

Abstract
P-glycoprotein (Pgp), a product of the multi-drug resistance gene MDR1a, is a broad specificity efflux ATP cassette transmembrane transporter that is predominantly expressed in epithelial tissues. Because mdr1a−/− mice tend to develop spontaneous colitis in bacteria-dependent manner, Pgp is believed to have a role in protection of the intestinal epithelium from luminal bacteria. Here we demonstrate that levels of Pgp in the small intestine of newborn rodents dramatically increase during breastfeeding, but not during formula feeding (FF). In rats and mice, levels of intestinal Pgp peak on days 3–7 and 1–5 of breastfeeding, respectively. The mdr1a−/− neonatal mice subjected to FF, hypoxia, and hypothermia have significantly higher incidence and pathology, as well as significantly earlier onset of necrotizing enterocolitis (NEC) than congenic wild type mice. Breast-fed mdr1a−/− neonatal mice are also more susceptible to intestinal damage caused by the opportunistic pathogen Cronobacter sakazakii that has been associated with hospital outbreaks of NEC. Breast milk, but not formula, induces Pgp expression in enterocyte cell lines in a dose- and time-dependent manner. High levels of ectopically expressed Pgp protect epithelial cells in vitro from apoptosis induced by C. sakazakii. Taken together, these results show that breast milk-induced expression of Pgp may have a role in the protection of the neonatal intestinal epithelium from injury associated with nascent bacterial colonization.
Keywords: bacteria, breast milk, intestinal epithelium, necrotizing enterocolitis, P-glycoprotein
Necrotizing enterocolitis (NEC) is a severe intestinal inflammation that primarily affects pre-term neonates. The risk factors for NEC include prematurity, formula feeding (FF), and bacterial colonization of the gut.1–4 Perinatal stresses such as hypoxia, intestinal ischemia, or hypothermia increase susceptibility to NEC.5–9 Despite advances in neonatal care, morbidity and mortality of NEC have not changed appreciably in the last three decades.10,11 According to the broadly accepted pathogenetic model of NEC, perinatal insults compromise the gut barrier in a susceptible host, leading to bacterial translocation across the barrier, and inflammatory responses in lamina propria immunocytes. These inflammatory responses further compromise the epithelium, establishing a vicious circle of inflammation-induced epithelial damage that culminates in exuberant inflammation, tissue necrosis, and sepsis.12–16 Breastfeeding, as opposed to FF, is known to provide protection against NEC.17–22 Interestingly, pasteurization of donor milk appears to abrogate this protection,23 suggesting heat lability of the protective factors. Breast milk have been reported to protect the epithelium in a variety of ways, including inhibition of bacterial attachment,24 attenuation of inflammatory cytokine production,25 and detoxification of NEC-inducing platelet-associated factor.26,27 Several factors found in milk, namely epidermal growth factor (EGF),28–31 heparin-binding EGF,32–35 lactoferrin,36,37 and oligosaccharides38 have been reported to protect against clinical or experimental NEC. Despite these insights, the definitive mechanisms by which breast milk protects immature intestinal epithelium remain unknown.
One of the key mechanisms that protect the intestinal epithelium from injury associated with intestinal microbiota involves P-glycoprotein (Pgp), a member of the ATP-binding-cassette transmembrane transporter family.39 Pgp, which is encoded by the multi-drug resistance gene MDR1 in humans and Mdr1a and Mdr1b genes in rodents,39–41 was first identified as a protein whose overexpression confers the refractory phenotype on chemotherapy-resistant tumors. The drug resistance phenotype of Pgp overexpression is associated with the ability of this transporter to facilitate the ATP-dependent efflux of a broad range of xenobiotics.42,43 In addition, Pgp is believed to function as a toxin efflux pump.40 Because Pgp has broad substrate specificity,43–45 its potential physiologic substrates may include short chain fatty acids, aldehydes, NO metabolites, products of lipid peroxidation, and bacterial toxins.46–48 Pgp is expressed in a variety of epithelial tissues.44,49 Mdr1a mRNA is expressed in the intestinal epithelium, blood brain barrier, and blood testis barrier.50,51 Mdr1b mRNA is highly expressed in the adrenal gland, pregnant uterus, and ovaries.50
Pgp levels are abnormally low in the intestine of patients with newly diagnosed or refractory ulcerative colitis.52 Expression of Mdr1a is also reduced in the dextran sulfate sodium-induced colitis in mice,53 and in IL-10-deficient mice that develop spontaneous colitis.54
Whereas mdr1b−/− mice appear normal, mdr1a−/− mice have a tendency to develop spontaneous colitis, which resembles human inflammatory bowel disease.55 Interestingly, mdr1a−/− mice reconstituted with wild type bone marrow still develop colitis,55 indicating that Mdr1a deficiency in the epithelium, but not in the hematopoietic compartment, is responsible for the disease. As the spontaneous colitis in mdr1a−/− mice is preventable by oral antibiotics, and does not develop in specific pathogen-free environment,55 Pgp appears to protect the intestine against luminal bacteria. This notion is further supported by the fact that Helicobacter bilis and H. hepaticus cause more severe colitis in mdr1a−/− mice than in wild-type mice.56,57 Based on the facts that bacterial colonization predisposes to NEC,10,58,59 and that Pgp protects the intestine from bacteria-associated inflammation, we hypothesized that decreased expression of Pgp in the neonatal small intestine may contribute to the development of NEC.
Here we report that (i) Pgp expression in the neonatal intestine and in enterocyte cell lines is induced by breast milk, (ii) that Pgp deficiency predisposes newborn mice to NEC, and (iii) that Pgp expression protects epithelial cells from bacteria-induced apoptosis. These data suggest that Pgp induction may contribute to the protective effect of breast milk in NEC.
MATERIALS AND METHODS
Reagents
Reagents used in this study were purchased from the following suppliers: colchicine, cycloheximide, mouse anti β-actin monoclonal antibody (Ab, Sigma-Aldrich, St. Louis, MO, USA); secondary horseradish peroxidase-conjugated Abs and normal sera (Jackson Immunoresearch, West Grove, PA, USA); anti-Pgp Ab C219 (Signet, Dedham, MA, USA); real-time PCR reagents (Roche, Indianapolis, IN, USA); anti-V5 Ab (Invitrogen, Carlsbad, CA, USA); EGF and rat TNF-α (Peprotech, Rocky Hill, NJ, USA); EGFR and phospho-EGFR Abs (Cell Signaling Technology, Beverly, MA, USA).
Rodent NEC
All animal experiments have been approved by the Animal Care and Use Committee and Biosafety Committee at CHLA. Timed pregnant Sprague-Dawley rats were purchased from Harlan (Madison, WI, USA). Timed pregnant mdr1a−/− (FVB background) and wild-type FVB mice were purchased from Taconic (Oxnard, CA, USA). Induction of NEC in rats by FF/hypoxia (FF/H) has been described previously.60,61 Briefly, pregnant rats were induced at term with 2 U Pitocin (American Partners, Los Angeles, CA, USA). Immediately after birth, newborn rats were randomly assigned to the breast-fed (BF) control group, or to the FF/H group. FF/H animals were fed three times daily, by oral gavage with 200 μl per feeding of formula (16% w/v Similac PM 60/40, (Ross Metabolics, Columbus, OH, USA) in the Esbilac canine milk replacer, (PetAg, Hampshire, IL, USA)), and subjected to hypoxia (5% O2, 95% N2 for 10 min) before each feeding. Induction of NEC in mice by FF/hypoxia + hypothermia (FF/H + H) was as described.62 Newborn mice were delivered by cesarean section of timed-pregnant females at E19.5, or by natural birth. Starting 2 h after delivery, neonates were fed every 3 h with 20 μl on day 1 or 30 μl thereafter, of formula (2 × strength Esbilac) using a 24-gauge (1.9Fr) polyurethane catheter, and subjected to hypoxia (100% N2 for 1 min) and hypothermia (8 °C for 10 min) three times daily, up to the day 3 of life. Newborn rodents were maintained at 35 °C and 70% relative humidity; control animals were allowed to nurse with their mothers. All animals were examined regularly, and those displaying signs of distress (cyanosis, lethargy profuse bleeding, severe abdominal distension) were killed. Surviving rats and mice were killed on day 4 and 3, respectively. NEC was macroscopically diagnosed by one of the following: abdominal distention, bloody stool, pneumatosis, and discoloration of a segment of small intestine. Paraffin sections of small intestinal samples of all the killed animals were stained with hematoxylin-eosin and graded by a pathologist blinded to groups. NEC histopathology grade (scale of 0–4, with increments of 1), from normal morphology (grade 0) to complete obliteration of the epithelium and intestinal perforation (grade 4), was based on extent of epithelial sloughing, submucosal edema, neutrophil infiltration, and villus architecture destruction in the worst-affected area. Macroscopic diagnosis of NEC was confirmed by a histopathology grade of 1 or higher.
Breast Milk
Postpartum rats were kept with their litters for 4 days before the first milking. Before 2–4 h of milking, animals were injected with 0.2 U Pitocin. Before 15 min of milking, animals were anesthetized by intramuscular injection of ketamine (50 mg/kg), with lidocaine (7 mg/kg). The mammary gland area was cleaned with 70% ethyl alcohol. Milk was collected by gentle application of pressure to each nipple/mammary gland,63 and aspiration of milk drops with a sterile pipette. Human breast milk was obtained from healthy anonymous volunteers according to the protocol approved by the Committee on Clinical Investigation at CHLA. Milk was cleared of lipids and insoluble matter by centrifugation at 100 000 g and 20 °C for 15 min in the A-95 rotor of Airfuge (Beckman Coulter, Fullerton, CA, USA).
Cell Culture
Rat enterocyte cell lines IEC-6 and IEC-18 were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). RIE-1 rat enterocytes were a gift from Dr Pawel Kiela (University of Arizona, Tucson, AZ, USA). Rat enterocyte cell lines were grown in Dulbecco-modified Eagle medium supplemented with 5% fetal calf serum and 0.5 U/ml insulin. Caco-2 human colon carcinoma cells were purchased from ATCC and grown in alpha-MEM medium supplemented with 20% serum. AuxB1 and CHrC5 cells originating from the CHO cell line were kindly provided by Dr Victor Ling (British Columbia Cancer Research Center, Vancouver, Canada), and were grown in alpha-MEM medium supplemented with 10% serum. All cell lines were grown at 37° C and 10% CO2.
Plasmids and Transfection
MDR1 cDNA coding sequence was amplified from MDR1-pGEM3Zf(−)64 using PCR, with primers CACCATGGATCTTGAAGGGGACCG, CTGGCGCTTTGTTCCAGCCTGGAC, and the Phusion high-fidelity PCR kit (New England Biolabs, Ipswich, MA, USA). The resulting DNA fragment was inserted into pcDNA3.1-V5-His (Invitrogen) to generate pcDNA3-MDR1-V5. The insertion added the V5 epitope to the C-terminus of Pgp. The entire MDR1 coding region was sequenced to ascertain the absence of mutations. For transfection, IEC-6 cells grown to 90% confluence were gently trypsinized and re-suspended at 108 cells/ml in the Transfection Reagent V (Lonza, Walkersville, MD, USA). Cell suspension aliquots of 100 μl were mixed with 8 μg of plasmid DNA, and electroporated using the T-030 program of Nucleofector (Lonza). After 10 min recovery in RPMI-1640 medium + 5% serum, cells were plated in their regular growth medium. Stable transfectants were selected in the presence of 1 mg/ml G418, and expression of Pgp was confirmed by western blots with anti-V5 Ab. Cells stably transfected with pcDNA3-MDR1-V5, but not pcDNA3.1-V5-His, were resistant to 10 μg/ml of colchicine, a known substrate of Pgp.
Western Blots
Cells or mucosal scrapings from small intestine were lysed on ice in RIPA buffer (20 mM Tris pH 8.0, 100 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% sodium dodecylsulfate) supplemented with protease and phosphatase inhibitors (0.5 mM PMSF, 10 μg/ml each of leupeptin, aprotinin, and pepstatin, 1 mM benzamidine, 0.1 mM sodium orthovanadate, 20 mM NaF, 0.1 mM phenylarsine oxide, 0.2 mM sodium pyrophosphate), and lysates were cleared by centrifugation at 9000 g at 4 °C for 15 min. Protein samples of 50 μg were separated on denaturing polyacrylamide gels, and the gels were electroblotted onto nitro-cellulose membranes. Membranes were blocked with 2% fish gelatin in phosphate-buffered saline (PBS) and 0.1% Tween 20. Following sequential incubation with primary Ab and horseradish peroxidase-conjugated secondary Abs, membranes were soaked in luminol-peroxide reagent, and exposed to X-ray film. Band densities on western blots were determined using GelDoc and Quantity One software (Bio-Rad, Hercules, CA, USA).
Immunofluorescence
Small intestine sections of 4 μM were deparaffinized and boiled for 10 min in 10 mM Na-citrate. After blocking with 5% normal donkey serum in PBS-Tween, sections were incubated for 1 h with 1:50 dilution of C219 Ab, followed by 1 h with 1:200 dilution of FITC-conjugated donkey anti-mouse Ab. Sections were washed with PBS-Tween and mounted in diamidino phenylindole (DAPI) medium. Images were acquired using BX51 microscope, S97809 camera, and Picture Frame software (Olympus, Center Valley, PA, USA). To minimize variability, samples were processed in parallel and photographed at the same camera settings; identical adjustments were applied to all images.
Real-Time RT-PCR
Total RNA was isolated using Trizol (Invitrogen). Oligo-dT-primed first strand cDNA was synthesized using First-Strand Beads (GE Healthcare, Piscataway, NJ, USA). Real-time PCR was performed using the Light-Cycler 1.5 (Roche), with primers listed in Table 1. Levels of acidic ribosomal phosphoprotein P0 (Arbp P0) mRNA (housekeeping gene transcript) were determined for each sample, and levels of other transcripts were normalized to those of Arbp P0. The absolute quantification method was utilized to determine specific cDNA content in each sample, which involved generation of a standard curve from reactions with serially diluted target DNA, and interpolation based on linear regression analysis.
Table 1.
Primers for RT-PCR analysis of gene expression
| Gene | Species | Orientation | Sequence |
|---|---|---|---|
| Arbp P0 | Rat | Forward Reverse |
5′-CCGTGATGCCCAGGGAAGACAG-3′ 5′-CTGGCACCTTATTGGCCAGCAG-3′ |
| Mdr1a | Rat | Forward Reverse |
5′-GCTTCTTCCAAAGTGTACATCTTG-3′ 5′-TGCCCATCTTTGAGAAGTTCTTG-3′ |
| Arbp P0 | Mouse | Forward Reverse |
5′-CCGTGATGCCCAGGGAAGACAG-3′ 5′-CCAACAGCATATCCCGAATCTCAG-3′ |
| Mdr1a | Mouse | Forward Reverse |
5′-ACAGCGGTTTCCAGGAGCTGCTG-3′ 5′-TGCCCATCTTTGAGAAGTTCTTG-3′ |
| IL-6 | Mouse | Forward Reverse |
5′-TACCACTTCACAAGTCGGAGGCTT-3′ 5′-TTGGATGGTCTTGGTCCTTAGCCA-3′ |
| COX-2 | Mouse | Forward Reverse |
5′-TCATGAAATATGTGCTGACATCC-3′ 5′-AGGAGGGTAGATCATCTCTACC-3′ |
| iNOS | Mouse | Forward Reverse |
5′-TGTGCTGTTCTCAGCCCAACAA-3′ 5′-AGCCTGAAGTCATGTTTGCCGT-3′ |
Induction of Apoptosis In Vitro
All experiments with C. sakazakii have been approved by the Institutional Biosafety Committee at CHLA. C. sakazakii 51329 was purchased from ATCC and grown overnight in Luria broth (1% tryptone, 0.5% yeast extract, 0.5% NaCl) at 37° C. Bacteria were collected by centrifugation, washed in 0.9% NaCl, re-suspended in appropriate cell culture medium, and kept on ice. Epithelial cell cultures were treated with C. sakazakii in antibiotic-free media at 107 cfu/ml. Bacteria-containing medium was changed every 2 h to avoid bacterial overgrowth. A fresh inoculum of bacteria was added at each medium change.65,66 For cytokine-induced apoptosis, cells were treated with 10 ng/ml rat tumor necrosis factor alpha (TNF-α) + 0.1 μg/ml cycloheximide. After 8 h treatment, apoptotic cells were stained using the ApopTag Red in situ apoptosis detection kit (Chemicon, Temecula, CA, USA) according to the manufacturer’s protocol, mounted in DAPI medium, and photographed using BX51 microscope and Picture Frame software. Percentages of apoptotic cells were determined by counting normal and apoptotic nuclei in multiple randomly selected fields.
Statistical Analysis
Mann–Whitney rank-sum, χ2-, Student’s t-test, or ANOVA, was used for data comparisons. Statistical significance was set at a two-sided 5% level. All quantitative data were expressed as mean ± s.e.m. Statistical analyses were performed using STATA/MP 10.0 software package (College Station, TX, USA).
RESULTS
FF is Associated With Low Levels of Pgp in the Small Intestine
As Pgp is known to protect the intestinal epithelium from bacteria-induced inflammation,55–57,67 we hypothesized that NEC may be associated with low levels of Pgp in the gut. To test this hypothesis, we compared Pgp expression in the ileal mucosa of neonatal rats that have been BF or subjected to the NEC-inducing FF/H regimen. Levels of Pgp protein and mRNA in the ileal mucosa of newborn and 4-day-old animals were determined using western blots and real time RT-PCR. Pgp protein was undetectable, and Mdr1a mRNA expressed at the low basal level in the neonatal mucosa. Pgp protein band density increased almost 100-fold in the ileum of BF animals, whereas there was no significant increase in Pgp protein expression in FF/H animals (Figures 1a and b). Mdr1a mRNA expression in the ileum followed the same pattern; its levels significantly increased in BF, but not in FF/H animals (Figure 1c). FF alone, without hypoxia, also failed to significantly induce Pgp protein or Mdr1a mRNA, and low levels of Pgp expression in the formula-fed animals were observed regardless of whether or not these animals developed NEC (data not shown). These results indicate that Pgp expression in rat small intestine is undetectable at birth, and increases significantly by day 4 of breastfeeding, but not FF.
Figure 1.

Breastfeeding increases expression of P-glycoprotein (Pgp) in the small intestine. (a) Western blots for Pgp protein levels in mucosal scrapings from terminal ileum of newborn (NB) rats immediately after birth, breast-fed (BF) for 4 days, and subjected to formula feeding/hypoxia (FF/H) for 4 days. β-actin re-probes are shown to demonstrate lane load. ‘ +’, positive control (lysate of CHrC5 cells). Pgp electromobility variations are presumably due to different degrees of glycosylation.82 Images are representative of five independent experiments. (b) Average Pgp protein band densities. Pgp levels were normalized to those of β-actin in the same sample. *Significant difference from NB or FF (P<0.05). (c) Average levels of Mdr1a mRNA in the terminal ileum of BF and FF/H rats as determined by real-time RT-PCR. Levels of Mdr1a mRNA were normalized to those of Arbp P0 mRNA in the same sample. *Significant difference from NB or FF (P<0.05). (d) Small intestinal sections from 4-day-old breast-fed (BF) or formula-fed (FF) rats immunostained with C219 antibody (Ab), or normal mouse serum as indicated. Green, Pgp immunofluorescence; blue, DAPI-stained nuclei. Bar = 100 μM. Images are representative of three independent experiments.
To identify the sites of Pgp expression, we immunostained sections of small intestine from formula-fed and BF rats with the anti-Pgp Ab C219. There was no appreciable Pgp staining in the intestine of formula-fed animals (Figure 1d, FF C219). In the intestine of BF rats, Pgp prominently localized at the apical aspect of the epithelium, whereas little, if any, Pgp staining was observed at the basolateral aspect or in the lamina propria (Figure 1d, BF C219). Pgp staining was specific, because it was not observed upon substitution of the C219 Ab with normal mouse serum (Figure 1d, BF Norm). Thus, breastfeeding induces Pgp predominantly in the apical aspect of the epithelium of the small intestine.
Pgp Levels in the Small Intestine Peak Shortly After the Start of Breastfeeding
As breastfeeding, which is known to protect from NEC, induces Pgp expression in the small intestine, Pgp induction may be one of the protective mechanisms. To examine relationship between Pgp expression and breastfeeding, we compared the timing of both events. Levels of Pgp protein in the ileal mucosa of rats were determined at various time points during breastfeeding, and after weaning. Rats normally wean at about 3 weeks;68 therefore, to obtain fully weaned animals, we separated a 21-day-old offspring from the dam, and allowed them to feed on chow for additional 7 days. In rats, Pgp was undetectable on days 0–1, significantly increased on day 2, peaked on days 3–7, decreased by day 14, and returned to background after weaning (Figures 2a and b). As the C219 Ab did not react with mouse Pgp, we were unable to perform western blots with mouse tissue. Instead, we examined levels of Mdr1a mRNA in the small intestine of BF and weaned (6 days post weaning) mice, using real-time RT-PCR. Mdr1a mRNA levels in mouse small intestine peaked on days 1–5, decreased thereafter, and returned to background after weaning (Figure 2c). In rats and mice, the incidence of NEC is highest on day 4 and days 2–3 of FF, respectively (YS Guner and HR Ford, unpublished observations). Thus, Pgp expression in the small intestine of BF rodents occurs before the time at which NEC develops in formula-fed animals, which is consistent with the role of Pgp in protection against NEC. These results also demonstrate that Pgp expression and breastfeeding temporally coincide, and that levels of Pgp peak shortly after the start of breastfeeding.
Figure 2.

Timing of P-glycoprotein (Pgp) expression in the terminal ileum of breast-fed (BF) rodents. (a) A representative western blot for Pgp and β-actin protein levels in mucosal scrapings from terminal ileum of BF rats at the indicated age. ‘ +’, positive control (CHrC5 lysate). (b) Average Pgp protein band densities. Pgp levels were normalized to those of β-actin in the same sample. *Significant difference from day 0 (n = 4, P<0.05). (c) Average levels of Mdr1a mRNA in small intestinal mucosa of FVB mice on the indicated days of breastfeeding. Levels of Mdr1a mRNA were normalized to those of Arbp P0 mRNA in the same sample. *Significant difference from day 0 (n = 3, P<0.05).
Mdr1a Deficiency Predisposes Mice to NEC
High levels of Pgp seen in BF, but not in formula-fed rodents, and time course of breastfeeding-induced Pgp expression are consistent with the protective role of Pgp in NEC. To further define the role of Pgp in the pathogenesis of NEC, we compared NEC timing and pathology in mdr1a−/− with congenic wild-type mice subjected to the FF/hypoxia plus hypothermia (FF/H + H) NEC-inducing regimen.62 Newborn mice were formula-fed every 3 h, and subjected to hypoxic and hypothermic stresses three times daily.
In the wild-type group, 12/29 animals (41%) developed NEC (median pathology grade 0; range 0–4; Figure 3a). Among the wild-type mice that developed NEC, 0, 2, 8, and 12 had the disease by 24, 36, 48, and 60 h, respectively. The average time for the development of NEC was 50 ± 2.8 h (range 36–60 h). In the mdr1a−/− group, 19/29 animals (66%) developed NEC (median pathology grade 2; range 0–3; Figure 3a). Among the mdr1a−/− mice that developed NEC, 3, 11 and 19 had the disease by 12, 24, and 36 h, respectively. The average time for the development of NEC was 27 ± 3.2 h. Pathology grades of NEC were significantly higher in the mdr1a−/− group (P<0.05, two-tailed Mann–Whitney test). Incidence of NEC was also significantly higher in the mdr1a−/− group (P<0.01, χ2 test). NEC developed significantly faster in the mdr1a−/− group (P<0.01, Student’s t-test). At the time of the onset and progression of NEC (40–72 h of the FF/H + H regimen), levels of mRNAs encoding IL-6, cyclooxygenase-2, and inducible nitric oxide synthase, the pro-inflammatory proteins upregulated in NEC, were also significantly higher in mdr1a−/− mice (Figure 3b). These data demonstrate that mdr1a−/− mice are more susceptible to NEC in the FF/H + H model, which supports the protective role of Pgp in the pathogenesis of NEC.
Figure 3.

Mdr1a deficiency predisposes mice to necrotizing enterocolitis (NEC). (a) Pathology grades in wild-type FVB and mdr1a−/− mice subjected to the FF/H +H regimen. Incidence and grades of NEC are significantly higher in the mdr1a−/− group (n = 29, P<0.01, χ2-test; P<0.05, two-tailed Mann–Whitney test, respectively). (b) Expression of IL-6, cyclooxygenase-2 (COX-2), and inducible nitric oxide synthase (iNOS) mRNAs in the small intestinal mucosa of wild-type (open bars) and mdr1a−/− (filled bars) mice on days 2–3 of the FF/H + H regimen. mRNA levels are relative to Arbp P0 transcript levels, which were assigned the value of 1. *Significant differences from wild type (n = 5, P<0.05). (c) Pathology grades in breast-fed (BF) wild-type FVB and mdr1a−/− mice, challenged with 105 cfu C. sakazakii on day 1. Samples of small intestine were collected on day 4. Pathology grades are significantly higher in the mdr1a−/− group (P = 0.02, n = 21, two-tailed Mann–Whitney test). (d) Representative pathology micrographs. Left, normal architecture, absence of pathology, grade 0; middle, epithelial sloughing, and partial destruction of villus tips, grade 2; right, submucosal edema, and severe disruption of villus architecture, grade 3. Bar = 100 μM.
Opportunistic pathogens are believed to have an important role in the pathogenesis of NEC. C. sakazakii is an emerging opportunistic pathogen that has been associated with hospital outbreaks of NEC.71–73 C. sakazakii is commonly found in dairy products,74,75 and NEC outbreaks have been traced to infant formula contaminated with these bacteria.73 C. sakazakii role as a causative agent in NEC is further supported by the fact that oral introduction of these bacteria exacerbates intestinal injury in experimental NEC.65,66 Thus, C. sakazakii may serve as a model microorganism that can be used to study the role of opportunistic Gram-negative bacteria in the pathogenesis NEC. If breast milk-induced expression of Pgp contributes to protection against opportunistic bacteria, one could expect higher intestinal pathology grades in BF mdr1a−/− mice compared with BF wild-type controls, upon challenge with C. sakazakii. To examine the effect of Mdr1a status on breast milk-dependent protection from pathogenic bacteria, mdr1a−/− and wild-type newborn mice were gavaged with or without a one-time dose of live C. sakazakii on day 1 after birth, and allowed to continue nursing with their mothers. Samples of small intestine collected on day 4 were graded by pathologist. In the wild-type group, 2/21 animals (9.5%) developed intestinal pathology of grade 1 or higher (median pathology grade 0; range 0–4; Figure 3c). In the mdr1a−/− group, 17/21 animals (81%) developed NEC (median pathology grade 4; range 0–4; Figure 3c). The mdr1a−/− mice displayed significantly higher pathology grades (P = 0.02, n = 21, two-tailed Mann–Whitney test), indicating that the Mdr1a gene product Pgp is required for the efficient breastfeeding-conferred protection against an opportunistic pathogen.
Representative pathology micrographs are shown in Figure 3d.
A possible explanation for higher susceptibility of mdr1a−/− mice to NEC was general health impairment in these knockout mice. However, this is unlikely, because mdr1a−/− mice bred normally, had normal litter size, and did not apparently differ from wild-type FVB mice in any aspect of growth, development, or vitality. Furthermore, there was no significant difference in birth weight between wild-type and mdr1a−/− neonates (average birth weights 1.16 ± 0.1 g and 1.20 ± 0.1 g, respectively; n = 50, P<0.05). Therefore, increased susceptibility of mdr1a−/− mice to NEC is unlikely to be a consequence of general health impairment in these animals.
Breast Milk Induces Pgp Expression in Enterocyte Cell Lines
As demonstrated by Figures 1 and 2, Pgp levels in the small intestine positively correlate with breastfeeding, which led us to hypothesize that factor(s) in breast milk directly induce expression of Pgp in the intestinal epithelium. To test this hypothesis, we examined changes in Pgp expression in enterocyte cell lines treated with breast milk or formula. Rat breast milk was obtained from dams starting at day 4 after delivery. Breast milk or formula was cleared of lipids and insoluble material by high-speed centrifugation. IEC-6, IEC-18 and RIE-1 cells, all untransformed enterocyte cell lines derived from rat small intestine, were treated with cleared breast milk or formula. The former, but not the latter, up-regulated Pgp expression in all three enterocyte cell lines (Figures 4a–c and e). The ability of breast milk to induce Pgp was abrogated by heating to 60° C for 10 min (Figures 4a–c and e). Pgp was also induced by cleared human breast milk in Caco-2 enterocytes of human origin in a dose- and time-dependent fashion (Figures 4d, f and g). Desalting cleared milk on a Sephadex G25 column (~10 kDa cut-off) did not decrease Pgp-inducing activity (data not shown). According to these data, water-soluble, heat-labile high molecular weight factor(s) present in breast milk, but not in formula, induce expression of Pgp in enterocytes, which suggests that breast milk may directly induce Pgp in the intestinal epithelium.
Figure 4.
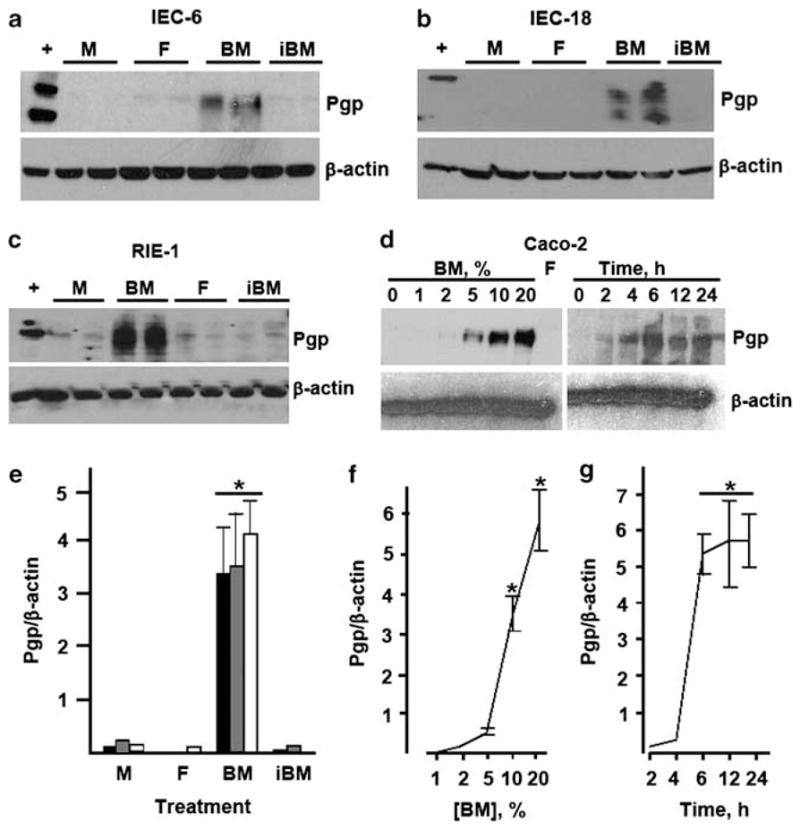
Breast milk induces P-glycoprotein (Pgp) in cultured enterocytes. (a–c) IEC-6, IEC-18, or RIE-1 cells were treated for 24 h with medium (M), 5% centrifugation-cleared formula (F), 5% cleared breast milk (BM), or 5% cleared breast milk incubated at 60° C for 10 min (iBM), as indicated. Levels of Pgp and β-actin were examined by western blotting. For each cell line and treatment, two independently treated samples are shown. (d) Dose response (left) and time course (right) of Pgp expression in Caco-2 cells treated with human breast milk for 24 h at indicated concentrations (left), or 10% human breast milk for indicated time. ‘ +’, positive control (CHrC5 cells). Data are representative of at least three independent experiments. (e) Average Pgp band densities on western blots represented by (a–c). *Significant differences from other treatment groups (P<0.01, n = 3). (f) Dose response of Pgp induction in Caco-2 cells by human breast milk. *Significant difference from other timepoints (P<0.05, n = 3 for each concentration). (g) Time course of breast milk-induced expression of Pgp in Caco-2 cells. *Significant differences from other time points (P<0.05, n = 3 for each time point).
EGF Does Not Induce Pgp Expression in IEC-6 Cells
Epidermal growth factor (EGF), which is found in breast milk,69 has been previously implicated in protection against experimental28–32 and clinical70 NEC. Due to its protective effect, EGF could be a candidate Pgp-inducing factor. To test Pgp induction by EGF, we examined changes in Pgp levels in cultured enterocytes, following treatment with this cytokine. IEC-6 cells were treated with EGF or equivalent amount of solvent, and Pgp expression was examined by western blotting. EGF had no effect on Pgp expression (Figure 5a). Failure to induce Pgp was not due to unresponsiveness of IEC-6 cells to EGF, as this cytokine induced time-dependent tyrosine phosphorylation of EGF receptor, as expected (Figure 5b). These results show that breast milk factor(s), other than EGF, are responsible for the induction of Pgp in enterocytes.
Figure 5.
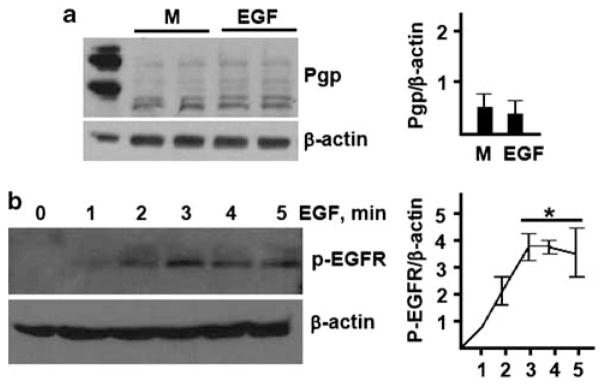
Epidermal growth factor (EGF) does not induce P-glycoprotein (Pgp) in IEC-6 cells. (a) Pgp and β-actin levels in IEC-6 cells treated with medium (M) or 100 ng/ml EGF, as indicated, for 24 h. ‘ +’, Positive control (CHrC5 cells). (b) Phospho-EGFR and β-actin levels in IEC-6 cells treated with 100 ng/ml EGF for indicated time. *Significantly higher phospho-EGFR levels compared with other time points (P<0.05, n = 3).
Pgp Protects Enterocytes Against C. Sakazakii-Induced Apoptosis
It is believed that Pgp protects the intestinal epithelium from harmful bacteria by facilitating removal of toxic bacterial products from enterocytes.40 To test whether Pgp protects epithelial cells from bacteria, we compared frequencies of C. sakazakii-induced apoptosis in cells expressing low or high levels of Pgp. High levels of Pgp expression were achieved by stable transfection of IEC-6 cells with pcDNA3-MDR1-V5; control cells were transfected with the empty pcDNA3 vector. Expression of Pgp in the resulting transfectants was verified by western blotting. As shown in Figure 6a, left, expression of Pgp in IEC-6 tranfectants is detectable with both anti-V5 and anti-Pgp Abs. In addition, we used the colchicine-resistant epithelial cell line CHrC5 that expresses high levels of Pgp due to Mdr1a gene amplification, and its parental line AuxB1, in which Pgp expression is undetectable (Figure 6a, right).76 Cells expressing low or high levels of Pgp were exposed either to live C. sakazakii, or to TNF-α + cycloheximide, and percentages of apoptotic cells were determined using terminal transferase deoxyuridine nick end labeling. MDR1-transfected IEC-6 cells displayed significantly lower levels of C. sakazakii-induced apoptosis, but not of TNF-α + cycloheximide-induced apoptosis, than vector-transfected cells. CHrC5 cells were also more resistant to C. sakazakii-induced apoptosis, but not to TNF-α + cycloheximide-induced apoptosis, than AuxB1 cells (Figures 6b and c). These data demonstrate that Pgp expression protects epithelial cells from C. sakazakii-induced apoptosis, but not from TNF-α-induced apoptosis.
Figure 6.
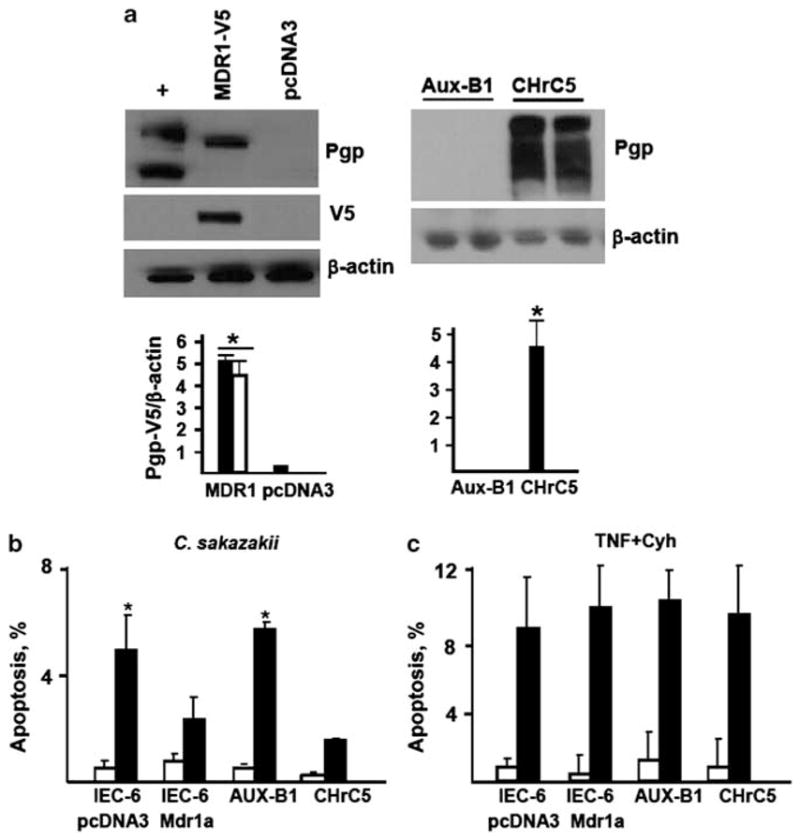
P-glycoprotein (Pgp) protects epithelial cells from C. sakazakii-induced apoptosis. (a) Expression of V5-tagged Pgp protein in IEC-6 cells stably transfected with pcDNA3-MDR1-V5 or pcDNA3 analyzed by Western blotting with Pgp and V5 epitope antibody (Abs; left); expression of Pgp protein in CHrC5 and AuxB1 cells (right). β-actin blots are shown to demonstrate lane load. (+), positive control (CHrC5 cells). Solid bars, Pgp immunoreactivity; open bars, V5 immunoreactivity. *Significant differences from cells transfected with pcDNA3 (P<0.05, n = 3) or from Aux-B1 cells (P<0.01, n = 3). (b) Average frequencies of spontaneous (open bars) and C. sakazakii-induced (filled bars) apoptosis in indicated cell lines. *Significant differences compared to the cell line expressing Pgp (n = 3, P<0.05). (c) Average frequencies of spontaneous (open bars) or tumor necrosis factor (TNF-α) + cycloheximide-induced apoptosis in indicated cell lines.
DISCUSSION
In this report, we examined whether the epithelial transmembrane efflux pump Pgp has a role in the pathogenesis of NEC. Our data demonstrate an inverse relationship between Pgp expression in the intestinal epithelium and the development of experimental NEC. Unlike formula-fed rats, which had high incidence of NEC and low levels of intestinal Pgp, BF rats had normal intestinal morphology and high levels of intestinal Pgp. To probe causal relationship between Pgp expression and NEC, we compared incidence, severity, and timing of NEC in wild-type and Mdr1a-deficient mice subjected to the NEC-inducing FF/hypoxia + hypothermia (FF/H + H) regimen. The mdr1a−/− mice had significantly higher incidence, significantly higher pathology grades, and significantly earlier onset of NEC than the congenic wild-type mice. The mdr1a−/− mice subjected to the FF/H + H regimen also had higher levels of expression of genes encoding inflammatory proteins associated with NEC at the time of onset and progression of the disease. Moreover, breastfeeding provided lesser protection from C. sakazakii-induced intestinal pathology in mdr1a−/− mice than in congenic wild-type mice. By demonstrating that Mdr1a deficiency predisposes mice to NEC, we establish causality between Pgp expression and protection from NEC.
Our in vivo experiments clearly demonstrate that breast-feeding induces Pgp in the small intestine. Pgp expression in the intestinal epithelium of neonatal rodents dramatically increases shortly after initiation of breastfeeding, but not FF. Pgp expression levels peak on days 3–7 and 1–5 of breastfeeding in rats and mice, respectively, and return to background levels after weaning. In the small intestine of BF rats, Pgp expression is largely confined to the apical aspect of the epithelium. As formula-fed rats do not show Pgp upregulation, our data indicate that Pgp expression that we observed is a result of breastfeeding, rather than gene expression program of postnatal development. During the first few days of life, the intestine is colonized predominantly by aerobic and facultative anaerobic bacteria,77 some of which may be opportunistic pathogens attacking the weak immature epithelium.78 As normal composition of the intestinal microbiota is established only after weaning,79,80 it is possible that breastfeeding-induced Pgp expression protects the epithelium during the period of increased vulnerability associated with succession of bacterial populations.
Although increased expression of Pgp in the intestine during postnatal development has been reported previously,81 our study is the first to demonstrate that this increase is due to breastfeeding. The underlying mechanisms of induction remain unclear. One possibility is that factors in breast milk induce Pgp directly, by binding to their cognate receptors on the surface of the enterocytes. An alternative is indirect induction, for example, via improved nutrition afforded by breastfeeding, or via interactions between suckling animals and their mother that do not involve breast milk. To gain an insight into the mechanisms of Pgp induction, we compared Pgp levels in breast milk-treated and formula-treated enterocyte cell lines. As breast milk, but not formula, induced Pgp in cultured enterocytes, we conclude that the former can induce Pgp expression directly. Although these results do not rule out indirect induction of Pgp by breast milk or other aspects of breastfeeding in vivo, they strongly argue for the direct induction.
The identity of physiologic substrates of Pgp in the intestine remains unknown. At least some of the Pgp substrates may be toxic bacterial products, or toxic products of epithelial origin induced by bacteria.40 Our observation of the protective role of Pgp in C. sakazakii-induced enterocyte apoptosis, but not in TNF-α-induced apoptosis supports the specific role of Pgp in protection against bacteria, rather than non-specific anti-apoptotic effect. Unfortunately, there is a paucity of published data regarding the putative physiologic substrates of Pgp in the intestinal epithelium whose removal may contribute to the protection.
Our data provide an insight into the nature of factor(s) in breast milk that is responsible for the induction of Pgp in enterocytes. As these factor(s) have high molecular weight and are heat-labile, they could be protein(s). Heat sensitivity of Pgp-inducing factor(s) is consistent with the report suggesting that pasteurization might abrogate protection against NEC by breast milk.23 Additional studies are needed to identify the Pgp-inducing factor(s). Pgp-inducing factor(s) of breast milk may find use as prophylactic supplements for baby formula.
Acknowledgments
We thank Pawel Kiela and Victor Ling for gifts of cell lines, Victoria Camerini and David Warburton for helpful discussion, Kerstin Goth, Patricia Boyle-Lockerbie, Diana Yu, Christina Mather, and Christian Ochoa for assistance with experiments. Our special thanks are for anonymous donors who provided samples of breast milk. This study was supported by Saban Institute Training Grant to YSG; NIH Loan Repayment Grant to SC; NIH grants AI014032, AI049473 to HRF; and AI083612 to AVG.
Footnotes
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Guthrie SO, Gordon PV, Thomas V, et al. Necrotizing enterocolitis among neonates in the United States. J Perinatol. 2003;23:278–285. doi: 10.1038/sj.jp.7210892. [DOI] [PubMed] [Google Scholar]
- 2.Hammerman C, Bin-Nun A, Kaplan M. Germ warfare: probiotics in defense of the premature gut. Clin Perinatol. 2004;31:489–500. doi: 10.1016/j.clp.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 3.Morowitz MJ, Poroyko V, Caplan M, et al. Redefining the role of intestinal microbes in the pathogenesis of necrotizing enterocolitis. Pediatrics. 125:777–785. doi: 10.1542/peds.2009-3149. [DOI] [PubMed] [Google Scholar]
- 4.Patole S. Prevention and treatment of necrotising enterocolitis in preterm neonates. Early Hum Dev. 2007;83:635–642. doi: 10.1016/j.earlhumdev.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 5.Hall NJ, Eaton S, Peters MJ, et al. Mild controlled hypothermia in preterm neonates with advanced necrotizing enterocolitis. Pediatrics. 2010;125:e300–e308. doi: 10.1542/peds.2008-3211. [DOI] [PubMed] [Google Scholar]
- 6.Hsueh W, Caplan MS, Qu XW, et al. Neonatal necrotizing enterocolitis: clinical considerations and pathogenetic concepts. Pediatr Dev Pathol. 2003;6:6–23. doi: 10.1007/s10024-002-0602-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nowicki PT. Ischemia and necrotizing enterocolitis: where, when, and how. Semin Pediatr Surg. 2005;14:152–158. doi: 10.1053/j.sempedsurg.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Powell RW, Dyess DL, Collins JN, et al. Regional blood flow response to hypothermia in premature, newborn, and neonatal piglets. J Pediatr Surg. 1999;34:193–198. doi: 10.1016/s0022-3468(99)90255-5. [DOI] [PubMed] [Google Scholar]
- 9.Sase M, Lee JJ, Ross MG, Buchmiller-Crair TL. Effect of hypoxia on fetal rabbit gastrointestinal motility. J Surg Res. 2001;99:347–351. doi: 10.1006/jsre.2001.6189. [DOI] [PubMed] [Google Scholar]
- 10.Guner YS, Friedlich P, Wee CP, et al. State-based analysis of necrotizing enterocolitis outcomes. J Surg Res. 2009;157:21–29. doi: 10.1016/j.jss.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 11.Salzman AL, Menconi MJ, Unno N, et al. Nitric oxide dilates tight junctions and depletes ATP in cultured Caco-2BBe intestinal epithelial monolayers. Am J Physiol. 1995;268(2 Part 1):G361–G373. doi: 10.1152/ajpgi.1995.268.2.G361. [DOI] [PubMed] [Google Scholar]
- 12.Anand RJ, Leaphart CL, Mollen KP, et al. The role of the intestinal barrier in the pathogenesis of necrotizing enterocolitis. Shock. 2007;27:124–133. doi: 10.1097/01.shk.0000239774.02904.65. [DOI] [PubMed] [Google Scholar]
- 13.Frost BL, Jilling T, Caplan MS. The importance of pro-inflammatory signaling in neonatal necrotizing enterocolitis. Semin Perinatol. 2008;32:100–106. doi: 10.1053/j.semperi.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neu J, Chen M, Beierle E. Intestinal innate immunity: how does it relate to the pathogenesis of necrotizing enterocolitis. Semin Pediatr Surg. 2005;14:137–144. doi: 10.1053/j.sempedsurg.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 15.Neu J, Mshvildadze M, Mai V. A roadmap for understanding and preventing necrotizing enterocolitis. Curr Gastroenterol Rep. 2008;10:450–457. doi: 10.1007/s11894-008-0084-x. [DOI] [PubMed] [Google Scholar]
- 16.Srinivasan PS, Brandler MD, D’Souza A. Necrotizing enterocolitis. Clin Perinatol. 2008;35:251–272. x. doi: 10.1016/j.clp.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 17.Boyd CA, Quigley MA, Brocklehurst P. Donor breast milk vs infant formula for preterm infants: systematic review and meta-analysis. Arch Dis Child Fetal Neonatal Ed. 2007;92:F169–F175. doi: 10.1136/adc.2005.089490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGuire W, Anthony MY. Donor human milk vs formula for preventing necrotising enterocolitis in preterm infants: systematic review. Arch Dis Child Fetal Neonatal Ed. 2003;88:F11–F14. doi: 10.1136/fn.88.1.F11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meinzen-Derr J, Poindexter B, Wrage L, et al. Role of human milk in extremely low birth weight infants’ risk of necrotizing enterocolitis or death. J Perinatol. 2009;29:57–62. doi: 10.1038/jp.2008.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sisk PM, Lovelady CA, Dillard RG, et al. Early human milk feeding is associated with a lower risk of necrotizing enterocolitis in very low birth weight infants. J Perinatol. 2007;27:428–433. doi: 10.1038/sj.jp.7211758. [DOI] [PubMed] [Google Scholar]
- 21.Stout G, Lambert DK, Baer VL, et al. Necrotizing enterocolitis during the first week of life: a multicentered case-control and cohort comparison study. J Perinatol. 2008;28:556–560. doi: 10.1038/jp.2008.36. [DOI] [PubMed] [Google Scholar]
- 22.Sullivan S, Schanler RJ, Kim JH, et al. An exclusively human milk-based diet is associated with a lower rate of necrotizing enterocolitis than a diet of human milk and bovine milk-based products. J Pediatr. 2010;156:562–567. e561. doi: 10.1016/j.jpeds.2009.10.040. [DOI] [PubMed] [Google Scholar]
- 23.Schanler RJ, Lau C, Hurst NM, et al. Randomized trial of donor human milk vs preterm formula as substitutes for mothers’ own milk in the feeding of extremely premature infants. Pediatrics. 2005;116:400–406. doi: 10.1542/peds.2004-1974. [DOI] [PubMed] [Google Scholar]
- 24.Brooks HJ, McConnell MA, Corbett J, et al. Potential prophylactic value of bovine colostrum in necrotizing enterocolitis in neonates: an in vitro study on bacterial attachment, antibody levels and cytokine production. FEMS Immunol Med Microbiol. 2006;48:347–354. doi: 10.1111/j.1574-695X.2006.00151.x. [DOI] [PubMed] [Google Scholar]
- 25.Claud EC, Savidge T, Walker WA. Modulation of human intestinal epithelial cell IL-8 secretion by human milk factors. Pediatr Res. 2003;53:419–425. doi: 10.1203/01.PDR.0000050141.73528.AD. [DOI] [PubMed] [Google Scholar]
- 26.Caplan MS, Simon D, Jilling T. The role of PAF, TLR, and the inflammatory response in neonatal necrotizing enterocolitis. Semin Pediatr Surg. 2005;14:145–151. doi: 10.1053/j.sempedsurg.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 27.Furukawa M, Narahara H, Yasuda K, et al. Presence of platelet-activating factor-acetylhydrolase in milk. J Lipid Res. 1993;34:1603–1609. [PubMed] [Google Scholar]
- 28.Caplan M. Is EGF the Holy Grail for NEC? J Pediatr. 2007;150:329–330. doi: 10.1016/j.jpeds.2007.01.027. [DOI] [PubMed] [Google Scholar]
- 29.Dvorak B. Milk epidermal growth factor and gut protection. J Pediatr. 2010;156(2 Suppl):S31–S35. doi: 10.1016/j.jpeds.2009.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nair RR, Warner BB, Warner BW. Role of epidermal growth factor and other growth factors in the prevention of necrotizing enterocolitis Milk epidermal growth factor and gut protection. Semin Perinatol. 2008;32:107–113. doi: 10.1053/j.semperi.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 31.Warner BW, Warner BB. Role of epidermal growth factor in the pathogenesis of neonatal necrotizing enterocolitis. Semin Pediatr Surg. 2005;14:175–180. doi: 10.1053/j.sempedsurg.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 32.Dvorak B, Khailova L, Clark JA, et al. Comparison of epidermal growth factor and heparin-binding epidermal growth factor-like growth factor for prevention of experimental necrotizing enterocolitis. J Pediatr Gastroenterol Nutr. 2008;47:11–18. doi: 10.1097/MPG.0b013e3181788618. [DOI] [PubMed] [Google Scholar]
- 33.Feng J, El-Assal ON, Besner GE. Heparin-binding epidermal growth factor-like growth factor decreases the incidence of necrotizing enterocolitis in neonatal rats. J Pediatr Surg. 2006;41:144–149. doi: 10.1016/j.jpedsurg.2005.10.018. discussion 144–149. [DOI] [PubMed] [Google Scholar]
- 34.Radulescu A, Yu X, Orvets ND, et al. Deletion of the heparin-binding epidermal growth factor-like growth factor gene increases susceptibility to necrotizing enterocolitis. J Pediatr Surg. 2010;4:729–734. doi: 10.1016/j.jpedsurg.2009.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Radulescu A, Zorko NA, Yu X, et al. Preclinical neonatal rat studies of heparin-binding EGF-like growth factor in protection of the intestines from necrotizing enterocolitis. Pediatr Res. 2009;65:437–442. doi: 10.1203/PDR.0b013e3181994fa0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mohan P, Abrams SA. Oral lactoferrin for the treatment of sepsis and necrotizing enterocolitis in neonates. Cochrane Database Syst Rev. 2009:CD007138. doi: 10.1002/14651858.CD007138.pub2. [DOI] [PubMed] [Google Scholar]
- 37.Venkatesh MP, Abrams SA. Oral lactoferrin for the prevention of sepsis and necrotizing enterocolitis in preterm infants. Cochrane Database Syst Rev. 2010;5:CD007137. doi: 10.1002/14651858.CD007137.pub2. [DOI] [PubMed] [Google Scholar]
- 38.Bode L, Rudloff S, Kunz C, et al. Human milk oligosaccharides reduce platelet-neutrophil complex formation leading to a decrease in neutrophil beta 2 integrin expression. J Leukoc Biol. 2004;76:820–826. doi: 10.1189/jlb.0304198. [DOI] [PubMed] [Google Scholar]
- 39.Borst P, Zelcer N, van Helvoort A. ABC transporters in lipid transport. Biochim Biophys Acta. 2000;1486:128–144. doi: 10.1016/s1388-1981(00)00053-6. [DOI] [PubMed] [Google Scholar]
- 40.Ho GT, Moodie FM, Satsangi J. Multidrug resistance 1 gene (P-glycoprotein 170): an important determinant in gastrointestinal disease? Gut. 2003;52:759–766. doi: 10.1136/gut.52.5.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Croop JM, Raymond M, Haber D, et al. The three mouse multidrug resistance (mdr) genes are expressed in a tissue-specific manner in normal mouse tissues. Mol Cell Biol. 1989;9:1346–1350. doi: 10.1128/mcb.9.3.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455:152–162. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 43.Sakaeda T, Nakamura T, Okumura K. MDR1 genotype-related pharmacokinetics and pharmacodynamics. Biol Pharm Bull. 2002;25:1391–1400. doi: 10.1248/bpb.25.1391. [DOI] [PubMed] [Google Scholar]
- 44.Arias IM, Gatmaitan Z, Mazzanti R, et al. Structure and function of P-glycoprotein in the normal liver and intestine. Princess Takamatsu Symp. 1990;21:229–239. [PubMed] [Google Scholar]
- 45.Sharma R, Awasthi YC, Yang Y, et al. Energy dependent transport of xenobiotics and its relevance to multidrug resistance. Curr Cancer Drug Targets. 2003;3:89–107. doi: 10.2174/1568009033482047. [DOI] [PubMed] [Google Scholar]
- 46.Chang XB. A molecular understanding of ATP-dependent solute transport by multidrug resistance-associated protein MRP1. Cancer Metastasis Rev. 2007;26:15–37. doi: 10.1007/s10555-007-9041-7. [DOI] [PubMed] [Google Scholar]
- 47.Musiek ES, Breeding RS, Milne GL, et al. Cyclopentenone isoprostanes are novel bioactive products of lipid oxidation which enhance neurodegeneration. J Neurochem. 2006;97:1301–1313. doi: 10.1111/j.1471-4159.2006.03797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sakurazawa T, Ohkusa T. Cytotoxicity of organic acids produced by anaerobic intestinal bacteria on cultured epithelial cells. J Gastroenterol. 2005;40:600–609. doi: 10.1007/s00535-005-1594-z. [DOI] [PubMed] [Google Scholar]
- 49.Schiengold M, Schwantes L, Schwartsmann G, et al. Multidrug resistance gene expression during the murine ontogeny. Mech Ageing Dev. 2001;122:255–270. doi: 10.1016/s0047-6374(00)00223-2. [DOI] [PubMed] [Google Scholar]
- 50.Schinkel AH, Mayer U, Wagenaar E, et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci USA. 1997;94:4028–4033. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brady JM, Cherrington NJ, Hartley DP, et al. Tissue distribution and chemical induction of multiple drug resistance genes in rats. Drug Metab Dispos. 2002;30:838–844. doi: 10.1124/dmd.30.7.838. [DOI] [PubMed] [Google Scholar]
- 52.Gutmann H, Hruz P, Zimmermann C, et al. Breast cancer resistance protein and P-glycoprotein expression in patients with newly diagnosed and therapy-refractory ulcerative colitis compared with healthy controls. Digestion. 2008;78:154–162. doi: 10.1159/000179361. [DOI] [PubMed] [Google Scholar]
- 53.Iizasa H, Genda N, Kitano T, et al. Altered expression and function of P-glycoprotein in dextran sodium sulfate-induced colitis in mice. J Pharm Sci. 2003;92:569–576. doi: 10.1002/jps.10326. [DOI] [PubMed] [Google Scholar]
- 54.Buyse M, Radeva G, Bado A, et al. Intestinal inflammation induces adaptation of P-glycoprotein expression and activity. Biochem Pharmacol. 2005;69:1745–1754. doi: 10.1016/j.bcp.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 55.Panwala CM, Jones JC, Viney JL. A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J Immunol. 1998;161:5733–5744. [PubMed] [Google Scholar]
- 56.Maggio-Price L, Bielefeldt-Ohmann H, Treuting P, et al. Dual infection with Helicobacter bilis and Helicobacter hepaticus in p-glycoprotein-deficient mdr1a−/− mice results in colitis that progresses to dysplasia. Am J Pathol. 2005;166:1793–1806. doi: 10.1016/S0002-9440(10)62489-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maggio-Price L, Shows D, Waggie K, et al. Helicobacter bilis infection accelerates and H. hepaticus infection delays the development of colitis in multiple drug resistance-deficient (mdr1a−/−) mice. Am J Pathol. 2002;160:739–751. doi: 10.1016/S0002-9440(10)64894-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hunter CJ, Petrosyan M, Ford HR, et al. Enterobacter sakazakii: an emerging pathogen in infants and neonates. Surg Infect (Larchmt) 2008;9:533–539. doi: 10.1089/sur.2008.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hunter CJ, Upperman JS, Ford HR, Camerini V. Understanding the susceptibility of the premature infant to necrotizing enterocolitis (NEC) Pediatr Res. 2008;63:117–123. doi: 10.1203/PDR.0b013e31815ed64c. [DOI] [PubMed] [Google Scholar]
- 60.Barlow B, Santulli TV. Importance of multiple episodes of hypoxia or cold stress on the development of enterocolitis in an animal model. Surgery. 1975;77:687–690. [PubMed] [Google Scholar]
- 61.Nadler EP, Dickinson E, Knisely A, et al. Expression of inducible nitric oxide synthase and interleukin-12 in experimental necrotizing enterocolitis. J Surg Res. 2000;92:71–77. doi: 10.1006/jsre.2000.5877. [DOI] [PubMed] [Google Scholar]
- 62.Jilling T, Simon D, Lu J, et al. The roles of bacteria and TLR4 in rat and murine models of necrotizing enterocolitis. J Immunol. 2006;177:3273–3282. doi: 10.4049/jimmunol.177.5.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luckey TD, Mende TJ, Pleasants J. The physical and chemical characterization of rat’s milk. J Nutr. 1954;54:345–359. doi: 10.1093/jn/54.3.345. [DOI] [PubMed] [Google Scholar]
- 64.Lincke CR, van der Bliek AM, Schuurhuis GJ, et al. Multidrug resistance phenotype of human BRO melanoma cells transfected with a wild-type human mdr1 complementary DNA. Cancer Res. 1990;50:1779–1785. [PubMed] [Google Scholar]
- 65.Hunter CJ, Singamsetty VK, Chokshi NK, et al. Enterobacter sakazakii enhances epithelial cell injury by inducing apoptosis in a rat model of necrotizing enterocolitis. J Infect Dis. 2008;198:586–593. doi: 10.1086/590186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hunter CJ, Williams M, Petrosyan M, et al. Lactobacillus bulgaricus prevents intestinal epithelial cell injury caused by Enterobacter sakazakii-induced nitric oxide both in vitro and in the newborn rat model of necrotizing enterocolitis. Infect Immun. 2009;77:1031–1043. doi: 10.1128/IAI.01192-08. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 67.Resta-Lenert S, Smitham J, Barrett KE. Epithelial dysfunction associated with the development of colitis in conventionally housed mdr1a−/− mice. Am J Physiol Gastrointest Liver Physiol. 2005;289:G153–G162. doi: 10.1152/ajpgi.00395.2004. [DOI] [PubMed] [Google Scholar]
- 68.Guner YS, Kiela PR, Xu H, et al. Differential regulation of renal sodium-phosphate transporter by glucocorticoids during rat ontogeny. Am J Physiol. 1999;277(5 Part 1):C884–C890. doi: 10.1152/ajpcell.1999.277.5.C884. [DOI] [PubMed] [Google Scholar]
- 69.McCleary MJ. Epidermal growth factor: an important constituent of human milk. J Hum Lact. 1991;7:123–128. doi: 10.1177/089033449100700317. [DOI] [PubMed] [Google Scholar]
- 70.Sullivan PB, Lewindon PJ, Cheng C, et al. Intestinal mucosa remodeling by recombinant human epidermal growth factor(1–48) in neonates with severe necrotizing enterocolitis. J Pediatr Surg. 2007;42:462–469. doi: 10.1016/j.jpedsurg.2006.10.039. [DOI] [PubMed] [Google Scholar]
- 71.Arseni A, Malamou-Ladas E, Koutsia C, et al. Outbreak of colonization of neonates with Enterobacter sakazakii. J Hosp Infect. 1987;9:143–150. doi: 10.1016/0195-6701(87)90052-1. [DOI] [PubMed] [Google Scholar]
- 72.Clark NC, Hill BC, O’Hara CM, et al. Epidemiologic typing of Enterobacter sakazakii in two neonatal nosocomial outbreaks. Diagn Microbiol Infect Dis. 1990;13:467–472. doi: 10.1016/0732-8893(90)90078-a. [DOI] [PubMed] [Google Scholar]
- 73.van Acker J, de Smet F, Muyldermans G, et al. Outbreak of necrotizing enterocolitis associated with Enterobacter sakazakii in powdered milk formula. J Clin Microbiol. 2001;39:293–297. doi: 10.1128/JCM.39.1.293-297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mullane N, Healy B, Meade J, et al. Dissemination of Cronobacter spp. (Enterobacter sakazakii) in a powdered milk protein manufacturing facility. Appl Environ Microbiol. 2008;74:5913–5917. doi: 10.1128/AEM.00745-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Torres-Chavolla E, Ramirez-Cerda E, Gutierrez-Rojo R. Isolation and identification of Enterobacter sakazakii in infant milk formulas. Foodborne Pathog Dis. 2007;4:164–168. doi: 10.1089/fpd.2006.0071. [DOI] [PubMed] [Google Scholar]
- 76.Ballinger JR, Sheldon KM, Boxen I, et al. Differences between accumulation of 99mTc-MIBI and 201Tl-thallous chloride in tumour cells: role of P-glycoprotein. J Nucl Med. 1995;39:122–128. [PubMed] [Google Scholar]
- 77.Adlerberth I, Wold AE. Establishment of the gut microbiota in Western infants. Acta Paediatr. 2009;98:229–238. doi: 10.1111/j.1651-2227.2008.01060.x. [DOI] [PubMed] [Google Scholar]
- 78.Weaver LT, Laker MF, Nelson R. Intestinal permeability in the newborn. Arch Dis Child. 1984;59:236–241. doi: 10.1136/adc.59.3.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Favier CF, Vaughan EE, De Vos WM, Akkermans AD. Molecular monitoring of succession of bacterial communities in human neonates. Appl Environ Microbiol. 2002;68:219–226. doi: 10.1128/AEM.68.1.219-226.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tapiainen T, Ylitalo S, Eerola E, Uhari M. Dynamics of gut colonization and source of intestinal flora in healthy newborn infants. APMIS. 2006;114:812–817. doi: 10.1111/j.1600-0463.2006.apm_488.x. [DOI] [PubMed] [Google Scholar]
- 81.Mahmood B, Daood MJ, Hart C, et al. Ontogeny of P-glycoprotein in mouse intestine, liver, and kidney. J Investig Med. 2001;49:250–257. doi: 10.2310/6650.2001.33969. [DOI] [PubMed] [Google Scholar]
- 82.Gribar JJ, Ramachandra M, Hrycyna CA, et al. Functional characterization of glycosylation-deficient human P-glycoprotein using a vaccinia virus expression system. J Membr Biol. 2000;173:203–214. doi: 10.1007/s002320001020. [DOI] [PubMed] [Google Scholar]