

Abstract
Background
We have shown that BNIP3 expression is significantly increased in HF. In this study, we tested the effects of BNIP3 manipulation in HF.
Methods and Results
In a rat model of pressure overload HF, BNIP3 knockdown significantly decreased LV volumes with significant improvement in LV diastolic and systolic function. There were significant decreases in myocardial apoptosis and LV interstitial fibrosis. Ultrastructurally, BNIP3 knockdown attenuated mitochondrial fragmentation and restored mitochondrial morphology and integrity. On the molecular level there were significant decreases in ER stress and mitochondrial apoptotic markers. One of the mechanisms by which BNIP3 mediates mitochondrial dysfunction is via the oligomerization of the VDAC channels causing a shift of calcium from the ER to mitochondrial compartments leading to the decrease in ER calcium content, mitochondrial damage, apoptosis and LV interstitial fibrosis and hence contributes to both systolic and diastolic myocardial dysfunction, respectively. In systolic HF, the downregulation of SERCA2a, along with an increased BNIP3 expression, further worsen myocardial diastolic and systolic function and contribute to the major remodeling seen in systolic HF as compared to diastolic HF with normal SERCA2a expression.
Conclusions
The increase in BNIP3 expression contributes mainly to myocardial diastolic dysfunction through mitochondrial apoptosis, LV interstitial fibrosis and to some extent to myocardial systolic dysfunction due to the shift of calcium from the ER to the mitochondria and to the decrease in ER calcium content. However, SERCA2a downregulation remains a prerequisite for the major LV remodeling seen in systolic HF.
Keywords: heart failure, hypertrophy, remodeling, apoptosis, gene therapy
In heart failure (HF), the crosstalk between the Endoplasmic-Sarcoplasmic reticulum ER/SR and the juxtaposed mitochondria is altered leading to the malfunction of the cardiomyocyte and to the decline in cardiac function. On the SR level, there is hyperphosphorylation of the ryanodine receptors (RYR), hypophosphorylation of phospholamban (PLN), downregulation and dysfunction of SERCA2a. These changes in the calcium cycling proteins lead to increases in SR Ca2+ release and to decreases in SR Ca2+ uptake and SR Ca2+ content. On the mitochondrial level there is an increase in the pro-apoptotic Bcl-2 and Bcl-2 like family proteins, such as Bax and BNIP3 respectively, in favor of the anti-apoptotic marker Bcl-2. BNIP3 is a mitochondrial death and mitophagy marker that has been shown to induce LV remodeling post myocardial infarction 1-9. In our previous study we highlighted the role of JNK in modulating FOXO3a for the expression of BNIP3 in pressure overload hypertrophy (POH) and in HF 10. This signaling pathway was further validated in human samples of HF 10. BNIP3 expression increased the first two weeks after POH and peaked at HF development 10. In this study, we show how BNIP3 knockdown, using tail vein injection of AAV9 Sh BNIP3, reversed cardiac remodeling and improved LV diastolic and systolic function in a pressure overload rat model of diastolic and systolic HF. Moreover, BNIP3 knockdown robustly attenuated LV apoptosis and interstitial fibrosis with major improvements of various cellular components, specifically with regard to mitochondrial morphology and integrity. Mechanistically, we find that BNIP3 exerts its destructive effects on the mitochondria via the oligomerization of the VDAC channels leading to mitochondrial Ca2+ overload, release of cytochrome C and mitochondrial apoptosis as shown below.
Methods
Isolation and Culture of Adult Rat Cardiomyocytes and In Vitro Experiments
Adult rat ventricular cardiomyocytes were isolated from male Sprague-Dawley rats weighing (250–350 g) as previously described 11, 12. The following experiments were performed (n=3 for each experiment in vitro): (1) cell viability, (2) mitochondrial membrane potential, (3) Immunofluorescence staining, (4) mitochondrial Ca2+ loading Supplement Figure 1 and (5) VDAC oligomerization. Details are in the supplement section.
Western Blotting
Please refer to the supplement section.
Co-Immunoprecipitation
The co-immunoprecipitation was performed using Pierce Classic IP Kit (Thermo scientific, Rockford, IL, USA). Details are in the supplement section.
Transmission Electron Microscopy
Samples were viewed under a transmission electron microscope (HITACHI H-7650, Japan). Details are in the supplement section.
Production of Recombinant Adenoviruses and Adeno-associated Virus (AAV)
Recombinant adenoviruses encoding green fluorescent protein (Ad-GFP) was prepared as previously described 13. Ad-BNIP3 and Ad-Sh BNIP3 were done at vector biolabs. We generated a recombinant cardiotropic adeno-associated virus of serotype 9 (AAV9), allowing for cardiomyocyte-targeted RNAi against BNIP3 (AAV9 Sh BNIP3) under control of the U6 promoter 14 Supplement Figure 2.
Apoptosis by Tunnel Staining
Tunnel staining was performed using the Apoptag red in situ detection kit (Millipore, Billerica, MA, USA). Details are in the supplement section.
Masson Trichrome Staining
Cryosections were stained using Masson’s Trichrome staining kit protocol (Sigma, St. Louis, MO, USA). Details are in the supplement section.
Measurement of Intracellular Calcium Kinetics and ER Calcium Content
Fura-2 AM (Molecular probes, Eugene, OR, USA) was used to detect intracellular Ca2+ concentration [Ca]i. Data are expressed as the 340/380 ratio following subtraction of background fluorescence. Details are in the supplement section.
Experimental Model of Ascending Aortic Banding, Cross Clamp with Ascending Aortic Banding and Study Design
All procedures involving the handling of animals were approved by the Animal Care and Use Committee of the Mount Sinai School of Medicine and adhered with the Guide for the Care and Use of laboratory Animals published by the National Institutes of Health. Sprague-Dawley rats weighing 180-200 g underwent ascending aortic banding (AAB), as previously described in detail 15. For the systolic HF model n=7 and for the diastolic HF model n=5 for each group. The cross clamp surgery with gene transfer and AAB was performed as previously described in detail 16, 17 (n=4 for each group). Details are in the supplement section.
Echocardiography
Transthoracic echocardiography was performed using a vivid 7 echocardiography apparatus with a 14 MHZ probe (i13L probe, General Electric, New York, NY). Details are in the supplement section.
Invasive Pressure-Volume Loop Measurements of the Left Ventricle
Hemodynamics were recorded using a Scisense P-V Control Unit (FY897B)18, 19,20. Details are in the supplement section.
Statistical Analysis
Results are shown as mean ± Standard Deviation. Statistical significance was determined using one-way ANOVA followed by Tukey. There was no adjustment for multiple comparisons across the variables being tested. For the pretreatment and one month after treatment data, mixed effect models with a random intercept; treatment, time and the interaction treatment * time were the predictors in the model. A significant interaction means that the change between before and after in either of the non-control groups was statistically different than the difference observed in the control group. A p-value of < 0.05 was considered statistically significant. The p-values presented in the figures are two sided.
Results
BNIP3 Overexpression Induces Mitophagy and Apoptosis in Cardiomyocytes In Vitro and In Vivo and Impairs Diastolic and Systolic Cardiac Function in a Rat Model of Early POH
BNIP3 localizes to the mitochondria in cardiomyocytes Supplement Figure 3-A. BNIP3 overexpressing cardiomyocytes showed significant increases in cell death, cytoplasmic cytochrome C and cleaved caspase 3 and significantly decreased mitochondrial membrane potential Supplement Figure 3 B-D.
Gene transfer of Ad-BNIP3 in a rat model of early POH was associated with a significantly lower body weight, LV weight, a significantly lower heart weight to body weight ratio and a significantly thinner septal and posterior wall, by echocardiography Figure 1 A-C and Supplement Table 1. There were significant increases in LV end systolic diameters and volumes and significant decreases in LV fractional shortening (LVFS) and LV ejection fraction (LVEF) in the Ad-BNIP3 group Figure 1 D-E. Baseline hemodynamics and tracings are shown in Supplement Table 2 and Figure 1-F, respectively. The Ad-BNIP3 group had significant increases in their LVEDP despite having a significantly lower maximum pressure and significantly decreased LVEF Figure 1 G-I.
Figure 1. BNIP3 overexpression increased cardiomyocyte death in vitro and impaired LV systolic and diastolic function in a rat model of early POH.
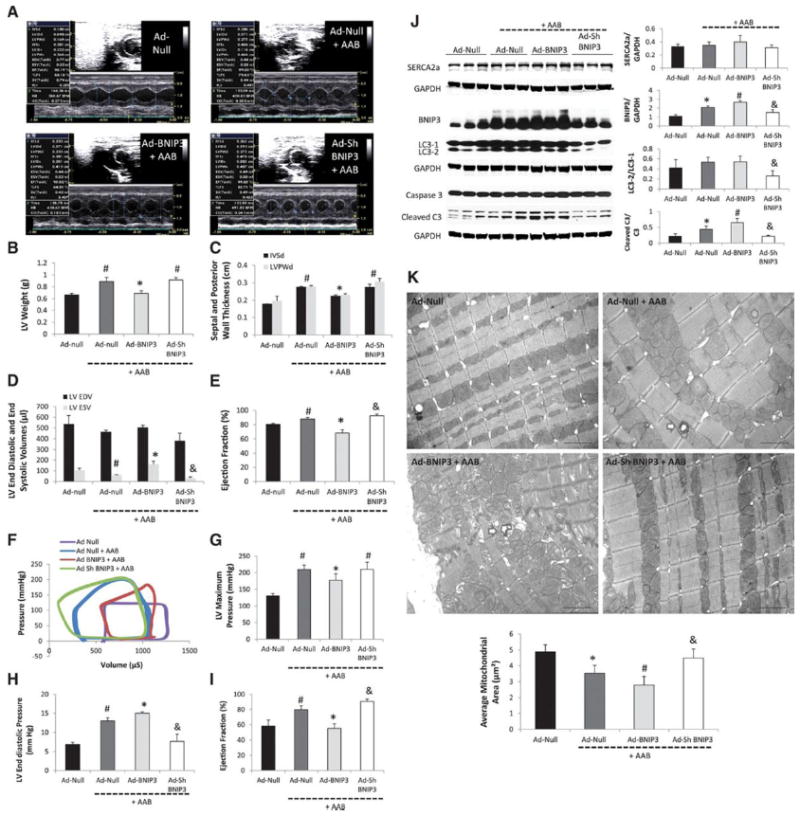
1 A-E: M-mode images two weeks after gene delivery via a cross clamp technique and AAB. There were significant decreases in LV weight, IVSd, LVPWd and LVEF and significant increase in LV end systolic volume in the Ad-BNIP3 + AAB group, *P<0.05 vs Ad-Null + AAB and Ad-Sh BNIP3 + AAB, #P<0.05 vs Ad-Null. There was slight but significant decrease in LV end systolic volume and increase in LVEF in the Ad-Sh BNIP3 + AAB group, &P<0.05 vs Ad-Null + AAB. 1 F-I: Pressure-volume loop measurements showed significant decreases in LV maximum pressure and LVEF and significant increases in LVEDP in the Ad-BNIP3 + AAB group, *P<0.05 vs Ad-Null + AAB and Ad-Sh BNIP3 + AAB, #P<0.05 vs Ad-Null. There was significant decrease in LVEDP and a slight but significant increase in LVEF in the Ad-Sh BNIP3 + AAB group, &P<0.05 vs Ad-Null + AAB. 1-J: BNIP3 expression as well as cleaved caspase 3 significantly increased in the Ad-Null + AAB and was the highest in Ad-BNIP3 + AAB, *P<0.05 vs Ad-Null, #P<0.05 vs all other groups. BNIP3 knockdown significantly attenuated the increase in BNIP3 expression, cleaved caspase 3 and the conversion of LC3-1 to LC3-2 in response to PO, &P<0.05 vs Ad-Null + AAB and Ad-BNIP3 + AAB. There was no difference in SERCA2a expression in all groups. 1-K: Ultrastructurally, there was significant decrease in mitochondrial area two weeks after PO, *P<0.05 vs Ad-Null and was the worst in Ad-BNIP3 group with significant mitochondrial fragmentation, cristae destruction and myofibrillar damage, #P<0.05 vs all other groups. BNIP3 knockdown prevented mitochondrial damage in PO, &P<0.05 vs Ad-Null + AAB and Ad-BNIP3 + AAB. Arrows are showing autophagosomes. Images 5,000X magnified, scale bar 2μm.
On the molecular level, there was no difference in SERCA2a expression between all groups. BNIP3 expression significantly increased in early POH (Ad-Null + AAB) and was further increased in the Ad-BNIP3 group Figure 1-J. The conversion of LC3-1 to LC3-2 and cleaved caspase 3 were significantly decreased in the Ad-Sh BNIP3 group Figure 1-J. Ultrastructurally, BNIP3 overexpression in PO accentuated mitochondrial fragmentation and cristae destruction. BNIP3 knockdown robustly attenuated mitochondrial fragmentation and the presence of autophagosomes in PO Figure 1-K. Of note there are differences in each group in terms of mitochondria size which may be due to the inhomogeneous nature of the gene transfer.
Therapeutic Gene Delivery of AAV9-Sh BNIP3 Reverses Cardiac Remodeling and Improves LV Diastolic and Systolic Function in a Pressure Overload Induced HF Rat Model of Diastolic and Systolic Dysfunction
To examine the effects of knocking down BNIP3 in a chronic model of HF, we used AAV vectors that afford us long-term expression. Echocardiography data of the diastolic heart failure with preserved ejection fraction animals are shown in the Supplement Table 3. M-mode images are shown in Figure 2-A. There were no significant differences in echocardiographic parameters between the Sh Luc and Sh BNIP3 groups before treatment. One month after treatment, there was significant decrease in LV end systolic diameter and significant increase in LVFS in the AAV9 Sh BNIP3 group. LV end diastolic and end systolic volumes were significantly decreased due to the significant decrease in LV length in diastole and systole and reflected a significant increase in LVEF Figure 2 B-C and Supplement Figure 4. Hemodynamic data are shown in the Supplement Table 4 and tracings are shown in Figure 2-D. There were significant decreases in LVEDP and EDPVR and significant increases in LV ejection fraction and ESPVR in the AAV9 Sh BNIP3 group Figure 2 E-H. Western blotting of LV tissue lysate showed significant decreases in BNIP3 expression as well as in Bax to Bcl-2 ratio, ER stress maker (p-eIF2a) and ER stress apoptotic marker (CHOP) and cleaved caspase 3 in the AAV9 Sh BNIP3 group Figure 2-I. However, there was no difference in SERCA2a expression between the AAV9 Sh Luc and AAV9 Sh BNIP3 groups, respectively Figure 2-I. Myocardial apoptosis and interstitial fibrosis were significantly decreased in the AAV9 Sh BNIP3 group Figure 2 J-K. Ultrastructurally, autophagosomes, mitochondrial fragmentation and myofibrillar damage were noted in the AAV9 Sh Luc group. BNIP3 knockdown robustly improved the aforementioned ultrastructural findings Figure 2-L and Supplement Figure 5. Similar results were noted for the systolic HF animals treated with AAV9 Sh Luc vs AAV9 Sh BNIP3 except for that the systolic HF animals had much higher LV volumes and significantly lower SERCA2a expression compared to sham operated animals Figure 3-I. Echocardiography and hemodynamic data are presented in Figure 3 A-H and Supplement Figure 6 and Supplement Tables 5-6. Myocardial apoptosis, interstitial fibrosis and ultrastructure data are presented in Figure 3 J-L and Supplement Figure 7.
Figure 2. Tail vein delivery of 5E10 vg/ml of AAV9 Sh BNIP3 reversed cardiac remodeling and improved LV diastolic function and contractility in a rat model of diastolic HF with preserved EF.
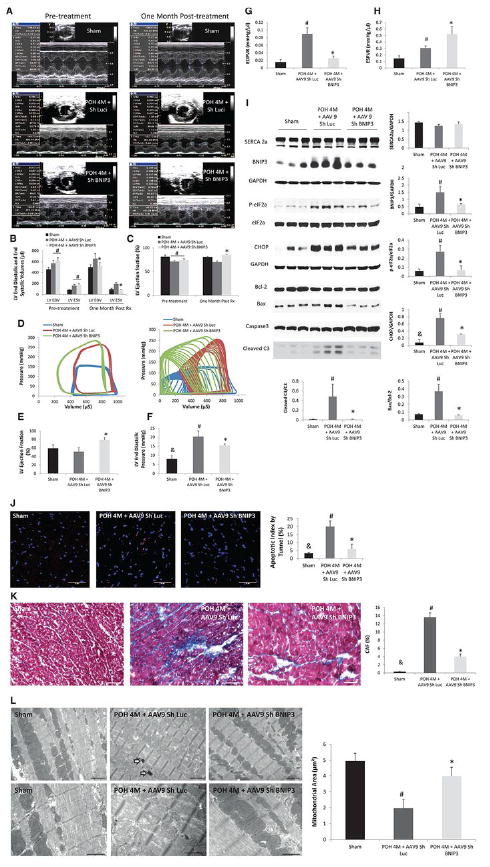
2 A-C: M-mode images of the above group of animals before and one month after treatment with AAV9 Sh Luc vs AAV9 Sh BNIP3. LV volumes were significantly decreased and the LVEF was significantly increased one month after treatment with AAV9 Sh BNIP3, *P<0.05 vs POH 4M + AAV9 Sh Luc, #P<0.05 vs sham. 2-D: P-V loop tracings in the different groups at baseline and during inferior vena cava occlusion. 2-E: LVEF significantly improved one month after Sh BNIP3 treatment, *P<0.05 vs other two groups. 2 F-G: LVEDP and EDPVR were significantly increased in the Sh Luc group, #P<0.05 vs sham, &P<0.05 vs other two groups. Those parameters were significantly decreased one month after Sh BNIP3 treatment, *P<0.05 vs POH 4M + AAV9 Sh Luc. 2-H: LV contractility significantly increased in the Sh BNIP3 group, *P<0.05 vs POH 4M + AAV9 Sh Luc. Note that the Sh Luc group has a falsely increased ESPVR compared to sham animals as their LVESP are higher, but their V0 is significantly shifted to the right as compared to the sham group. 2-I: Western blot analysis of LV tissue lysate showed robust decrease in BNIP3 expression, ER stress (p-eIF2a) and ER stress apoptotic marker (CHOP) as well as in Bax to Bcl-2 ratio and in cleaved caspase 3 in the Sh BNIP3 group, P<0.05 vs POH 4M + AAV9 Sh Luc, #P<0.05 vs sham and &P<0.05 vs other two groups. There was no difference in SERCA2a expression between all groups. 2 J-K: There were robust decreases in myocardial apoptosis and in LV interstitial fibrosis in the Sh BNIP3 group, *P<0.05 vs POH 4M + AAV9 Sh Luc, #P<0.05 vs sham and &P<0.05 vs other two groups. 2-L: Ultrastructurally, there were significant mitochondrial fragmentation and cristae destruction with dilated T tubules and damaged myofibrils. BNIP3 knockdown robustly attenuated mitochondrial fragmentation and cristae destruction with the mitochondrial area almost back to control level and attenuated myofibrillar damage, #P<0.05 vs sham and *P<0.05 vs POH 4M + AAV9 Sh Luc. Arrows showing the presence of autophagosomes. Above images are 5,000X magnified, scale bar 2μm. Lower images are 12,000X magnified, scale bar 1μm.
Figure 3. Tail vein delivery of 5E10 vg/ml of AAV9 Sh BNIP3 reversed cardiac remodeling and improved LV diastolic function and contractility in a rat model of systolic HF.
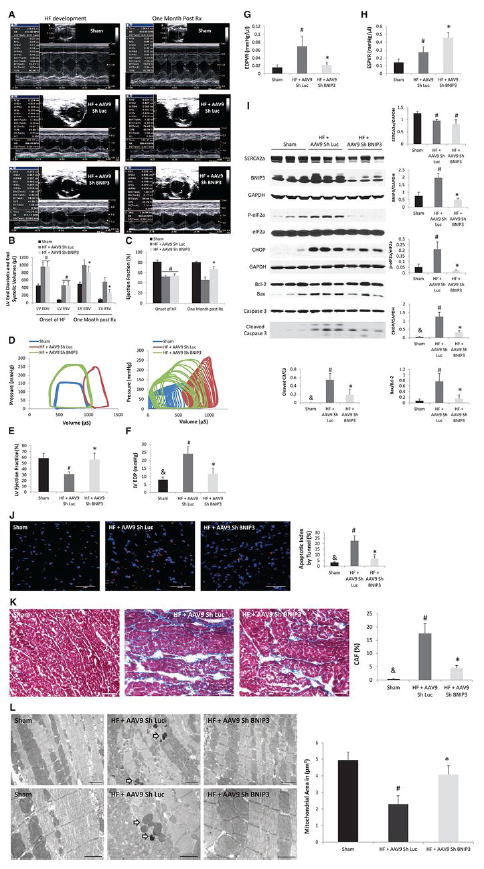
3 A-C: M-mode images of the above group of animals before and one month after treatment with AAV9 Sh Luc vs AAV9 Sh BNIP3. LV volumes were significantly decreased and the LVEF was significantly increased one month after treatment with AAV9 Sh BNIP3, *P<0.05 vs HF + AAV9 Sh Luc, #P<0.05 vs sham. 3-D: P-V loop tracings in the different groups at baseline and during inferior vena cava occlusion. 3-E: LVEF significantly improved one month after Sh BNIP3 treatment, *P<0.05 vs HF + AAV9 Sh Luc and #P<0.05 vs sham. 3 F-G: LVEDP and EDPVR were significantly increased in the Sh Luc group, #P<0.05 vs sham, &P<0.05 vs other two groups. Those parameters were significantly decreased one month after Sh BNIP3 treatment, *P<0.05 vs HF + AAV9 Sh Luc. 3-H: LV contractility significantly increased in the Sh BNIP3 group, *P<0.05 vs HF + AAV9 Sh Luc. Note that the Sh Luc group has a falsely increased ESPVR compared to sham animals as their LVESP are higher, but their V0 is significantly shifted to the right as compared to the sham group. 3-I: Western blot analysis of LV tissue lysate showed robust decrease in BNIP3 expression, ER stress (p-eIF2a) and ER stress apoptotic marker (CHOP) as well as in Bax to Bcl-2 ratio and in cleaved caspase 3 in the Sh BNIP3 group, P<0.05 vs HF + AAV9 Sh Luc, #P<0.05 vs sham and &P<0.05 vs other two groups. There was significant decrease in SERCA2a expression in the HF groups, #P<0.05 vs sham. 3 J-K: There were robust decreases in myocardial apoptosis and in LV interstitial fibrosis in the Sh BNIP3 group, *P<0.05 vs HF + AAV9 Sh Luc, #P<0.05 vs sham and &P<0.05 vs other two groups. 3-L: Ultrastructurally, there was significant mitochondrial fragmentation and cristae destruction with dilated T tubules and damaged myofibrils. BNIP3 knockdown robustly attenuated mitochondrial fragmentation and cristae destruction with the mitochondrial area almost back to control level and attenuated myofibrillar damage, #P<0.05 vs sham and *P<0.05 vs HF + AAV9 Sh Luc. Arrows showing the presence of autophagosomes. Above images are 5,000X magnified, scale bar 2μm. Lower images are 12,000X magnified, scale bar 1μm.
The Increase in BNIP3 Expression Decreases ER Calcium and Increases Mitochondrial Calcium
BNIP3 overexpressing cardiomyocytes had significantly decreased beat-to-beat Ca2+ release due to the significant decrease in ER Ca2+ with marked impairment in cardiomyocyte relaxation and threefold increase in mitochondrial Ca2+, Figure 4 A-B. Hypertrophic cardiomyocytes showed significant increase in beat-to-beat Ca2+ release and no change in ER Ca2+ content two weeks after PO. However, five weeks after PO, beat-to-beat Ca2+ transients were significantly decreased due to a significant decrease in ER Ca2+ content. Beat-to-beat Ca2+ transients and ER Ca2+ content were significantly higher and mitochondrial Ca2+ was significantly lower in hypertrophic cardiomyocytes isolated from animals five weeks after PO treated with AAB and tail vein injection of AAV9 Sh BNIP3 Figure 4 C-D.
Figure 4. BNIP3 regulates ER and mitochondrial calcium homeostasis.
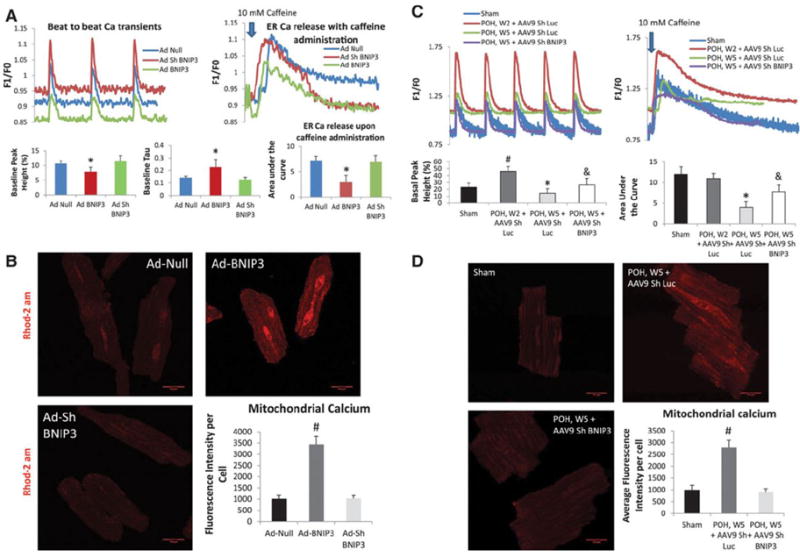
4-A: BNIP3 overexpression in normal cardiomyocytes significantly decreased beat-to-beat Ca2+ release and ER Ca2+ content with significant increase in Tau, *P<0.05 vs Ad-Null and Ad-Sh BNIP3. There were no differences between the Ad-Null and Ad-Sh BNIP3 treated cardiomyocytes. 4-B: There was threefold increase in mitochondrial Ca2+ in the Ad-BNIP3 group, *P<0.05 vs Ad-Null and Ad-Sh BNIP3. 4-C: In hypertrophic cardiomyocytes, there is robust increase in beat-to-beat Ca2+ release with no change in ER Ca2+ content two weeks after PO, #P<0.05 vs sham. However, five weeks after PO beat-to-beat Ca2+ transients are significantly decreased due to a significant decrease in ER Ca2+ content, *P<0.05 vs all other groups. BNIP3 knockdown significantly increased beat to beat Ca2+ transients and ER Ca2+ content in hypertrophic cardiomyocytes five weeks after PO, &P<0.05 vs POH, W5 + AAV9 Sh Luc 4-D: Mitochondrial Ca2+ significantly increased in hypertrophic cardiomyocytes five weeks after PO, #P<0.05 vs other two groups.
BNIP3 Regulates Mitochondrial Calcium via the Modulation of the VDAC Channels
Co- immunoprecipitation did not show a complex formation between BNIP3 and VDAC channels Figure 5-A. There was modulation of the VDAC channels by BNIP3. The increase in BNIP3 expression increases the oligomerization of the VDAC channels, mainly in the form of a dimer, with and without the presence of a cross linking reagent (EGS) leading to the increase in mitochondrial Ca2+ and to cytochrome C release Figure 5-B. DIDS, an anion channel inhibitor, inhibited the oligomerization of the VDAC channels and significantly attenuated mitochondrial Ca2+ and cell death in Ad-BNIP3 infected cardiomyocytes Figure 5 C-E. Ultrastructurally, DIDS significantly attenuated mitochondrial fragmentation and autophagosome formation in Ad-BNIP3 infected cardiomyocytes. Overall, the mitochondrial area was significantly higher in cardiomyocytes treated with DIDS Figure 5-F and Supplement Figure 8.
Figure 5. BNIP3 modulates the VDAC channels and shifts the Ca from the ER into the mitochondria.
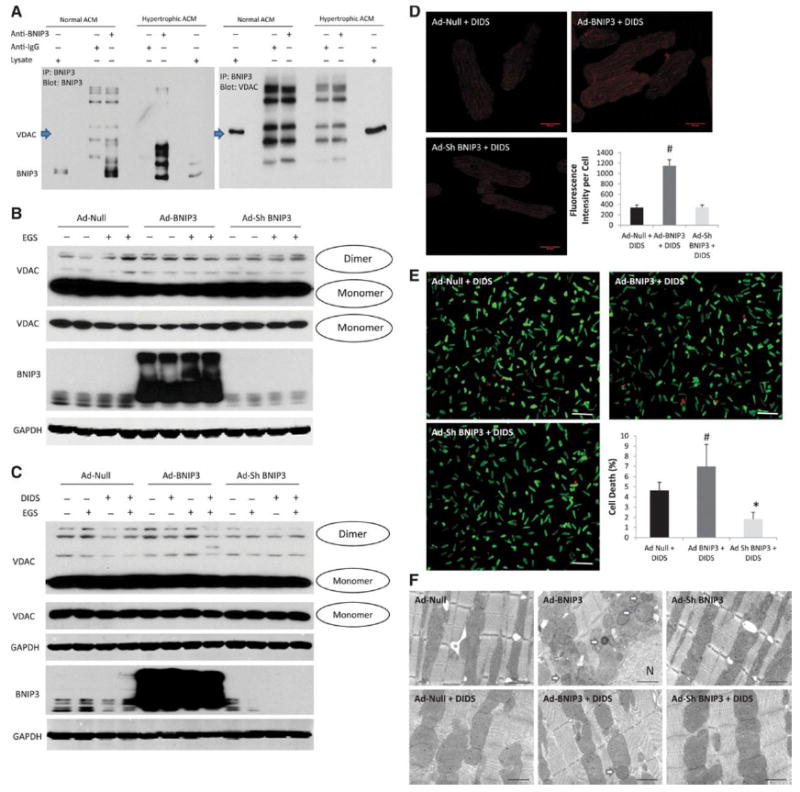
5-A: Co-immunoprecipitation did not show that BNIP3 and VDAC channels form a complex. 5-B: Rather there is modulation of the VDAC channels by BNIP3. The increase in BNIP3 expression causes oligomerization of the VDAC channels, mainly in the form of a dimer, with and without the presence of a cross linking reagent (EGS) leading to the increase in mitochondrial Ca2+. 5 C-D: DIDS, an anion channel inhibitor, inhibited the oligomerization of the VDAC channels in Ad-BNIP3 infected cardiomyocytes. There was threefold decrease in mitochondrial Ca2+ in all groups with significant decrease in the Ad-BNIP3 infected cardiomyocytes treated with DIDS, #P<0.05 vs Ad-Null + DIDS and Ad-Sh BNIP3 + DIDS. 5-E DIDS significantly attenuated cell death in Ad-BNIP3 infected cardiomyocytes, #P<0.05 vs Ad-Null + DIDS and *P<0.05 vs other two groups. 5-F: Ultrastructurally, DIDS significantly attenuated mitochondrial fragmentation and autophagosome formation in Ad-BNIP3 infected cardiomyocytes. The mitochondrial area was significantly higher in cardiomyocytes treated with DIDS compared to no DIDS treatment. Images 12,000X magnified, scale bar 1 μm.
The Expression of a Constitutively Active FOXO3a Increases Cell Death In Vitro and Impairs Cardiac Diastolic and Systolic Function in a Rat Model of Early POH
Cardiomyocytes overexpressing constitutively active FOXO3a (Ad-FX3) showed significant increases in BNIP3 expression10 and fourfold increases in cell death Supplement Figure 9. Gene transfer of Ad-FX3 in a rat model of early POH was associated with a significantly lower body weight, LV weight, a significantly lower heart weight to body weight ratio and a significantly thinner septal and posterior wall by echocardiography Figure 6 A-C and Supplement Table 7. There were significant increases in LV end systolic diameters and volumes and subsequently reflected significant decreases in LVFS and LVEF in the Ad-FX3 + AAB group Figure 6 D-E. Baseline hemodynamics and tracing are shown in Supplement Table 8 and Figure 6-F, respectively. The Ad-FX3 + AAB group had significant increase in their LV end diastolic pressures (LVEDP) despite having a significantly lower maximum pressure and significant decrease in LVEF Figure 6 G-I. On the molecular level, Ad-DN-FX3 significantly attenuated the increase in BNIP3 expression in response to PO Figure 6-J.
Figure 6. The expression of a constitutively active FOXO3a impairs cardiac diastolic and systolic function in a rat model of early POH.
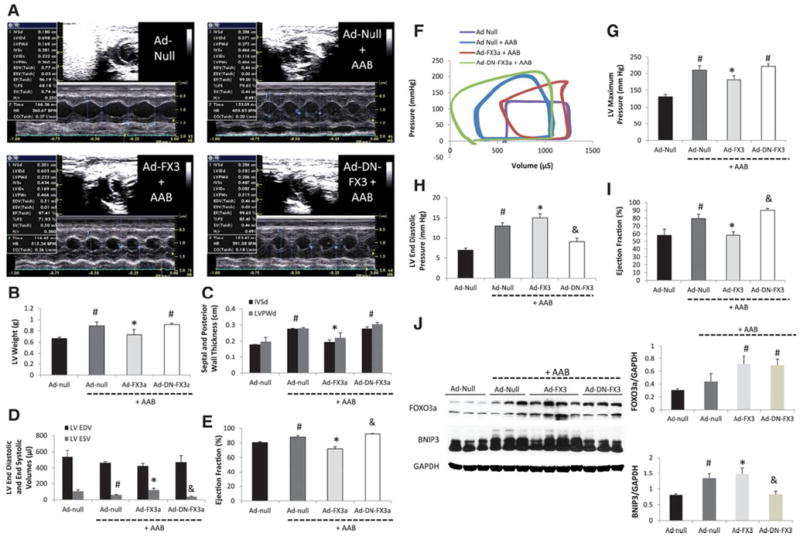
6-A: M-mode images two weeks after gene delivery via a cross clamp technique and AAB. 6 B-E: There were significant decreases in LV weight, IVSd, LVPWd and LV EF and significant increase in LV end systolic volume in the Ad-FX3a + AAB group, *P<0.05 vs Ad-Null + AAB and Ad-DN-FX3a + AAB, #P<0.05 vs Ad-Null. There was slight but significant decrease in LV end systolic volume and increase in LVEF in the Ad-DN-FX3 + AAB group, &P<0.05 vs Ad-Null + AAB. 6 F-I: Pressure-volume loop measurements showed significant decreases in LV maximum pressure and EF and significant increases in LVEDP in the Ad-FX3a + AAB group, *P<0.05 vs Ad-Null + AAB and Ad-DN-FX3a + AAB, #P<0.05 vs Ad-Null. There was significant decrease in LVEDP and a slight but significant increase in LVEF in the Ad-DN-FX3a + AAB group, &P<0.05 vs Ad-Null + AAB. 6-J: FOXO3a expression increased in the Ad-FX3a and Ad-DN-FX3a groups, #P<0.05 vs Ad-Null and Ad-Null + AAB. BNIP3 expression increased in the Ad-Null + AAB and in the Ad-FX3a + AAB groups, #P<0.05 vs Ad-Null and *P<0.05 vs all other groups. The delivery of Ad-DN-FX3a significantly attenuated the increase in BNIP3 expression in response to PO, &P<0.05 vs Ad-Null + AAB and Ad-FX3a + AAB.
Discussion
BNIP3 is a mitochondrial death and mitophagy marker located at the outer mitochondrial membrane (OMM) with the N terminus oriented into the cytoplasm and the C terminus inside the mitochondria. It has been shown that an increase in BNIP3 expression under hypoxemic conditions increases cell death and mitochondrial autophagy and attributes to LV remodeling and myocardial apoptosis post myocardial infarction 1-4, 6-9, 21. Also, it has been shown how BNIP3 and NIX play role in ER/SR-mitochondrial Ca2+ homeostasis 22, 23. In our previous study, we have shown that BNIP3 expression increases in POH and peaks at the time of HF development and highlighted the critical role of JNK in the modulation of FOXO3a for the expression of BNIP3. We also validated the JNK-FOXO3a-BNIP3 pathway in human samples of HF 10. In this study we show the critical role of BNIP3 in inducing LV remodeling and myocardial stiffness in PO rat model of HF and we highlight a novel mechanism through which the chronic increase in BNIP3 expression induces mitochondrial damage, cytochrome C release, apoptosis and thus myocardial fibrosis. In cardiac myocytes, BNIP3 is the effector of the transcription factor FOXO3a. The expression of constitutively active FOXO3a in cardiac myocytes, in vitro, was associated with significant increase in BNIP3 expression and cell death 10. Gene transfer of constitutively active FOXO3a in a rat model of POH was associated with worsening in LV diastolic function and a decline in LV systolic function. Similar results were noted with the overexpression of BNIP3 in a rat model of early POH. BNIP3 knockdown by gene transfer of Ad-Sh BNIP3 robustly improved diastolic function and LV relaxation and significantly attenuated mitochondrial fragmentation in POH. Moreover, BNIP3 knockdown in two models of HF, a diastolic and a systolic dysfunction, was associated with significant decreases in LV end diastolic and LV end systolic volumes, myocardial apoptosis and interstitial fibrosis and a significant increase in LVEF. Hemodynamically, there were significant improvements in LV relaxation and contractility. Ultrastructurally, BNIP3 knockdown in HF, markedly improved mitochondrial morphology and integrity with significant decrease in autophagosomes formation. From the results listed above, it seems that the chronic increase in BNIP3 expression mainly contributes to the diastolic dysfunction with moderate increases in LV volumes and decline in LVEF as seen in the HF model of diastolic dysfunction. However, the full-blown dilatation and remodeling of the LV chamber with marked decline in LVEF is only seen when SERCA2a expression is downregulated along with an abnormally increased BNIP3 expression. This contributes to the significant decline in LV systolic function, fluid retention and volume overload with major increases in LV end diastolic and end systolic volumes. We find that one of the mechanisms by which BNIP3 exerts these deleterious effects on the mitochondria is via the modulation of the VDAC channels. The increase in BNIP3 expression enhances the oligomerization of the VDAC channels shifting Ca2+ from the ER into the mitochondria 24-27. DIDS, an anion channel inhibitor, attenuated VDAC oligomerization, mitochondrial damage and cell death in BNIP3 overexpressing cardiomyocytes. The decrease in ER Ca2+ content creates an ER stress condition with significant increases in ER stress and ER stress apoptotic markers, p-eIF2a and CHOP, respectively. On the other hand, the increase in mitochondrial Ca2+ leads to their fragmentation, cristae destruction and to the release of cytochrome C, either from the mPTP or from the oligomers of the VDAC channels27, leading to the increase in myocardial apoptosis and LV interstitial fibrosis. Hence the stiffness of the myocardium seen with the increase in BNIP3 expression is a multifactorial process and to a large scale is clearly linked to mitochondrial calcium overload and decline in cardiac energetics, SERCA2a malfunction and to the increase in LV apoptosis and interstitial fibrosis. SERCA2a downregulation worsens the deleterious effect exerted by BNIP3 on the mitochondria as well as ER stress and becomes the core of two vicious cycles as shown in Figure 7. SERCA2a overexpression in systolic HF improved LV systolic function and attenuated BNIP3 mediated mitochondrial destruction and ER stress by shuffling the Ca2+ from the cytoplasm into the ER Supplement Figure 10. The simultaneous overexpression of SERCA2a and BNIP3 in cardiomyocytes in vitro attenuated mitochondrial fragmentation compared to BNIP3 overexpression alone. However, DIDS was superior in preventing BNIP3 induced mitochondrial destruction in BNIP3 overexpressing cardiomyocytes. Morphologically, the largest mitochondria were seen in cardiomyocytes in vitro by simultaneous overexpression of SERCA2a and BNIP3 knockdown and with BNIP3 knockdown plus DIDS treatment, Supplement Figure 10.
Figure 7. Schematic illustration of the mechanism by which BNIP3 induces mitochondrial damage and cell death.
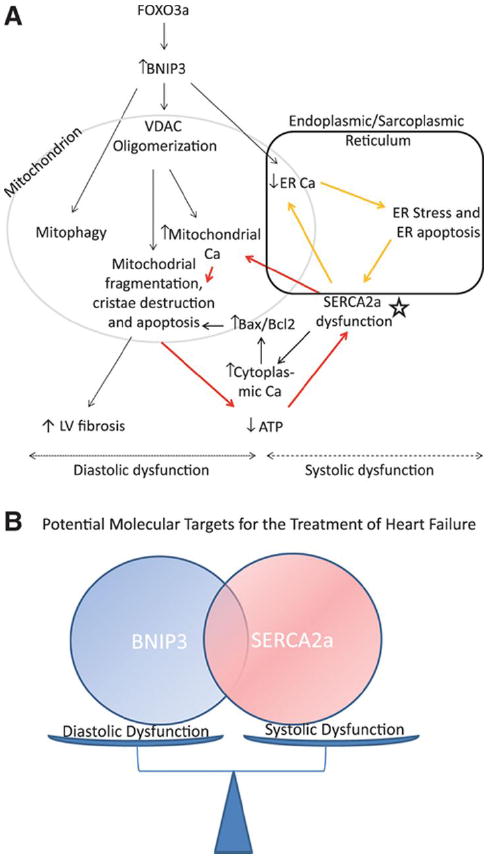
7-A: The increase in BNIP3 expression leads to the oligomerization of the VDAC channels causing the shift in ER Ca2+ towards the mitochondria. This shift of ER Ca2+ has two sequelae, the first being the increase in mitochondrial Ca2+ and the second is the decrease in ER Ca2+ content. The increase in mitochondrial Ca2+ leads to mitochondrial fragmentation, mitophagy and apoptosis, decline in cardiac energetics and LV interstitial fibrosis (diastolic dysfunction). Whereas, the decrease in ER Ca2+ leads to ER stress and ER stress induced apoptosis (systolic dysfunction). In systolic HF, SERCA2a downregulation (star), which is an independent process from the increase in BNIP3 expression, contributes to the further decline in ER Ca2+ and to the further increase in mitochondrial Ca2+ leading to two vicious cycles and hence becomes the core of these two cycles as shown in the figure above (red and yellow arrows). 7-B: Cartoon highlighting the interplay between BNIP3 and SERCA2a in modulating diastolic and systolic function of the cardiomyocyte, respectively.
In conclusion, the increase in BNIP3 expression, which is highest in HF, primarily contributes to diastolic dysfunction through the intracellular destruction exerted by BNIP3 on the mitochondria leading to mitophagy, apoptosis, decline in cardiac energetics and myocardial fibrosis and stiffness and to the systolic dysfunction via the ER-mitochondria Ca2+ shift and decline in ER Ca2+ content. However, the major remodeling of the LV chamber, which is seen in systolic HF, takes place when SERCA2a expression is downregulated, which along with the increase in BNIP3 expression contribute to the systolic and diastolic dysfunction of the cardiomyocyte with significant increases in LV end-diastolic and end-systolic volumes and significant decline in LV contractile function and LVEF.
Supplementary Material
Clinical Perspective.
Heart failure (HF), is a syndrome with complex pathophysiological disturbances and with major alterations in myocardial signaling pathways leading to changes in calcium handling proteins, mitochondrial dysfunction and heightened levels of myocardial apoptotic and autophagic cell death. Moreover, although there is a clear mortality benefit with beta-blockers, angiotensin converting enzyme inhibitors and mineralocorticoid receptor antagonists in systolic HF, no therapies have shown promise in treating diastolic HF with preserved ejection fraction in different randomized clinical trials. In this study we showed that the increase in BNIP3 expression correlated with diastolic dysfunction, mitochondrial apoptosis and autophagy that were evident as early as two weeks after pressure overload hypertrophy. Those parameters worsened with BNIP3 overexpression, in vivo, and peaked at HF development, whether diastolic or systolic. The downregulation of SERCA2a contributed to worsening in LV systolic function and cardiac remodeling leading to systolic heart failure. BNIP3 knockdown in HF robustly improved LV end diastolic pressure, myocardial relaxation, myocardial contractility and cardiac remodeling and significantly decreased myocardial apoptosis and LV interstitial fibrosis. The effects of increased BNIP3 expression on cardiomyocyte diastolic dysfunction is mediated by the calcium shift from the endoplasmic reticulum to the mitochondria leading to mitochondrial calcium overload, mitochondrial dysfunction and decline in cardiac energetics. This study highlights a novel role of BNIP3 as a potential therapeutic target for the treatment of diastolic HF.
Acknowledgments
Statistical revision was conducted via the support of NIH grant UL1TR000067 by Dr. Bagiella Emilia.
Sources of Funding
This work is supported by NIH R01 HL083156, HL080498, HL093183, and P20HL100396 to (R. J. H.) and by a 2011 Research Fellowship Award from the Heart Failure Society of America to (A.H.C).
Footnotes
Disclosures
A patent application has been submitted for this work.
References
- 1.Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, Li H, Kirshenbaum LA, Hahn HS, Robbins J, Jones WK, Dorn GW. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of bnip3 restrains postinfarction remodeling in mice. J Clin Invest. 2007;117:2825–2833. doi: 10.1172/JCI32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dorn GW., 2nd Mitochondrial pruning by nix and bnip3: An essential function for cardiac-expressed death factors. J Cardiovasc Transl Res. 2010;3:374–383. doi: 10.1007/s12265-010-9174-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gang H, Hai Y, Dhingra R, Gordon JW, Yurkova N, Aviv Y, Li H, Aguilar F, Marshall A, Leygue E, Kirshenbaum LA. A novel hypoxia-inducible spliced variant of mitochondrial death gene bnip3 promotes survival of ventricular myocytes. Circ Res. 2011;108:1084–1092. doi: 10.1161/CIRCRESAHA.110.238709. [DOI] [PubMed] [Google Scholar]
- 4.Kubli DA, Quinsay MN, Huang C, Lee Y, Gustafsson AB. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2008;295:H2025–2031. doi: 10.1152/ajpheart.00552.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Landes T, Emorine LJ, Courilleau D, Rojo M, Belenguer P, Arnaune-Pelloquin L. The bh3-only bnip3 binds to the dynamin opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep. 2010;11:459–465. doi: 10.1038/embor.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quinsay MN, Lee Y, Rikka S, Sayen MR, Molkentin JD, Gottlieb RA, Gustafsson AB. Bnip3 mediates permeabilization of mitochondria and release of cytochrome c via a novel mechanism. J Mol Cell Cardiol. 2010;48:1146–1156. doi: 10.1016/j.yjmcc.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quinsay MN, Thomas RL, Lee Y, Gustafsson AB. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy. 2010;6:17–24. doi: 10.4161/auto.6.7.13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Regula KM, Ens K, Kirshenbaum LA. Inducible expression of bnip3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res. 2002;91:226–231. doi: 10.1161/01.res.0000029232.42227.16. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J, Ney PA. Role of bnip3 and nix in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–946. doi: 10.1038/cdd.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaanine AH, Jeong D, Liang L, Chemaly ER, Fish K, Gordon RE, Hajjar RJ. Jnk modulates foxo3a for the expression of the mitochondrial death and mitophagy marker bnip3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012;3:265. doi: 10.1038/cddis.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piper HM, Probst I, Schwartz P, Hutter FJ, Spieckermann PG. Culturing of calcium stable adult cardiac myocytes. J Mol Cell Cardiol. 1982;14:397–412. doi: 10.1016/0022-2828(82)90171-7. [DOI] [PubMed] [Google Scholar]
- 12.Wold LE, Ren J. Mechanical measurement of contractile function of isolated ventricular myocytes. Methods Mol Med. 2007;139:263–270. doi: 10.1007/978-1-59745-571-8_17. [DOI] [PubMed] [Google Scholar]
- 13.Kim M, Oh JK, Sakata S, Liang I, Park W, Hajjar RJ, Lebeche D. Role of resistin in cardiac contractility and hypertrophy. J Mol Cell Cardiol. 2008;45:270–280. doi: 10.1016/j.yjmcc.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suckau L, Fechner H, Chemaly E, Krohn S, Hadri L, Kockskamper J, Westermann D, Bisping E, Ly H, Wang X, Kawase Y, Chen J, Liang L, Sipo I, Vetter R, Weger S, Kurreck J, Erdmann V, Tschope C, Pieske B, Lebeche D, Schultheiss HP, Hajjar RJ, Poller WC. Long-term cardiac-targeted rna interference for the treatment of heart failure restores cardiac function and reduces pathological hypertrophy. Circulation. 2009;119:1241–1252. doi: 10.1161/CIRCULATIONAHA.108.783852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Del Monte F, Butler K, Boecker W, Gwathmey JK, Hajjar RJ. Novel technique of aortic banding followed by gene transfer during hypertrophy and heart failure. Physiol Genomics. 2002;9:49–56. doi: 10.1152/physiolgenomics.00035.2001. [DOI] [PubMed] [Google Scholar]
- 16.Hajjar RJ, Schmidt U, Matsui T, Guerrero JL, Lee KH, Gwathmey JK, Dec GW, Semigran MJ, Rosenzweig A. Modulation of ventricular function through gene transfer in vivo. Proc Natl Acad Sci U S A. 1998;95:5251–5256. doi: 10.1073/pnas.95.9.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.del Monte F, Hajjar RJ. Efficient viral gene transfer to rodent hearts in vivo. Methods Mol Biol. 2003;219:179–193. doi: 10.1385/1-59259-350-x:179. [DOI] [PubMed] [Google Scholar]
- 18.Pacher P, Nagayama T, Mukhopadhyay P, Batkai S, Kass DA. Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nat Protoc. 2008;3:1422–1434. doi: 10.1038/nprot.2008.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Porterfield JE, Kottam AT, Raghavan K, Escobedo D, Jenkins JT, Larson ER, Trevino RJ, Valvano JW, Pearce JA, Feldman MD. Dynamic correction for parallel conductance, gp, and gain factor, alpha, in invasive murine left ventricular volume measurements. J Appl Physiol. 2009;107:1693–1703. doi: 10.1152/japplphysiol.91322.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baan J, van der Velde ET, de Bruin HG, Smeenk GJ, Koops J, van Dijk AD, Temmerman D, Senden J, Buis B. Continuous measurement of left ventricular volume in animals and humans by conductance catheter. Circulation. 1984;70:812–823. doi: 10.1161/01.cir.70.5.812. [DOI] [PubMed] [Google Scholar]
- 21.Gustafsson AB. Bnip3 as a dual regulator of mitochondrial turnover and cell death in the myocardium. Pediatric cardiology. 2011;32:267–274. doi: 10.1007/s00246-010-9876-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diwan A, Matkovich SJ, Yuan Q, Zhao W, Yatani A, Brown JH, Molkentin JD, Kranias EG, Dorn GW., 2nd Endoplasmic reticulum-mitochondria crosstalk in nix-mediated murine cell death. J Clin Invest. 2009;119:203–212. doi: 10.1172/JCI36445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L, Li L, Liu H, Borowitz JL, Isom GE. Bnip3 mediates cell death by different pathways following localization to endoplasmic reticulum and mitochondrion. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2009;23:3405–3414. doi: 10.1096/fj.08-124354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keinan N, Tyomkin D, Shoshan-Barmatz V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol Cell Biol. 2010;30:5698–5709. doi: 10.1128/MCB.00165-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, Tuft RA, Fogarty KE, Rizzuto R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of ca2+ microdomains to mitochondria. J Cell Biol. 2002;159:613–624. doi: 10.1083/jcb.200205091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rizzuto R, Marchi S, Bonora M, Aguiari P, Bononi A, De Stefani D, Giorgi C, Leo S, Rimessi A, Siviero R, Zecchini E, Pinton P. Ca(2+) transfer from the er to mitochondria: When, how and why. Biochim Biophys Acta. 2009;1787:1342–1351. doi: 10.1016/j.bbabio.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. Vdac, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med. 2010;31:227–285. doi: 10.1016/j.mam.2010.03.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.