and antiretroviral therapy, facilitating macrophage particle uptake and the subsequent slow release of the drug.
MRI assessment, drug pharmacokinetics & biodistribution
In mice, magnetite and drug biodistribution parallel one another, as readily seen after parenteral injections.
MRI T2 maps and 3D spoiled gradient recalled echo image sets can assess drug tissue distribution in the spleen, liver, kidney and lungs.
Drug tissue levels correlate with changes in tissue relaxivity.
In vivo MRI results demonstrate that SMART particles can facilitate evaluations of drug pharmacokinetics and biodistribution, and provide rapid assessments for the next-generation cell and tissue ligand-decorated particles.
SMART technology utilizes MRI scans to rapidly assess tissue drug biodistribution and shows the potential of improving clinical outcomes for HIV/AIDS.
Acknowledgments
The authors would like to thank Han Chen and You Zhou of the University of Nebraska-Lincoln (NE, USA) electron microscopy core facility for supplying the transmission electron microscopy images. The authors would also like to thank Larisa Poluektova for advice on immunohistochemical staining, and Daphne Baber and Nathan Smith for their expert technical assistance.
Footnotes
For reprint orders, please contact: reprints@futuremedicine.com
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
Financial & competing interests disclosure
The work was supported by the NIH grants 1P01 DA028555, 2R01 NS034239, 2R37 NS36126, P01 NS31492, P20RR 15635, P01MH64570 and P01 NS43985 (to HE Gendelman) and support of the Nebraska Research Initiative (to MD Boska). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1.Wainberg MA. AIDS: drugs that prevent HIV infection. Nature. 2011;469(7330):306–307. doi: 10.1038/469306a. [DOI] [PubMed] [Google Scholar]
- 2.Pretorius E, Klinker H, Rosenkranz B. The role of therapeutic drug monitoring in the management of patients with human immunodeficiency virus infection. Ther Drug Monit. 2011;33(3):265–274. doi: 10.1097/FTD.0b013e31821b42d1. [DOI] [PubMed] [Google Scholar]
- 3.Guthi JS, Yang SG, Huang G, et al. MRI-visible micellar nanomedicine for targeted drug delivery to lung cancer cells. Mol Pharm. 2010;7(1):32–40. doi: 10.1021/mp9001393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lebel RM, Menon RS, Bowen CV. Relaxometry model of strong dipolar perturbers for balanced-SSFP: application to quantification of SPIO loaded cells. Magn Reson Med. 2006;55(3):583–591. doi: 10.1002/mrm.20799. [DOI] [PubMed] [Google Scholar]
- 5.Liu W, Dahnke H, Rahmer J, Jordan EK, Frank JA. Ultrashort T2* relaxometry for quantitation of highly concentrated superparamagnetic iron oxide (SPIO) nanoparticle labeled cells. Magn Reson Med. 2009;61(4):761–766. doi: 10.1002/mrm.21923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Girard OM, Ramirez R, Mccarty S, Mattrey RF. Toward absolute quantification of iron oxide nanoparticles as well as cell internalized fraction using multiparametric MRI. Contrast Media Mol Imaging. 2012;7(4):411–417. doi: 10.1002/cmmi.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7▪▪.Kabanov AV, Gendelman HE. Nanomedicine in the diagnosis and therapy of neurodegenerative disorders. Prog Polym Sci. 2007;32(8–9):1054–1082. doi: 10.1016/j.progpolymsci.2007.05.014. Demonstrated the idea of theranostics by combining polylactic-co-glycolic (PLGA) and magnetite in one formulation. One single MRI scan could be used to assess drug biodistribution and pharmacokinetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8▪.Nowacek AS, Balkundi S, McMillan J, et al. Analyses of nanoformulated antiretroviral drug charge, size, shape and content for uptake, drug release and antiviral activities in human monocyte-derived macrophages. J Control Release. 2011;150(2):204–211. doi: 10.1016/j.jconrel.2010.11.019. In vitro analysis demonstrated that nanoART is rapidly taken up in macrophages and then slowly released serving as a potential depot for long-acting drug formulations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mascheri N, Dharmakumar R, Zhang Z, Paunesku T, Woloschak G, Li D. Fast low-angle positive contrast steady-state free precession imaging of USPIO-labeled macrophages: theory and in vitro experiment. Magn Reson Imaging. 2009;27(7):961–969. doi: 10.1016/j.mri.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beduneau A, Ma Z, Grotepas CB, et al. Facilitated monocyte-macrophage uptake and tissue distribution of superparmagnetic iron-oxide nanoparticles. PLoS One. 2009;4(2):4343. doi: 10.1371/journal.pone.0004343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gendelman HE, Orenstein JM, Martin MA, et al. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J Exp Med. 1988;167(4):1428–1441. doi: 10.1084/jem.167.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang J, Gautam N, Bathena SP, et al. UPLC-MS/MS quantification of nanoformulated ritonavir, indinavir, atazanavir, and efavirenz in mouse serum and tissues. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879(23):2332–2338. doi: 10.1016/j.jchromb.2011.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balkundi S, Nowacek AS, Veerubhotla RS, et al. Comparative manufacture and cell-based delivery of antiretroviral nanoformulations. Int J Nanomedicine. 2011;6:3393–3404. doi: 10.2147/IJN.S27830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nowacek AS, McMillan J, Miller R, Anderson A, Rabinow B, Gendelman HE. Nanoformulated antiretroviral drug combinations extend drug release and antiretroviral responses in HIV-1-infected macrophages: implications for neuroAIDS therapeutics. J Neuroimmune Pharmacol. 2010;5(4):592–601. doi: 10.1007/s11481-010-9198-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15▪▪.Nowacek AS, Miller RL, McMillan J, et al. NanoART synthesis, characterization, uptake, release and toxicology for human monocyte-macrophage drug delivery. Nanomedicine (Lond) 2009;4(8):903–917. doi: 10.2217/nnm.09.71. Demonstrated that nanoART is taken up by blood-borne macrophages at a level of >95% total drug content by 8 h for the antiretroviral drug atazanavir. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roy U, McMillan J, Alnouti Y, et al. Pharmacodynamic and antiretroviral activities of combination nanoformulated antiretrovirals in HIV-1-infected human peripheral blood lymphocyte-reconstituted mice. J Infect Dis. 2012;206(10):1577–1588. doi: 10.1093/infdis/jis395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ganz T, Nemeth E. Iron metabolism: interactions with normal and disordered erythropoiesis. Cold Spring Harb Perspect Med. 2012;2(5):a011668. doi: 10.1101/cshperspect.a011668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andrews NC. Forging a field: the golden age of iron biology. Blood. 2008;112(2):219–230. doi: 10.1182/blood-2007-12-077388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dash PK, Gendelman HE, Roy U, et al. Long-acting nanoART elicits potent antiretroviral and neuroprotective responses in HIV-1-infected humanized mice. AIDS. 2012;26(17):2135–2144. doi: 10.1097/QAD.0b013e328357f5ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dou H, Grotepas CB, McMillan JM, et al. Macrophage delivery of nanoformulated antiretroviral drug to the brain in a murine model of neuroAIDS. J Immunol. 2009;183(1):661–669. doi: 10.4049/jimmunol.0900274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roy U, McMillan J, Alnouti Y, et al. Pharmacodynamic and antiretroviral activities of combination nanoformulated antiretrovirals in HIV-1-infected human peripheral blood lymphocyte-reconstituted mice. J Infect Dis. 2012;206(10):1577–1588. doi: 10.1093/infdis/jis395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kadiu I, Nowacek A, McMillan J, Gendelman HE. Macrophage endocytic trafficking of antiretroviral nanoparticles. Nanomedicine (Lond ) 2011;6(6):975–994. doi: 10.2217/nnm.11.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gendelman HE, Gelbard H, Swindells S. The Neurological Manifestations Of HIV-1 Infection. Lippincott-Raven Publishers; PA, USA: 2003. [Google Scholar]
- 24.Choi KY, Liu G, Lee S, Chen X. Theranostic nanoplatforms for simultaneous cancer imaging and therapy: current approaches and future perspectives. Nanoscale. 2012;4(2):330–342. doi: 10.1039/c1nr11277e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gorantla S, Dou H, Boska M, et al. Quantitative magnetic resonance and SPECT imaging for macrophage tissue migration and nanoformulated drug delivery. J Leukoc Biol. 2006;80(5):1165–1174. doi: 10.1189/jlb.0206110. [DOI] [PubMed] [Google Scholar]
- 26.Kibuule D, Dou H, Uberti M, et al. Magnetic labeled macrophages migrate across the blood brain barrier in mice with HIV-1 encephalitis. Presented at. The 12th Annual Meeting of the Society on NeuroImmune Pharmacology; Santa Fe, New Mexico, USA. 6 April 2006; p. Abstract TP-17. [Google Scholar]
- 27.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 28.Lasic DD. Doxorubicin in sterically stabilized liposomes. Nature. 1996;380(6574):561–562. doi: 10.1038/380561a0. [DOI] [PubMed] [Google Scholar]
- 29.Barenholz Y. Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 30.Petre CE, Dittmer DP. Liposomal daunorubicin as treatment for Kaposi’s sarcoma. Int J Nanomedicine. 2007;2(3):277–288. [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Pan J, Feng SS. Nanoparticles of lipid monolayer shell and biodegradable polymer core for controlled release of paclitaxel: effects of surfactants on particles size, characteristics and in vitro performance. Int J Pharm. 2010;395(1–2):243–250. doi: 10.1016/j.ijpharm.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 32▪.Avgoustakis K. Pegylated poly(lactide) and poly(lactide-co-glycolide) nanoparticles: preparation, properties and possible applications in drug delivery. Curr Drug Deliv. 2004;1(4):321–333. doi: 10.2174/1567201043334605. Demonstrated the drug release profiles of PLGA nanoparticles can be modulated for days to weeks. Supported the idea that PLGA nanoparticles can be used to synthesize long-acting nanoformulations. [DOI] [PubMed] [Google Scholar]
- 33.Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55(3):329–347. doi: 10.1016/s0169-409x(02)00228-4. [DOI] [PubMed] [Google Scholar]
- 34.Cho K, Wang X, Nie S, Chen ZG, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14(5):1310–1316. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- 35.Chan JM, Zhang L, Yuet KP, et al. PLGA-lecithin-PEG core-shell nanoparticles for controlled drug delivery. Biomaterials. 2009;30(8):1627–1634. doi: 10.1016/j.biomaterials.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 36.Li B, Xu H, Li Z, et al. Bypassing multidrug resistance in human breast cancer cells with lipid/polymer particle assemblies. Int J Nanomedicine. 2012;7:187–197. doi: 10.2147/IJN.S27864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wessels E, Simpson JC. Impact of live cell imaging on coated vesicle research. Semin Cell Dev Biol. 2007;18(4):412–423. doi: 10.1016/j.semcdb.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 38▪.Kingsley J, Dou H, Morehead J, Rabinow B, Gendelman H, Destache C. Nanotechnology: a focus on nanoparticles as a drug delivery system. J Neuroimmune Pharmacol. 2006;1(3):340–350. doi: 10.1007/s11481-006-9032-4. Review of cell-based nanoformulated drug delivery. Specific discussions are made in areas of drug formulation, distribution and bioactivity. Discusses bioimaging that allows 3D whole-body MRI at high spatial resolution. [DOI] [PubMed] [Google Scholar]
- 39.Bareford LM, Swaan PW. Endocytic mechanisms for targeted drug delivery. Adv Drug Deliv Rev. 2007;59(8):748–758. doi: 10.1016/j.addr.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kanmogne G, Singh S, Roy U, et al. Mononuclear phagocyte intercellular crosstalk facilitates transmission of cell-targeted nanoformulated antiretroviral drugs to human brain endothelial cells. Int J Nanomedicine. 2012;7:2373–2378. doi: 10.2147/IJN.S29454. [DOI] [PMC free article] [PubMed] [Google Scholar]
Website
- 101.ImageJ. Image processing and analysis in Java. http://imagej.nih.gov/ij.