

Abstract
Employing a genetically modified yeast strain as a screening tool, 4-dimethylaminobenzoic acid (5) was isolated from the marine sediment-derived Streptomyces sp. CP27-53 as a weak yeast sirtuin (Sir2p) inhibitor. Using this compound as a scaffold, a series of disubstituted benzene derivatives were evaluated to elucidate the structure activity relationships for Sir2p inhibition. The results suggested that 4-alkyl or 4-alkylaminobenzoic acid is the key structure motif for Sir2p inhibitory activity. The most potent Sir2p inhibitor, 4-tert-butylbenzoic acid (20), among the tested compounds in this study turned out to be a weak but selective SIRT1 inhibitor. The calculated binding free energies between the selected compounds and the catalytic domain of SIRT1 were well correlated to their measured SIRT1 inhibitory activities.
Keywords: Sirtuin, Class III HDAC inhibitor, marine-derived Streptomyces sp, 4-dimethylaminobenzoic acid, 4-tert-butylbenzoic acid
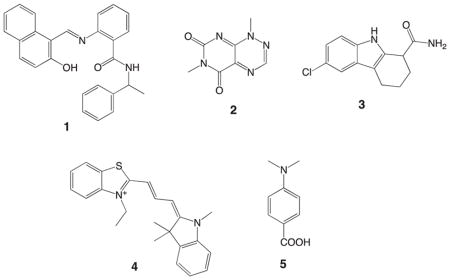
Sirtuins are a group of NAD+-dependent histone deacetylases (HDACs) that are evolutionally conserved from bacteria to mammals.1 This group of enzymes regulates the key biological functions including gene silencing and cell cycle by removing acetyl groups from lysine residues in the histones or non-histone substrates. To date, seven human isoforms (SIRT1-7) have been found and categorized as class III HDACs that are different from classical zinc-dependent class I/II/IV HDACs.2 Recently, SIRT1 and SIRT2 have been considered as molecular targets for cancer chemotherapy. The physiological functions of SIRT1 with regard to tumorigenesis include the negative regulation of the tumor suppressor gene P53,3 the positive regulation of the oncoprotein B-cell lymphoma 6 protein (BCL6),4 and induction of the FOXO-1-dependent vascular growth factor-C (VEGF-C).5 On the other hand, SIRT2 promotes cancer cell proliferation due to the enhanced stability of the Myc oncoproteins.6 It has been reported that apoptosis caused by p53 accumulation in HeLa cells7 and granulocytic differentiation in the acute promyelocytic leukemia (APL)8 can be induced based on down-regulation and inhibition of SIRT2. Though SIRT1 and SIRT2 are also reported to show tumor-suppressing effects9–11 and to be down-regulated in specific cancer types,12 the tumor promoting effects listed above strongly suggest SIRT1/2 inhibitors have great potential to be anticancer drugs. Encouraging evidence includes significant cytotoxic properties of the two SIRT1/2 inhibitors, sirtinol (1: synthetic13) and toxoflavin (2: natural product14), against the MCF-7 and A549 cancer cell lines, respectively.15,16 It is interesting that the potent and selective SIRT1 inhibitor, EX-527 (3: synthetic17), required at least 100 μM to effectively repress MCF-7 cell proliferation.18 However, the selective SIRT2 inhibitor, AC-93253 (4: synthetic), exhibited potent cytotoxic effects (IC50 10–100 nM) against the prostate (DU-145), lung (A549, NCI-H460) and pancreas (MiaPaCa2) cancer cell lines with a great therapeutic window (up to 200-fold).19
We have recently reported the HDAC screening method using the genetically modified yeast strain DMY2843 to identify SIRT1/2 inhibitors from marine-sediment derived actinomycetes.20 The primary screening for the extract library using the yeast assay selected a total of 19 actinomycete strains that would produce yeast sirtuin (Sir2p) inhibitors. A new polyketide tetramic acid derivative designated as streptosetin A with weak Sir2p and SIRT1/2 inhibitory activities has been isolated from one of the active strains, Streptomyces sp. CP13-10.20 Our continued search for sirtuin inhibitors from the remaining active strains led to the identification of 4-dimethylaminobenzoic acid (5) produced by the Streptomyces sp. CP27-53 strain as a Sir2p inhibitor. In this article, we report the identification of 5 and structure activity relationships (SARs) and SIRT inhibitory activities of benzoic acid derivatives and related analogues of 5.
The marine sediment-derived Streptomyces sp. CP27-53 was cultured in a liquid medium (15 L) containing soluble starch (1%), yeast extract (0.4%), peptone (0.2%), CaCO3 (0.1%) and FeSO4·7H2O (40 mg) in artificial seawater adjusted to pH 7.4 for 10 days at 30 C at 200 rpm. The culture was separated to broth and pellet by centrifugation. The broth was treated with HP20 to absorb organic compounds, which were eluted with MeOH, whereas the pellet was extracted with MeOH three times. The combined MeOH extract was cleaned by liquid-liquid partition between EtOAc and H2O to give an organic extract. Yeast screening of the HPLC peak library created from the organic extract revealed that the compound eluting at 14.3 min in the HPLC chromatogram was responsible for the Sir2p inhibitory activity (Figure S1). Furthermore, this active compound was purified by reversed-phase HPLC and identified as 4-dimethylaminobenzoic acid (5) based on the spectroscopic data (see supporting information). The structure was further confirmed by direct comparison of the 1H and 13C NMR data with those of an authentic sample. This compound showed Sir2p inhibitory activity with an MIC of 200 μM after 48 h against the yeast strain.
To elucidate the SARs for Sir2p inhibition by 5, a series of substituted benzoic acid derivatives and related analogues of 5 were evaluated using the yeast strain DMY2843 (Table 1 and Figure 1). All the compounds in Group A (6–9) were inactive against the yeast strain, which suggested that the two functional groups, dimethylamino group and carboxylic acid, must be on para-positions to show Sir2p inhibitory activity. The MIC values of 400 μM and 800 μM for compounds 10 and 11 in Group B, respectively, indicated that more methyl groups on the nitrogen enhanced the Sir2p activities. Next, the activity data for Group C including the weaker activity for compound 12 with methyl ester than that of compound 5 and the lack of activity for compounds 13–16 indicated that the carboxylic acid appeared to be required for inhibition activity. It is interesting that sulfonic acid 14 was inactive despite the similarity between the functional groups. In Group D, compound 17, in which the dimethylamino group was replaced by a methyl group, showed diminished activity whereas compound 18, in which the dimethylamino group was replaced by a similarly sized and shaped isopropyl group, showed similar inhibitory activity to that of compound 5. The results indicated that the size of the substituent is very important for enhancement of inhibitory activity. Therefore, we evaluated the two derivatives in Group E, compounds 19 and 20 with trimethylammonium and tert-butyl groups as respective substituents. Compound 20 showed the best activity (MIC 50 μM) among the derivatives tested, which supported the importance of the size of the substituent as suggested above. The lack of activity seen in compound 19 suggested that bulky but not charged groups are required for enhanced inhibitory activity for benzoic acid derivatives.
Table 1.
Minimum Inhibition Concentration (MIC) Values of Disubstituted Benzene Derivatives Based on Sir2p Inhibition Against the Yeast Strain DMY2844
| Structural group | R1 | R2 | Location | MIC (μM) | |
|---|---|---|---|---|---|
| A | 5 | (CH3)2N− | −COOH | p | 200 |
| 6 | (CH3)2N− | −COOH | m | NAa | |
| 7 | (CH3)2N− | −COOH | o | NA | |
| 8 | (CH3)2N− | none | NA | ||
| 9 | none | −COOH | NA | ||
|
| |||||
| B | 10 | CH3NH− | −COOH | p | 400 |
| 11 | NH2− | −COOH | p | 800 | |
|
| |||||
| C | 12 | (CH3)2N− | −COOCH3 | p | 400 |
| 13 | (CH3)2N− | −CONH2 | p | NA | |
| 14 | (CH3)2N− | −SO3H | p | NA | |
| 15 | (CH3)2N− | −Br | p | NA | |
| 16 | (CH3)2N− | NO2 | p | NA | |
|
| |||||
| D | 17 | CH3− | −COOH | p | 800 |
| 18 | (CH3)2CH− | −COOH | p | 200 | |
|
| |||||
| E | 19 | (CH3)3N+− | −COO- | p | NA |
| 20 | (CH3)3C− | −COOH | p | 50 | |
Not active at 800 μM
Figure 1.

Yeast HDAC screening results (48 h) for 5, 19 and 20. (A) YPD media with 5-fluorouracil (5-FOA) and (B) YPD media. Sir2p inhibition was defined as selective activity when the tested compound resulted in the death of yeast cells (clear) only in the presence of 5-FOA. Cytotoxicity was observed as the death of yeast cells in both media A and B.
In light of their Sir2p inhibitory activities, compounds 5 and 20 were further evaluated for SIRTs inhibitory activities. Compounds 5 inhibited SIRT1 and SIRT2 with 25.3% and 30.3% at 1.6 mM whereas compound 20 inhibited SIRT1 and SIRT2 with 54.8% and 28.0% at 1.6 mM, respectively. These results suggested that compound 20 was a weak but selective SIRT1 inhibitor (IC50 1.0 mM). Furthermore, the SIRT1 inhibitory activity was enhanced twofold by replacing the dimethylamino group with tert-butyl group, which supported the SAR pattern described above.
The crystal structure of the SIRT1 catalytic domain with NAD+ and an EX-527 analogue [(S)-2-chloro-5,6,7,8,9,10-hexahydrocyclohepta[b]indole-6-carboxamide (21)] has recently been reported.21 Based on the SIRT1/NAD+/21 coordinates, we constructed SIRT1/NAD+/5 and SIRT1/NAD+/20 complexes to evaluate binding free energies (ΔGbind) of compounds 5 and 20 relative to compound 21. The ΔGbind energies were calculated by using the MM-GBSA approach for post processing of explicit solvent molecular dynamics (MD) trajectories. This approach has been proven to provide absolute free energies in good agreement with experimental data.22–28 The potent and selective SIRT1 inhibitor 21 (IC50 60–100 nM17) showed a binding energy of −37.5 kcal/mol whereas compounds 5 and 20 were found to have binding energies of −26.8 kcal/mol and −31.6 kcal/mol, respectively. Conformational ensembles for compounds 5, 20 and 21 complexes produced by MD are depicted in Figure 2. The more favorable binding energy for compound 21 can be explained by its tighter conformational ensemble observed for this ligand in the binding domain than those of compounds 5 and 20. For compound 5, MD simulations produced two distinct conformational families, as clearly seen by the position of the carbonyl group. One conformational family was similar to those of compounds 20 and 21, in which the polar functional groups positioned towards the pocket entrance. In the second conformational family, compound 5 rotated by approximately 90 degrees. This rotation placed the carbonyl group adjacent to the nicotinamide group of the NAD+, resulting in slight displacement of the NAD+ from the active site and thus creating a possible escape route from the binding pocket. This behavior was observed after first 100 ns of simulation, and could also support weaker binding of 5 than 20. This conformational flexibility resulted in the less favorable binding energy of this ligand as compared to 20 and 21. The smaller size of compound 20 (as compared to 21) allowed it more freedom to move inside the binding pocket than compound 21. Based on this conformational analysis, it is possible to propose that increase in conformational flexibility of the compound in the SIRT1 binding pocket leads to decrease in binding affinity. The results obtained from the MD calculations suggest that bulkier aromatic acid derivatives may be better SIRT1 inhibitors by complementing the size and hydrophobic environment of the enzyme binding pocket, and we plan to test such derivatives in the near future.
Figure 2.
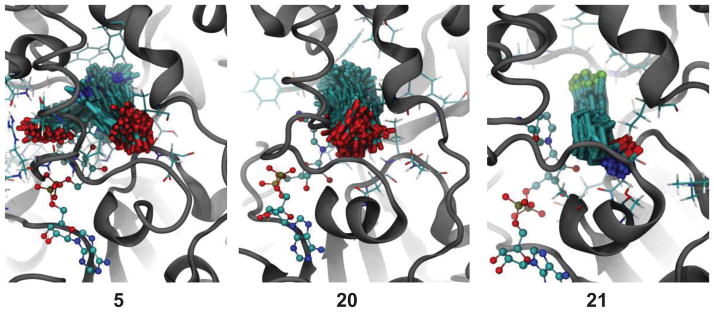
The representative conformational snapshots of compounds 5, 20 and 21 in the catalytic domain of SIRT1 produced by 0.5 μs explicit solvent MD simulations.
In this study, 4-tert-butylbenzoic acid (20) was discovered as a new Sir2p inhibitor based on the SAR study on the naturally occurring weak Sir2p inhibitor, 4-dimethylaminobenzoic acid (5), isolated from the Streptomyces sp. CP27-53. Compound 20 also showed a weak but selective inhibitory activity against SIRT1. It is quite interesting that the structure of 5 was identical to the capping group of the potent class I/II HDAC inhibitor trichostatin A.29 This study also demonstrated a reasonable correlation between the calculated binding energy and potency of SIRT1 inhibition activity, suggesting that it would be possible to establish a SIRT1 virtual screening method by collecting more data points. The SAR study and MD calculations implied that the size of the substituent in benzoic acid appears to be important for enhanced activity and we thus plan to evaluate large aromatic acid derivatives to identify superior sirtuin inhibitors.
Supplementary Material
Acknowledgments
This investigation was supported by the grants from the National Institutes of Health, SC2GM088057 (T.A.), SC2GM095448 (A.B.G.) and SC1GM095419 (W.W.) and the Beckman Scholarship (J.T.B). The Cell and Molecular Image Center (CMIC) at the College of Science and Engineering, San Francisco State University was funded by the grant (P20MD000544) from the National Center on Minority Health and Health Disparities.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Brachmann CB, Sherman JM, Devine SE, Cameron EE, Pillus L, Boeke JD. Genes Dev. 1995;9:2888. doi: 10.1101/gad.9.23.2888. [DOI] [PubMed] [Google Scholar]
- 2.Yamamoto H, Schoonjans K, Auwerx J. Mol Endocrinol. 2007;21:1745. doi: 10.1210/me.2007-0079. [DOI] [PubMed] [Google Scholar]
- 3.Yi J, Luo J. Biochim Biophys Acta. 2010;1804:1684. doi: 10.1016/j.bbapap.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tiberi L, van den Ameele J, Dimidschstein J, Piccirilli J, Gall D, Herpoel A, Bilheu A, Bonnefont J, Iacovino M, Kyba M, Bouschet T, Vanderhaeghen P. Nat Neurosci. 2012;15:1627. doi: 10.1038/nn.3264. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Wang E, Rinaldo F, Datta K. Oncogene. 2005;24:5510. doi: 10.1038/sj.onc.1208693. [DOI] [PubMed] [Google Scholar]
- 6.Liu PY, Xu N, Malyukova A, Scarlett CJ, Sun YT, Zhang XD, Ling D, Su SP, Nelson C, Chang DK, Koach J, Tee AE, Haber M, Norris MD, Toon C, Rooman I, Xue C, Cheung BB, Kumar S, Marshall GM, Biankin AV, Liu T. Cell Death Differ. 2013;20:503. doi: 10.1038/cdd.2012.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li YZ, Matsumori H, Nakayama Y, Osaki M, Kojima H, Kurimasa A, Ito H, Mori S, Katoh M, Oshimura M, Inoue T. Genes Cells. 2011;16:34. doi: 10.1111/j.1365-2443.2010.01460.x. [DOI] [PubMed] [Google Scholar]
- 8.Sunami Y, Araki M, Hironaka Y, Morishita S, Kobayashi M, Liew EL, Edahiro Y, Tsutsui M, Ohsaka A, Komatsu N. PLoS One. 2013;8:e57633. doi: 10.1371/journal.pone.0057633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fraga MF, Agrelo R, Esteller M. In: Biogerontology: Mechanisms and Interventions. Rattan SIS, Akman S, editors. Vol. 1100. Blackwell Publishing; Oxford: 2007. p. 60. [Google Scholar]
- 10.Guarente L. New Engl J Med. 2011;364:2235. doi: 10.1056/NEJMra1100831. [DOI] [PubMed] [Google Scholar]
- 11.Kim HS, Vassilopoulos A, Wang RH, Lahusen T, Xiao Z, Xu X, Li C, Veenstra TD, Li B, Yu H, Ji J, Wang XW, Park SH, Cha YI, Gius D, Deng CX. Cancer Cell. 2011;20:487. doi: 10.1016/j.ccr.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rotili D, Tarantino D, Nebbioso A, Paolini C, Huidobro C, Lara E, Mellini P, Lenoci A, Pezzi R, Botta G, Lahtela-Kakkonen M, Poso A, Steinkuhler C, Gallinari P, De Maria R, Fraga M, Esteller M, Altucci L, Mai A. J Med Chem. 2012;55:10937. doi: 10.1021/jm3011614. [DOI] [PubMed] [Google Scholar]
- 13.Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL. J Biol Chem. 2001;276:38837. doi: 10.1074/jbc.M106779200. [DOI] [PubMed] [Google Scholar]
- 14.Daves GD, Cheng CC, Robins RK. J Am Chem Soc. 1961;83:3904. [Google Scholar]
- 15.Wang J, Kim TH, Ahn MY, Lee J, Jung JH, Choi WS, Lee BM, Yoon KS, Yoon S, Kim HS. Int J Oncol. 2012;41:1101. doi: 10.3892/ijo.2012.1534. [DOI] [PubMed] [Google Scholar]
- 16.Choi G, Lee J, Ji JY, Woo J, Kang NS, Cho SY, Kim HR, Ha JD, Han SY. Int J Oncol. 2013;43:1205. doi: 10.3892/ijo.2013.2035. [DOI] [PubMed] [Google Scholar]
- 17.Napper AD, Hixon J, McDonagh T, Keavey K, Pons JF, Barker J, Yau WT, Amouzegh P, Flegg A, Hamelin E, Thomas RJ, Kates M, Jones S, Navia MA, Saunders J, DiStefano PS, Curtis R. J Med Chem. 2005;48:8045. doi: 10.1021/jm050522v. [DOI] [PubMed] [Google Scholar]
- 18.Peck B, Chen CY, Ho KK, Di Fruscia P, Myatt SS, Coombes RC, Fuchter MJ, Hsiao CD, Lam EW. Mol Cancer Ther. 2010;9:844. doi: 10.1158/1535-7163.MCT-09-0971. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Au Q, Zhang M, Barber JR, Ng SC, Zhang B. Biochem Biophys Res Commun. 2009;386:729. doi: 10.1016/j.bbrc.2009.06.113. [DOI] [PubMed] [Google Scholar]
- 20.Amagata T, Xiao J, Chen YP, Holsopple N, Oliver AG, Gokey T, Guliaev AB, Minoura K. J Nat Prod. 2012;75:2193. doi: 10.1021/np300640g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao X, Allison D, Condon B, Zhang F, Gheyi T, Zhang A, Ashok S, Russell M, MacEwan I, Qian Y, Jamison JA, Luz JG. J Med Chem. 2013;56:963. doi: 10.1021/jm301431y. [DOI] [PubMed] [Google Scholar]
- 22.Huo SH, Wang JM, Cieplak P, Kollman PA, Kuntz ID. J Med Chem. 2002;45:1412. doi: 10.1021/jm010338j. [DOI] [PubMed] [Google Scholar]
- 23.Gohlke H, Kiel C, Case DA. J Mol Biol. 2003;330:891. doi: 10.1016/s0022-2836(03)00610-7. [DOI] [PubMed] [Google Scholar]
- 24.Gohlke H, Case DA. J Comput Chem. 2004;25:238. doi: 10.1002/jcc.10379. [DOI] [PubMed] [Google Scholar]
- 25.Gouda H, Yanai Y, Sugawara A, Sunazuka T, Omura S, Hirono S. Bioorg Med Chem. 2008;16:3565. doi: 10.1016/j.bmc.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 26.Lee J, Kim JS, Seok C. J Phys Chem B. 2010;114:7662. doi: 10.1021/jp1017289. [DOI] [PubMed] [Google Scholar]
- 27.Gokey T, Baird TT, Guliaev AB. J Mol Modeling. 2012;18:4941. doi: 10.1007/s00894-012-1541-x. [DOI] [PubMed] [Google Scholar]
- 28.Homeyer N, Gohlke H. Mol Inf. 2012;31:114. doi: 10.1002/minf.201100135. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida M, Horinouchi S, Beppu T. BioEssays. 1995;17:423. doi: 10.1002/bies.950170510. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.