

Abstract
Coxsackievirus B3 (CVB3) is a common pathogen of myocarditis. We previously synthesized a siRNA targeting the CVB3 protease 2A (siRNA/2A) gene and achieved reduction of CVB3 replication by 92% in vitro. However, like other drugs under development, CVB3 siRNA faces a major challenge of targeted delivery. In this study, we investigated a novel approach to deliver CVB3 siRNAs to a specific cell population (e.g. HeLa cells containing folate receptor) using receptor ligand (folate)-linked packaging RNA (pRNA) from bacterial phage phi29. pRNA monomers can spontaneously form dimers and multimers under optimal conditions by base-pairing between their stem loops. By covalently linking a fluorescence-tag to folate, we delivered the conjugate specifically to HeLa cells without the need of transfection. We further demonstrated that pRNA covalently conjugated to siRNA/2A achieved an equivalent antiviral effect to that of the siRNA/2A alone. Finally, the drug targeted delivery was further evaluated by using pRNA monomers or dimers, which carried both the siRNA/2A and folate ligand and demonstrated that both of them strongly inhibited CVB3 replication. These data indicate that pRNA as a siRNA carrier can specifically deliver the drug to target cells via its ligand and specific receptor interaction and inhibit virus replication effectively.
Keywords: Packaging RNA, Coxsackievirus B3, Drug delivery, Gene therapy, Folate receptor, siRNA
1. Introduction
Epidemiological studies reported that coxsackie group B viruses, particularly the B3 strain (CVB3), are primary causal agents of human myocarditis (Kim et al., 2001). This disease often enters its late phase, dilated cardiomyopathy and patients with dilated cardiomyopathy usually progress to terminal heart failure and require heart transplantation as a last therapeutic option (Martino et al., 1994). At present, there is no specific treatment for this disease. CVB3 is a positive single-stranded RNA virus and infects host cells through a coxsackie and adenovirus receptor (CAR)(Bergelson et al., 1997). This viral genome encodes four capsid proteins VP1-VP4 and seven nonstructural proteins including two proteases 2A and 3C, and a RNA-dependent RNA polymerase 3D. CVB3 RNA can be directly translated into a single polyprotein, which is subsequently processed mainly by viral proteases 2A and 3C to produce mature structural and nonstructural proteins for viral replication (Klump et al., 1990). Thus, viral protease 2A plays a crucial role in the viral life cycle and pathogenesis (Chau et al., 2007, Xiong et al., 2007). For this reason, viral proteases are one of the major targets for antiviral drug design.
Recently, the development of therapeutics for viral myocarditis has focused on small interfering RNAs (siRNAs) that target viral mRNA to inhibit translation. siRNAs are short, double-stranded RNA (dsRNA) molecules that can target a specific sequence of mRNA for degradation via a cellular process known as RNA interference (RNAi) (Dorsett and Tuschl, 2004). In this process, siRNA incorporates into an RNA-induced silencing complex (RISC) that recognizes and cleaves the target sequence (Elbashir et al., 2001). Previously, we synthesized a siRNA targeting the CVB3 protease 2A gene, which achieved 92% inhibition of CVB3 replication (Yuan et al., 2005). Although this new strategy has exciting potential, like other parallel drug developments, one of the major barriers for clinical application is the non-specific distribution of the drugs in the body after administration. In this study, we investigated a novel approach to deliver siRNAs to CVB3 susceptible target cells using the ligand-linked pRNA (packaging RNA) as a vehicle.
The pRNA, one of the six copies of the RNA components within the nanomotor of bacterial phage 29, was discovered more than 20 years ago (Guo et al., 1987); however its potential application in drug packaging and delivery attracted scientists’ attention only recently. pRNA is 117 nucleotides (nts) long and has been found to play an essential role in phage DNA packaging. pRNA molecules can form dimers, trimers, and ultimately hexamer arrays through hand-in-hand interactions of the right and left interlocking loops (Guo, 2002, Guo et al., 1998). The structural features of pRNA allow for easy manipulation and permit the conversion of pRNA into a gene targeting and delivery vehicle. Since this molecule is smaller than any other presently used DNA vectors, it facilitates its cellular internalization and confers its lower immunogenicity than other large vectors. The multimer structure of pRNA complex allows it to carry both therapeutic molecules and a ligand simultaneously, which may enable it to deliver drug specifically to a target cell population (Guo, 2005). The pRNA molecule contains two independent folding domains with distinct functions. Replacement or insertion of oligonucleotides preceding nt #23 or following nt #97 does not interfere with the formation of dimers as long as the strands are paired. Therefore, the 5′/3′ proximate double-stranded helical region of the pRNA can be redesigned to carry additional sequences without altering its secondary structure or intermolecular interactions (Guo, 2002). Based on these characteristics, we replaced this helical region with our double stranded siRNA/2A and delivered this therapeutic molecule to the target cells via the pRNA vehicle.
In search of ligands guiding drug specific delivery, many molecules, such as RNA aptamers, peptides and antibodies have been tested (Guo et al., 2005, Kumar et al., 2007, Maruyama et al., 1995, Terada et al., 2006). In this study, we used folic acid as a ligand for delivery of siRNA/2A to HeLa cells, the CVB3 susceptible host cells expressing folate receptors. Folic acid is a vitamin that is essential for the biosynthesis of nucleotides and is not produced by mammalian cells. It is consumed in elevated quantities by proliferating cells and is transported across the plasma membrane using either the membrane-associated reduced folate carrier or the folate receptor (FR) (Reddy and Low, 1998). The former is found in virtually all cells; the latter is found primarily on polarized epithelial cells and activated macrophage. The reduced folate carrier is probably capable of internalizing the necessary folate in normal cells; however FR is frequently overexpressed on a wide range of tumor cells as a consequence of increased folate requirements (Lu and Low, 2002, Toffoli et al., 1997). Therefore, FR's natural ligand, folic acid, has become a popular molecule for targeting attached drugs to cancer cells. In this study, we constructed folate-labeled pRNA monomers and formed heterodimers with chimeric pRNA-siRNA/2A. One subunit of the dimer contained a folate-AMP at its 5′ end, which was used for target cell recognition, and the other harbored a moiety of therapeutic siRNA/2A. By this design, we specifically delivered the pRNA-siRNA/2A by incubation rather than transfection to the HeLa cells via folate-FR interactions. These pRNA-carried siRNA/2A molecules strongly inhibited CVB3 replication and achieved an antiviral efficiency equivalent to that of the free siRNA/2A delivered by transfection, indicating that functional siRNA/2A was properly processed and released after entering the target cells. Importantly, this approach specifically delivered the drug to the target cells but not to the folate receptor negative cells, indicating its promising role in drug targeted delivery for treating cancers and infectious diseases.
2. Materials and methods
2.1. Preparation of siRNA/2A and pRNA-siRNAs
siRNA targeting CVB3 protease 2A gene was synthesized by Qiagen-Xeragon (Germantown, MD) as described previously (Yuan et al., 2005). The final concentration of the siRNAs was 40 μM in the provided buffer. The pRNA-siRNA/2A or control pRNA-siRNAs were synthesized by in vitro transcription of cDNA fragments encoding the chimeric pRNA-siRNA. The cDNA fragments were produced by PCR following the standard procedures using the previously constructed plasmid pRNA(A-b′) DNA or nts 23/97 pRNA(A′-b) DNA and pRNA(B′-a′) DNA as templates and specifically designed primers (Yang et al., 1999, Zhang et al., 1995). The 5′ end primer contains the T7 phage 2.5 promoter sequence followed by sense sequence of the siRNA and the 3′ end primer contains the antisense sequence of the siRNA. All the primer sequences used are listed in Table 1 . The underlined sequences are T7 phage 2.5 promoter. The sequences in the shaded area are sense and antisense strands of siRNA sequences. The bold nt indicates the single point mutation in the middle region of the siRNA/2A sequence, which was used to generate the pRNA-siRNA/mut (control). Another control is the pRNA-siRNA/scr generated using primers containing a scrambled siRNA sequence. The folate-AMP used for labeling the pRNA was synthesized by TriLink BioTechnologies (USA).
Table 1.
Primers for synthesis of cDNA fragments encoding pRNA vector and pRNA-siRNAsa.
| DNA | Primer sequences (5′ → 3′) |
|---|---|
| pRNA vector (nts 7–106) | Forward: TAATACGACTCACTATTAGGGTACGGTACTTCCATTGTCATGTGTATGTTGGGGATTA |
| Reverse: TGCACTTTTGCCATGATTGACGGACAATCAAC | |
| pRNA-siRNA/2A | Forward: TAATACGACTCACTATTAGGTCCAAGAGAGTGAATACTTTGTATGTTGGGGATTA |
| Reverse: GGTCCAAGAGAGTGAATACAAATTGACACGCAATCAAC | |
| pRNA-siRNA/mut | Forward: TAATACGACTCACTATTAGGTCCAAGAGAGTGAATACTTTGTATGTTGGGGATTA |
| Reverse: GGTCCAAGACAGTGAATACAAATTGACACGCAATCAAC | |
| pRNA-siRNA/scr | Forward: TAATACGACTCACTATTATTCTCCGAACGTGTCACGTTTTGTATGTTGGGGATTA |
| Reverse: TTCTCCGAACGTGTCACGTAAATTGACACGCAATCAAC | |
The underlined sequences are T7 phage 2.5 promoter. The sequences in the shaded area are siRNA sequences. The remaining sequences are vector sequences. The bold nt indicates the single point mutation (mut). siRNA containing scrambled (scr) sequence is another control.
To synthesize pRNA-siRNA/2A and various control pRNA-siRNAs, in vitro transcription was conducted using T7-MegaShortscript kit (Ambion) as described previously (Guo et al., 2006). Briefly, the PCR amplified cDNA templates containing the T7 phage 2.5 promoter were transcribed with T7 RNA polymerase in the presence of ATP, GTP, UTP, and CTP (7.5 mM each). Transcription products were purified by urea-8% polyacrylamide gel electrophoresis (PAGE) and eluted with 0.5 M sodium acetate, 0.1 mM EDTA, and 0.1% sodium dodecyl sulfate (SDS). RNAs were ethanol precipitated and resuspended in sterilized DEPC-treated water. To label the 5′-end of pRNA with folate, both 4 mM folate-AMP and 0.25 mM ATP were included in a transcription reaction, together with 1 mM UTP, CTP, and GTP. The solution also contains 40 mM Tris (pH 8.0), 6 mM MgCl2, 2 mM spermidine, 0.01% Triton X-100, 5 mM DTT, 0.2 mM DNA templates, and 5 U/ml T7 RNA polymerase (Promega). As folate has been linked to the 5′ of the AMP, it can only be used for initiation but not for chain extension of transcription, thus ensuring that labeling occurs only at the 5′ end of the pRNA transcript. Magnesium (10 mM) was included in all buffers to maintain the folding of pRNA and the formation of dimers.
2.2. pRNA heterodimer formation
The in vitro transcription products of pRNA and chimeric pRNA-siRNA/2A were analyzed by denaturing PAGE as described previously (Guo, 2005). In brief, 12% denaturing polyacrylamide gel was prepared and 1 × TBE was used as gel running buffer. To prepare the RNA samples, an equal volume of gel loading buffer II (Ambion.) was added to each sample containing 1 μg RNA, and heated for 3–5 min at 80–90 °C before loading. Gels were run at 40–50 V for 3–3.5 h and then gently agitated for 10 min in 10 × GelRed (Biotium, CA) in ddH2O.
pRNA heterodimers were prepared by mixing folate-pRNA(nt7–106)(B-a’) and pRNA-siRNA/2A(A-b’) or other control pRNA-siRNAs from in vitro transcription products (0.5 μM) in a 1:1 molar ratio in a solution containing 10 mM MgCl2, 5 μl of RNAse OUT RNAse inhibitor, 15 μl of 0.1 mM DTT and 20 μl of 10 × RT buffer containing 50 mM Tris–HCl pH 8.0, 100 mM NaCl and 5 mM EDTA. The pRNA heterodimer formation was confirmed by native-PAGE, in which the polyacrylamide gel was poured without SDS and running in 1 × electrode buffer (0.025 M Tris–HCl, 0.192 M glycine pH 8.3).
2.3. Virus, cell culture, infection, and transfection
CVB3 was produced from a full-length cDNA clone (GenBank accession no. M33854) (provided by Reinhardt Kandolf, University of Tubingen, Germany) and amplified in HeLa cells (American Type Culture Collection) by transfection. Virus titer was routinely determined at the beginning of the experiment by plaque assay. HeLa cells and mouse embryo fibroblast (MEF) cells were cultured in a folate-free RPMI1640 medium (Gibco) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin (Invitrogen) in a 5% CO2 incubator. The serum provided a normal complement of endogenous folate for cell growth.
The transfection of siRNA/2A and chimeric pRNA-siRNA/2A was performed under optimal conditions (Yuan et al., 2005). Briefly, 2 × 105 cells were grown at 37 °C overnight. When cells reached 50–60% confluency, they were washed and overlaid with 380 μl of transfection complexes containing 240 pmol of siRNAs and 12 μl of oligofectamine (Invitrogen) overnight. Following transfection, cells were washed and infected with CVB3 at 0.01 MOI (multiplicity of infection) and incubated at 37 °C in 5% CO2 for 12 h. The cell lysates were collected for assay of inhibition of CVB3 replication.
2.4. Chimeric pRNA-siRNA processing by Dicer
Chimeric pRNA-siRNA was labeled with [γ-32P]ATP at the 5′ end using T4 polynucleotide kinase following the supplier's instructions (New England Biolabs). The labeled pRNA-siRNA products were processed by Dicer digestion. Briefly, 50 μl of reaction solution containing 0.5 μg of siRNA and 1.0 unit of recombinant RNA-specific endonuclease Dicer (Genlantis, San Diago, CA) was incubated at 37 °C for different periods of time. The processed siRNAs were confirmed by denaturing polyacrylamide/urea gel electrophoresis.
2.5. Specific delivery of folate-FITC conjugates to HeLa cells
HeLa cells and MEF cells (non-cancer cell, with very low number of folic acid receptors) were maintained in folic acid-deficient RPMI 1640 medium for two weeks to consume the remaining folate inside cells and then sub-cultured into two plates containing and without containing folate, respectively. After incubation overnight, the FITC-folic acid conjugates were added into each cell culture medium at a final concentration of 100 μM. After 4 h incubation at 37 °C, green fluorescent signal distribution in the cells was observed under a fluorescent microscope. To localize the binding and distribution of the FITC-folate on the surface or inside of the cells, cell nuclei were counterstained with 4′, 6-diamidino-2-phenylindole (DAPI, Molecular Probes).
2.6. Specific viral gene silencing with pRNA-siRNA/2A delivered through folate receptor
HeLa cells were maintained in folate-free RPMI 1640 medium for two weeks. One day before the experiment, HeLa cells were seeded in a six-well plate in folate-free RPMI 1640 medium. After being washed with phosphate-buffered saline (PBS) containing 10 mM MgCl2, the premade folate-pRNA-siRNA/2A monomers or folate-pRNA and pRNA-siRNA/2A heterodimers (0.5 μM) containing RNase inhibitor (Ambion) were then added to the cell culture and incubated for 8 h at 37 °C. After incubation, free RNA was washed off and the cells were infected with CVB3 at 0.01 MOI for 12 h. The cell lysates and supernatants were collected and analyzed for CVB3 replication efficiency by RT-PCR, Western blot and viral plaque assay. pRNA vector, pRNA-siRNA/mut and pRNA-siRNA/scr were used as controls. As a cell line that is CVB3 susceptible but folic acid receptor negative is not available thus far, we were not able to include one such cell line as an additional control for antiviral evaluation.
2.7. RT-PCR
Total RNA was extracted from cells at indicated time points post-transfection (pt) or post incubation using a RNeasy kit (Qiagen). The extracted RNA was treated with 1 μl of DNase I (20 units/μl) to remove residual DNA. Reverse transcription (RT) was conducted according to the manufacturer's instructions (Invitrogen) using 1 μg of extracted RNA and 1 μl of 3 μM hexamer primer and followed by PCR to amplify CVB3 2A cDNAs. The PCR reaction mixture contained 10 μl of RT product and 1 μl of 15 μM sense and antisense primers and the reaction was run for 28 cycles with standard parameters (Yang et al., 1999). The PCR primer sequences for 2A are the same as that for cloning of these genes mentioned above. The PCR product from each sample was analyzed by 0.8% agarose gel electrophoresis.
2.8. Western blot analysis
Protein concentration for each sample was quantified by the Bradford assay using fat-free-bovine serum albumin (BSA) as the standard. Western blot analysis was performed by standard protocols as previously described (Zhang et al., 2005). Equal amounts of proteins were subjected to SDS-PAGE and then were transferred onto nitrocellulose membranes (Hybond EDL™, Amersham). The molecular weight of proteins was estimated by comparison with the BenchMark™ pre-stained protein ladder (Invitrogen) loaded. The membranes were blocked with 5% skim milk containing 0.1% Tween 20 for 1 h. The blots were probed with primary mouse antibody against CVB3 capsid protein VP1 (DAKO) or β-actin (Sigma) for 1 h, followed by incubation with horseradish-peroxidase-conjugated goat secondary antibody to anti-mouse immunoglobulin G (1:1000 dilution, BD biosciences). Finally, VP1 and β-actin expression were detected by ECL reagents (Amersham).
2.9. Viral plaque assay
The virus titer was determined by plaque assay as described previously (Yuan et al., 2006). Briefly, HeLa cells were seeded into 6-well plates (8 × 105 cells/well) and incubated at 37 °C for 20 h. When cell confluency reached approximately 90%, cells were washed with PBS and then overlaid with 500 μl of diluted supernatants from the culture. The cells were incubated at 37 °C for 60 min, and the supernatants were removed. Finally, cells were overlaid with 2 ml of sterilized soft Bacto-agar-minimal essential medium. The cells were incubated at 37 °C for 72 h, fixed with Carnoy's fixative for 30 min, and then stained with 1% crystal violet. The plaques were counted, and the amount of plaque forming unit (PFU/ml) was calculated.
2.10. Cell viability assay
Cell viability was measured by using a 3-(4,5-dimethylthiazol-2-yl)-5- (3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (MTS) assay kit (Promega) according to the manufacturer's instructions. HeLa cells were grown in 6-well plates overnight and incubated with pRNA-siRNA monomers or heterodimers at the concentration described above for 8 h. These cells were then infected with CVB3 overnight, which was followed by adding MTS reagent using the protocol as per the manufacturer's instructions (Clontech). Cells were incubated for 2 h, and the absorbance of formazan was measured at 492 nm using an enzyme-linked immunosorbent assay (ELISA) reader. The quantity of formazan product is directly proportional to the number of viable cells in the culture medium. The survival value for absorbance of non-treated sham-infected cells were defined as 100%, and the remaining data of CVB3-infected cells, including that for siRNA-treated, control-treated and nontreated cells, were converted to the ratio of the non-treated sham-infected sample. In addition, morphological changes of cells following CVB3 infection were evaluated by phase-contrast microscopy.
3. Results
3.1. Construction of chimeric pRNA harboring siRNA/2A
pRNA contains a double-stranded helical domain at the 5′/3′ end and an intermolecular binding domain, which fold independently of each other (Zhang et al., 1995)(Fig. 1A). Complementary modification studies have revealed that altering the primary sequences of any nucleotide of the helical region of the pRNA does not affect its structure and folding as long as the two strands are base-paired (Chen et al., 1999, Zhang et al., 1995). As siRNA is a dsRNA helix, it can replace the helical region of the pRNA and is carried by the pRNA molecule. To construct these chimeric pRNA-siRNAs, a plasmid encoding the pRNA backbone sequence was constructed previously by cloning of the synthesized dsDNA fragments into a plasmid vector (Zhang et al., 1994). This plasmid was used as a template to conduct PCR to amplify cDNA fragments encoding chimeric pRNA-siRNA containing, in order, sense sequence of siRNA, a poly-A liker, pRNA sequence, a ploy-A linker and antisense sequence of the siRNA (Fig. 1B). The cDNA fragments were analyzed by agarose gel electrophoresis (Fig. 2A). In vitro transcription using these PCR-amplified cDNA fragments containing the T7 phage 2.5 promoter upstream of the insert was conducted using T7 RNA polymerase. The chimeric pRNA transcripts harboring the siRNA/2A sequences were shown in Fig. 1C. These chimeric pRNAs synthesized include (i) pRNA-siRNA/2A, a pRNA chimera that harbors an siRNA targeting the CVB3 2A protease gene, (ii) pRNA-siRNA/mut, a pRNA chimera containing a point mutation in the middle region of siRNA/2A, and (iii) pRNA-siRNA/scr, carrying a scrambled siRNA. In addition, a pRNA vector (nts 29–97) was also synthesized, which was used as a control. All the chimeric pRNA-siRNAs were analyzed by denaturing urea-PAGE (Fig. 2B).
Fig. 1.
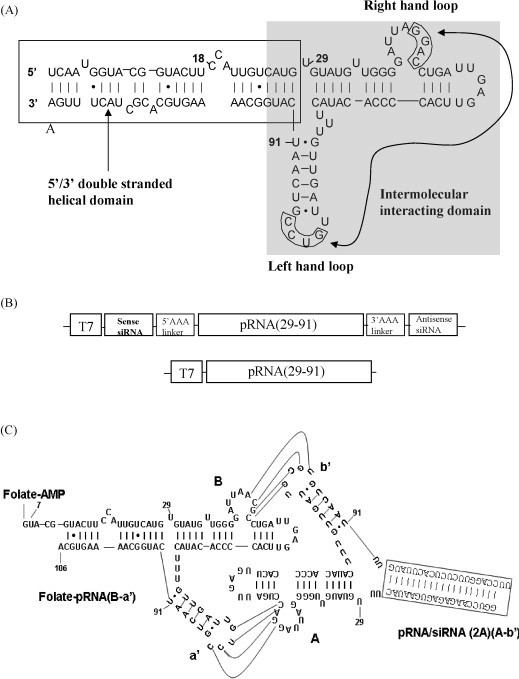
Design of cDNA encoding chimeric pRNA-siRNA/2A. (A) Phage 29 pRNA sequence and secondary structure. The left- and right-hand loops for intermolecular interacting (base-pairing) are circled. The curved line with arrows points to the two interacting loops. The double-stranded helical domain on the 5′/3′ ends is framed and the domain for dimer formation is shaded. (B) Schematic structures of cDNA encoding pRNA vector and chimeric pRNA-siRNA. The DNA contains a T7 phage 2.5 promoter followed by the chimeric pRNA-siRNA/2A sequence as indicated on the diagram. The cDNA encoding the pRNA (29–91) lacks the nts of 5′ 1–28 and 3′ 92–117, which are replaced by sense and antisense siRNA/2A, respectively. Poly-A linkers are used to link the siRNA/2A and the pRNA sequence. (C) Folate-labeled chimeric pRNA heterodimer complex harboring siRNA/2A. The 21-mer siRNA/2A marked by a frame is covalently linked to the 5′/3′(29/91) paired ends of pRNA connecting by poly-U linkers. The poly-U linkers are used to facilitate folding of the chimeric pRNA-siRNA/2A and enhance the processing by Dicer to release functional siRNA/2As. The ligand folate is covalently linked to the 5′end of pRNA. To increase the accessibility of the folate to its receptor, folate-labeled RNA is designed to be a 5′overhang, in which nts 107–117 at the 3′end of pRNA are truncated. The dimer is formed by hand-in-hand connections between the left- and right-hand loops. Curved lines indicate the base-pairing between two loops (a′-A and B-b′) of chimeric pRNA monomers.
Fig. 2.
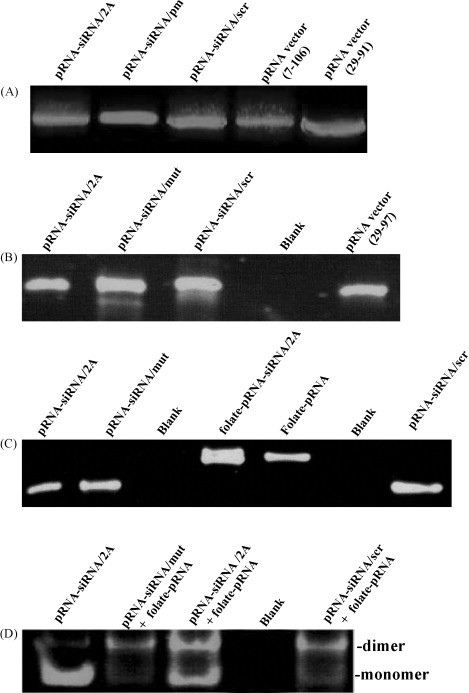
Synthesis of monomers and heterodimers of pRNA-siRNAs. (A) Agarose gel electrophoresis of DNA fragments used for in vitro transcription. cDNA fragments were amplified by PCR using the plasmid encoding Ab′ pRNA sequence as a template and primers listed in Table 1 (Zhang et al., 1995). (B) Analysis of chimeric pRNAs transcripts by denaturing PAGE. These pRNA chimera were synthesized by in vitro transcription using cDNA fragments demonstrated in (A) above. pRNA-siRNA/2A containing a point mutation (mut) in the middle of the siRNA/2A and a scrambled (scr) siRNA are indicated, respectively. (C) Analysis of folate-labeled chimeric pRNAs by denaturing PAGE. Folate-pRNA and folate-pRNA-siRNA/2A was synthesized by in vitro transcription in the presence of folate-AMP. (D) Demonstration of pRNA heterodimer complex formation by non-denaturing-PAGE. The pRNA-siRNA and folate-pRNA were dimerized by mixing folate-pRNA and pRNA-siRNA/2A or control pRNA-siRNAs in a buffer containing 10 mM MgCl2.
3.2. Synthesis of pRNA harboring a folate ligand
The folate labeling of pRNA was achieved by utilizing folate-AMP as an initiator of RNA transcription with a T7 phage 2.5 promoter under the published conditions (Huang et al., 2008). This promoter produced superior 5′ homogeneity of RNA over the T7 phage 6.5 promoter with comparable total RNA yields (Huang et al., 2008, Huang et al., 2003). As folate-AMP can only be used for initiation but not for chain extension, this ensures that labeling occurs only at the 5′-end of the transcripts.
In vitro transcription of 5′folate-pRNA was performed in the presence of both folate-AMP and ATP, together with CTP, UTP and GTP. In our study, the best molar ratio of folate-AMP to ATP is 20:1 in considering both total yield of the RNA production and the percentage of pRNA carrying the folate labeling. Comparing with the 16:1 ratio used previously (Guo et al., 2006), transcription using the 20:1 molar ratio produced even higher yield of RNA transcripts, which required no gel purification and could be directly used for dimer formation. The RNA labeled with folate had a slower migration rate than the nonlabeled RNA on denaturing polyacrylamide gel. pRNA-siRNA/2A DNA was also used as the template for in vitro transcription in the presence of folate-AMP to generate chimeric folate-pRNA-siRNA/2A monomer. To increase the accessibility of the folate molecule to folate receptor on the cell surface, folate-labeled pRNA was designed to be a 5′ overhang, in which nts 107–117 of pRNA were truncated (Guo et al., 2006). The folate-labeled pRNA vector could form heterodimers with a pRNA-siRNA/2A chimera to achieve specific delivery. Gel electrophoresis results indicated that under the optimal transcription conditions, most of the pRNA contains a folate moiety (Fig. 2C).
3.3. Assembly of pRNA heterodimers composed of chimeric monomers harboring either a siRNA or a folate ligand
Extensive studies of pRNA have revealed that two pRNA subunits form a dimer through the interaction of the complementary left- and right-hand loops of pRNA monomers (Guo et al., 1998). The sequence responsible for intermolecular pRNA/pRNA interaction in this study is located between nts 29 and 91 (Fig. 1C). pRNA(A-b′) contains a right-hand loop A (5′G45G46A47C48) and a left-hand loop b′ (3′U84G83C82G81), which together can pair with the left-hand loop a′ (3′C85C84U83G82) and the right-hand loop B (5′A45C46G47C48) of pRNA(B-a′), respectively. Chimeric monomer subunits were intentionally designed to possess either A-b′ or B-a′ to match with each other. We used pRNA(A-b′) DNA as a template to synthesize pRNA-siRNA(A-b′) and pRNA(B-a′) DNA as a template to synthesize folate-pRNA(B-a′), respectively. When chimeric pRNA-siRNA(A-b′) and folate-pRNA(B-a′) were mixed in a 1:1 molar ratio in the presence of 10 mM Mg2+, pRNA heterodimers were produced with high efficiency and confirmed by non-denaturing PAGE (Fig. 2D). Chimeric heterodimers were generated from different monomer subunits despite the replacement of the 5′/3′ helix with a ds-siRNA/2A or other siRNA controls.
3.4. Processing of chimeric pRNA-siRNA complex into ds-siRNA by dicer
To determine whether the chimeric complex could be processed into functional siRNA, chimeric pRNA-siRNA was subjected to treatment with recombinant Dicer, which is known for its function of processing long double-stranded pre-miRNA into a ∼21-nt siRNA in vitro and in vivo (Carmell and Hannon, 2004). The 5′-end 32P-labeled chimeric pRNA-siRNA complex was used as the substrate in this study, which harbored a 21-base ds-siRNA connected to the pRNA intermolecular interacting domain spanning nts 29 to 91 (Fig. 1C). In addition, poly-“U” bases were used to link the siRNA/2A and the pRNA backbone sequence to increase the ΔG and facilitate the folding of the pRNA-siRNA/2A. Incubation of the pRNA-siRNA complex with Dicer resulted in the processing complex into a ∼21-nt ds-siRNA as revealed by denaturing urea PAGE (Fig. 3 ).
Fig. 3.
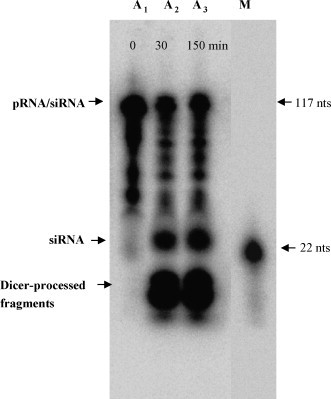
Autoradiogram showing the processing of chimeric pRNA-siRNA by Dicer. The chimeric pRNA-siRNAs labeled with 32P at the 5′ends were incubated with Dicer for 0 min (A1), 30 min (A2) and 150 min (A3), and subjected to electrophoresis in a 12% denatured urea polyacrylamide gel. Lane M is the 32P-labeled 22-nt RNA molecular weight marker moved from the same gel. Note that siRNA moved slower than the marker. This is probably due to the extra U linkers, which made the Dicer cleavage products more than 22 nts.
3.5. Specific delivery of folate-FITC conjugates to HeLa cells via interactions between folic acid and its receptor
Folate receptors are overexpressed in various types of cancer cells such as HeLa cells (Claycomb et al., 1998, Holm et al., 2000), but are generally absent in non-cancer cells such as MEF cells. To test whether folate conjugated to a pRNA molecule can guide the drug delivery specifically to target cells, we incubated the HeLa cells in the presence of folate-FITC conjugates and used MEF cells as a control. After 4 h incubation at 37 °C, intracellular distribution of fluorescent signals was observed under a fluorescent microscope. We found that HeLa cells sub-cultured in folate deficient medium had much stronger green fluorescent signals than their counterparts sub-cultured in folate-containing growth medium; while MEF cells, which are non-cancer cells and considered to have no folate receptors were not affected by either medium and showed no signal (Fig. 4 ).
Fig. 4.
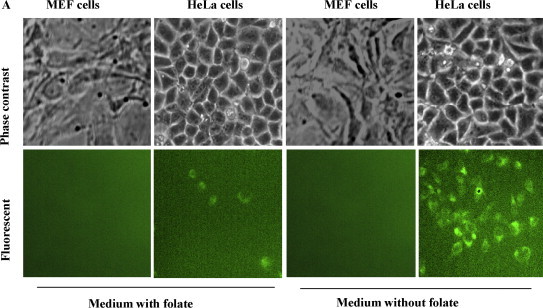
Specific delivery of folate-FITC conjugates to HeLa cells via the interactions between folic acid and its receptor. (A) HeLa cells and MEF cells (control) were maintained in folic acid-deficient RPMI 1640 medium for two weeks and then sub-cultured in media containing and without containing folate, respectively, but all containing folate-FITC conjugates. After incubation, green fluorescent signals were observed under a fluorescent microscope. The two images are not the same field. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
3.6. Antiviral evaluation of chimeric pRNA-siRNA monomers delivered by transfection
As HeLa cells are folate receptor-expressing tumor cells and also susceptible to CVB3 infection, these cells were used as a model system to evaluate our chimeric pRNA-siRNA/2A in this antiviral study. To test whether the antiviral activity of siRNA/2A was affected after linking to a pRNA, HeLa cells were transfected with pRNA-siRNA/2A or other control pRNA-siRNAs and then infected with CVB3. To make a comparison, free siRNA/2A was also used to transfect the HeLa cells with lipofectamine. Antiviral activity of pRNA-siRNA/2A was measured by different methods. As shown in Fig. 5 , both RT-PCR to detect 2A mRNA (Fig. 5B) and Western blot to detect CVB3 VP1 protein (Fig. 5C) demonstrated that siRNA/2A delivered by pRNA strongly inhibited CVB3 replication as compared to the controls including pRNA(nts 29–91) vector only, pRNA-siRNA/2A mut, pRNA-siRNA/scr and siRNA/2A. This antiviral activity was correlated well to data obtained from the morphology observation, showing that cells treated with pRNA-siRNA/2A had less cell death than that treated with a control pRNA-siRNA (Fig. 5A).
Fig. 5.
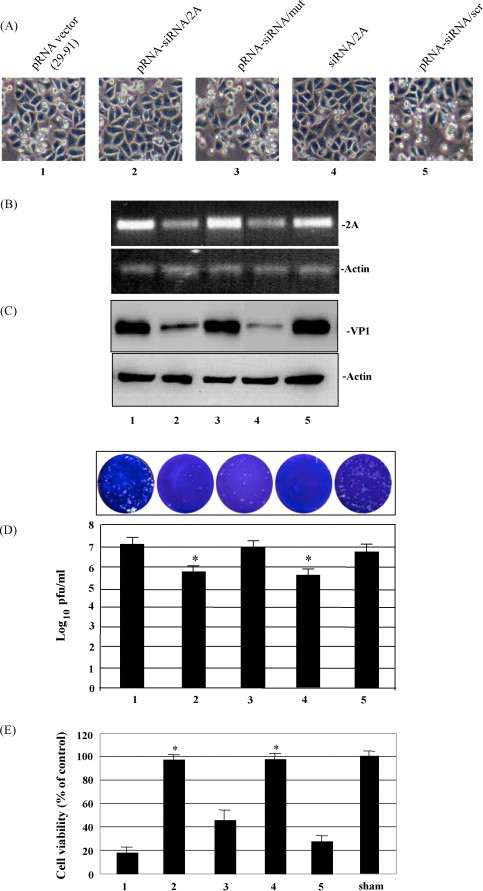
Antiviral evaluation of siRNA/2A delivered by transfection of pRNA monomers. HeLa cells were transfected by Lipofectimine 2000 with the indicated pRNA-siRNA/2A or control pRNA-siRNAs, and then infected with CVB3. The antiviral activity of siRNA/2A was evaluated by morphology observation of HeLa cell death (A), RT-PCR to detect CVB3 2A RNA (B), Western blot analysis to detect CVB3 VP1 protein (C) and viral plaque assay to detect infectious viral particles (D). The numbering of the samples or the lanes of the gels are the same as that in (A) above. The values shown represent the means ± SD of the data from three separated experiments, and the significance was determined by the Student's t test, *P < 0.05 (D). (E) MTS assays to determine the cell viability. HeLa cells were grown in a 6-well plate overnight and transfected with pRNA-siRNAs as indicated in (A) above. After transfection, cells were infected with CVB3 overnight. Cell viability was measured by the MTS assay. Data were normalized with background reading. The control is the cell culture that was not treated and sham-infected, which was defined as 100% survival. The values shown represent the means ± SD of the data from three separated experiments, *P < 0.05.
These data were further solidified by the viral plaque assay to measure infectious viral particles and the MTS assay to measure the cell viability. Fig. 5D shows that comparing to the levels of controls, an approximate 1–2 log10 decrease in PFU production was detected in these cultures treated with pRNA-siRNA/2A or free siRNA/2A, while no or only little inhibition of CVB3 replication occurred when mutated or scrambled siRNAs were introduced into the cells. Fig. 5E demonstrated that treatment with pRNA-siRNA/2A could maintain ∼95% of the cell survival as compared with cells treated with the pRNA vector control, which only had <20% cell survival. These results indicate that chimeric pRNA-siRNA/2A remains the high efficient antiviral activity as that of the free siRNA/2A and that pRNA is a reliable vehicle to deliver therapeutic molecules.
The above data imply that the chimeric pRNA-siRNA/2A was correctly processed by Dicer and functional ds-siRNAs were released. This speculation is supported by the in vitro processing experiment, which demonstrated that 32P-labeled pRNA-siRNA/2A were processed into 21-bp siRNAs by recombinant RNAse III-like ribonuclease Dicer after incubation at 37 °C (Fig. 3).
3.7. Antiviral evaluation of pRNA-siRNA/2A heterodimers delivered via folate and receptor interactions
The strategy of pRNA heterodimer-mediated gene silencing is the ligand-mediated cell recognition, subsequent internalization, and release of functional siRNAs to knockdown specific genes. pRNA heterodimers containing both folate and siRNA against CVB3 protease 2A were incubated with, rather than transfected into, HeLa cells followed by CVB3 infection for 12 h. Western blot analysis indicated that both folate-pRNA-siRNA/2A monomer and pRNA heterodimer complex (folate-pRNA + pRNA-siRNA/2A) strongly inhibited CVB3 replication as compared to the controls (Fig. 6A). Viral plaque assay showed a ∼1.5 log10 decrease in PFU/ml as compared with the controls (Fig. 6B). In addition, the cell viability assay demonstrated that cells treated with either folate-pRNA-siRNA/2A or pRNA heterodimers complexes could maintain over 90% cells survival as compared to cells treated with the controls, which showed ∼25% cell survival (Fig. 6C). These results suggest that specific knockdown of the protease 2A gene was achieved by folate receptor-mediated internalization of folate-pRNA-siRNA/2A monomer or pRNA heterodimer complex in the absence of transfection reagents.
Fig. 6.
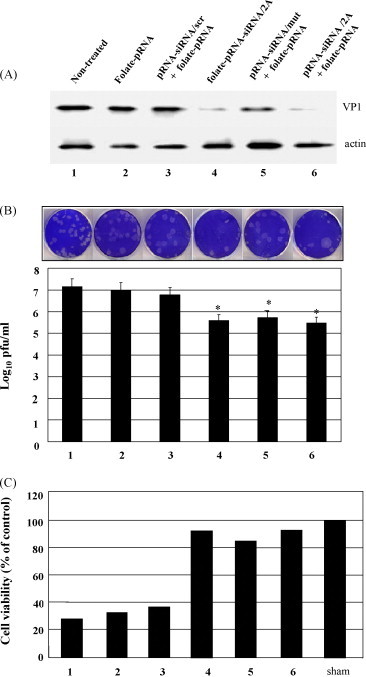
Antiviral evaluation of siRNA/2A delivered by folate-labeled pRNA monomers or heterodimer complexes by incubation. HeLa cells are incubated in the folic acid deficient RPMI 1640 medium for two weeks. One day before incubation, HeLa cells were passaged into 6-well plates. On the second day, HeLa cells were incubated with heterodimers of folate-pRNA and pRNA-siRNA/2A or with folate-pRNA-siRNA/2A monomers and then infected with CVB3. Antiviral activity of siRNA/2A was evaluated by Western blot analysis to detect CVB3 VP1 protein (A), viral plaque assay to detect infectious viral particles (B) and MTS assays to determine the cell viability (C) as described in Fig. 5. The data are the mean values of two independent experiments.
4. Discussion
In drug development, one of the most critical barriers for clinical application is the drug non-specific distribution in the body after administration. Although various methods have been described for delivering drug to the cells, most of these methods accomplish delivery nonspecifically. The non-specific delivery not only reduces therapeutic efficiency and causes harmful side effects but also wastes drug, resulting in high cost. Therefore, development of an effective system for targeted delivery of therapeutic molecules is the main goal of the present study.
We chose siRNA targeting CVB3 2A gene, a critical protease involved in viral life cycle and pathogenesis, as an effective drug to test a novel delivery system. We first determined whether covalent linkage of siRNA/2A, by replacement of double helix region, to the 5′/3′ ends of the backbone sequence of the pRNA affected the function of siRNA/2A. We demonstrated that pRNA-siRNA/2A achieved a similar strong anti-CVB3 activity as that of the free siRNA/2A, indicating that siRNA/2A sequences carried by pRNA can be properly folded into dsRNA helix, and processed by Dicer to release functional siRNA/2A after entering the cells. To increase the efficiency of the active siRNA/2A release from pRNA-siRNA/2A by Dicer, two additional uridines were introduced at the 3-way junction to increase the free energy for folding of the junction area into a single-stranded loop. These experiments indicate that pRNA molecule can be employed as a novel category of RNA vector for delivery of therapeutic RNA molecules.
Specific cell recognition and the silencing of target gene(s) in cells are two features required by effective gene therapy. Construction of pRNA dimers carrying both drugs and ligands simultaneously can satisfy this goal. As our main goal is to develop a new system for siRNA specific delivery to target cells, we selected HeLa cells, a CVB3 susceptible host and a cancer cell line, to test this system. Since almost all cancer cells are folate receptor positive cells (Holm et al., 2000, Toffoli et al., 1997), we used folate as a ligand to guide the receptor binding and internalization of siRNAs. By construction of folate-labeled chimeric pRNA-siRNA/2A monomers and heterodimer complexes, we demonstrated for the first time that ligand-mediated delivery of siRNA carried by pRNA vector could successfully inhibit viral replication. In 5′-labeling of the pRNA with folate, we found that the molar ratio of 5′folate-AMP:ATP at 20:1 could result in high yield and increase the labeling efficiency. For assembly of heterodimer complexes with pRNA-siRNA/2A and folate-pRNA, magnesium concentration is crucial. Mg2+ at 10 mM could result in high yield of dimers, which confirmed the previous report (Guo et al., 2006). This concentration may also be important for stabilizing the dimers during delivery. Thus, how to maintain this concentration during delivery in vivo needs to be investigated.
The phage-29 pRNA has been reported to deliver siRNA to treat cancer in several studies (Guo et al., 2006, Guo et al., 2005). However, it is used for delivery of siRNAs to treat viral infections has never been reported. Our data have showed the great potential of this small RNA molecule as an effective vector for targeted delivery of antiviral drugs. This is largely attributed to its unique characteristics (Guo, 2002, Guo, 2005): besides its low immunogenicity resulted from its smaller size than any other often used viral vectors thus far, its property to spontaneously form dimers, trimers and heximer array renders its capability to carry both drugs and ligands in one multimer (Shu et al., 2003). In case that the multimer carries a combination of 3–6 therapeutic molecules and/or ligands, it may render a synergistic effect on gene silencing and specific targeting. More potentially, simultaneously delivering multiple therapeutic sequences may be effective for reducing mutation escape (drug resistance) of the virus. This uniqueness is a critical requirement for antiviral drug development because viruses, particularly the RNA viruses such as CVB3, HIV, SARS-CoV, and influenza virus, all have a high mutation rate. That is why the drug resistance is a major barrier for treatment of the viral infections and why it is difficult to develop effective vaccines for preventing these diseases from frequent reemerging.
Another challenge to achieve targeted drug delivery using this system is the search for specific ligands, which will be conjugated to the pRNA. Here we used folate as a ligand for the in vitro study because the HeLa cells, an established cell line for the study of CVB3 infection, are happened to be cancer cells and express a very high level of folate receptors. As CVB3 is a primary pathogen of human myocarditis (Kim et al., 2001), we aim to delivery the drug to the heart to inhibit CVB3 replication. To date, there is no effective vector to specifically deliver therapeutics to this organ even though the AAV vector and retrovirus vector have achieved promising outcome, they are big DNA vector and have high immunogenicity (Fechner et al., 2008, Kim et al., 2008). The present study has shown promise to explore the possibility of using the small pRNA as a vehicle to transport the drug to the heart through ligand-receptor interactions. The ligands for targeting the heart are either specific myocardium-binding peptides or CVB3 receptor ligand, such as the peptides of viral capsid proteins. Currently, the search for such ligands in our laboratory is in progress.
Acknowledgements
This work was supported by a grant-in-aid from the Heart and Stroke Foundation of British Columbia and Yukon. Ji Yuan is a recipient of the Doctoral Research Award from the Canadian Institutes of Health Research and Michael Smith Foundation of Health Research
References
- Bergelson J.M., Cunningham J.A., Droguett G., Kurt-Jones E.A., Krithivas A., Hong J.S., Horwitz M.S., Crowell R.L., Finberg R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- Carmell M.A., Hannon G.J. RNase III enzymes and the initiation of gene silencing. Nat. Struct. Mol. Biol. 2004;11:214–218. doi: 10.1038/nsmb729. [DOI] [PubMed] [Google Scholar]
- Chau D.H., Yuan J., Zhang H., Cheung P., Lim T., Liu Z., Sall A., Yang D. Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis. 2007;12:513–524. doi: 10.1007/s10495-006-0013-0. [DOI] [PubMed] [Google Scholar]
- Chen C., Zhang C., Guo P. Sequence requirement for hand-in-hand interaction in formation of RNA dimers and hexamers to gear phi29 DNA translocation motor. RNA. 1999;5:805–818. doi: 10.1017/s1355838299990350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claycomb W.C., Lanson N.A., Jr., Stallworth B.S., Egeland D.B., Delcarpio J.B., Bahinski A., Izzo N.J., Jr. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. U. S. A. 1998;95:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett Y., Tuschl T. siRNAs: applications in functional genomics and potential as therapeutics. Nat. Rev. Drug Discov. 2004;3:318–329. doi: 10.1038/nrd1345. [DOI] [PubMed] [Google Scholar]
- Elbashir S.M., Lendeckel W., Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fechner H., Sipo I., Westermann D., Pinkert S., Wang X., Suckau L., Kurreck J., Zeichhardt H., Muller O., Vetter R., Erdmann V., Tschope C., Poller W. Cardiac-targeted RNA interference mediated by an AAV9 vector improves cardiac function in coxsackievirus B3 cardiomyopathy. J. Mol. Med. 2008;86:987–997. doi: 10.1007/s00109-008-0363-x. [DOI] [PubMed] [Google Scholar]
- Guo P. Structure and function of phi29 hexameric RNA that drives the viral DNA packaging motor: review. Prog. Nucleic Acid Res. Mol. Biol. 2002;72:415–472. doi: 10.1016/s0079-6603(02)72076-x. [DOI] [PubMed] [Google Scholar]
- Guo P. Bacterial virus phi29 DNA-packaging motor and its potential applications in gene therapy and nanotechnology. Methods Mol. Biol. 2005;300:285–324. doi: 10.1385/1-59259-858-7:285. [DOI] [PubMed] [Google Scholar]
- Guo P., Zhang C., Chen C., Garver K., Trottier M. Inter-RNA interaction of phage phi29 pRNA to form a hexameric complex for viral DNA transportation. Mol. Cell. 1998;2:149–155. doi: 10.1016/s1097-2765(00)80124-0. [DOI] [PubMed] [Google Scholar]
- Guo P.X., Erickson S., Anderson D. A small viral RNA is required for in vitro packaging of bacteriophage phi 29 DNA. Science. 1987;236:690–694. doi: 10.1126/science.3107124. [DOI] [PubMed] [Google Scholar]
- Guo S., Huang F., Guo P. Construction of folate-conjugated pRNA of bacteriophage phi29 DNA packaging motor for delivery of chimeric siRNA to nasopharyngeal carcinoma cells. Gene Ther. 2006;13:814–820. doi: 10.1038/sj.gt.3302716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S., Tschammer N., Mohammed S., Guo P. Specific delivery of therapeutic RNAs to cancer cells via the dimerization mechanism of phi29 motor pRNA. Hum. Gene. Ther. 2005;16:1097–1109. doi: 10.1089/hum.2005.16.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm J., Hansen S.I., Hoier-Madsen M., Korsbaek L., Beckmann H., Josefsen K. Ligand binding characteristics of a glycosylphosphatidyl inositol membrane-anchored HeLa cell folate receptor epitope-related to human milk folate binding protein. Biosci. Rep. 2000;20:109–118. doi: 10.1023/a:1005567417123. [DOI] [PubMed] [Google Scholar]
- Huang F., He J., Zhang Y., Guo Y. Synthesis of biotin-AMP conjugate for 5′ biotin labeling of RNA through one-step in vitro transcription. Nat. Protoc. 2008;3:1848–1861. doi: 10.1038/nprot.2008.185. [DOI] [PubMed] [Google Scholar]
- Huang F., Wang G., Coleman T., Li N. Synthesis of adenosine derivatives as transcription initiators and preparation of 5′ fluorescein- and biotin-labeled RNA through one-step in vitro transcription. RNA. 2003;9:1562–1570. doi: 10.1261/rna.5106403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.S., Hufnagel G., Chapman N.M., Tracy S. The group B coxsackieviruses and myocarditis. Rev. Med. Virol. 2001;11:355–368. doi: 10.1002/rmv.326. [DOI] [PubMed] [Google Scholar]
- Kim Y.J., Ahn J., Jeung S.Y., Kim D.S., Na H.N., Cho Y.J., Yun S.H., Jee Y., Jeon E.S., Lee H., Nam J.H. Recombinant lentivirus-delivered short hairpin RNAs targeted to conserved coxsackievirus sequences protect against viral myocarditis and improve survival rate in an animal model. Virus Genes. 2008;36:141–146. doi: 10.1007/s11262-007-0192-y. [DOI] [PubMed] [Google Scholar]
- Klump W.M., Bergmann I., Muller B.C., Ameis D., Kandolf R. Complete nucleotide sequence of infectious Coxsackievirus B3 cDNA: two initial 5′ uridine residues are regained during plus-strand RNA synthesis. J. Virol. 1990;64:1573–1583. doi: 10.1128/jvi.64.4.1573-1583.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P., Wu H., McBride J.L., Jung K.E., Kim M.H., Davidson B.L., Lee S.K., Shankar P., Manjunath N. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- Lu Y., Low P.S. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv. Drug Deliv. Rev. 2002;54:675–693. doi: 10.1016/s0169-409x(02)00042-x. [DOI] [PubMed] [Google Scholar]
- Martino T.A., Liu P., Sole M.J. Viral infection and the pathogenesis of dilated cardiomyopathy. Circ. Res. 1994;74:182–188. doi: 10.1161/01.res.74.2.182. [DOI] [PubMed] [Google Scholar]
- Maruyama K., Takizawa T., Yuda T., Kennel S.J., Huang L., Iwatsuru M. Targetability of novel immunoliposomes modified with amphipathic poly(ethylene glycol)s conjugated at their distal terminals to monoclonal antibodies. Biochim. Biophys. Acta. 1995;1234:74–80. doi: 10.1016/0005-2736(94)00263-o. [DOI] [PubMed] [Google Scholar]
- Reddy J.A., Low P.S. Folate-mediated targeting of therapeutic and imaging agents to cancers. Crit. Rev. Ther. Drug Carrier Syst. 1998;15:587–627. [PubMed] [Google Scholar]
- Shu D., Huang L.P., Hoeprich S., Guo P. Construction of phi29 DNA-packaging RNA monomers, dimers, and trimers with variable sizes and shapes as potential parts for nanodevices. J. Nanosci. Nanotechnol. 2003;3:295–302. doi: 10.1166/jnn.2003.160. [DOI] [PubMed] [Google Scholar]
- Terada T., Mizobata M., Kawakami S., Yabe Y., Yamashita F., Hashida M. Basic fibroblast growth factor-binding peptide as a novel targeting ligand of drug carrier to tumor cells. J. Drug Target. 2006;14:536–545. doi: 10.1080/10611860600849498. [DOI] [PubMed] [Google Scholar]
- Toffoli G., Cernigoi C., Russo A., Gallo A., Bagnoli M., Boiocchi M. Overexpression of folate binding protein in ovarian cancers. Int. J. Cancer. 1997;74:193–198. doi: 10.1002/(sici)1097-0215(19970422)74:2<193::aid-ijc10>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Xiong D., Yajima T., Lim B.K., Stenbit A., Dublin A., Dalton N.D., Summers-Torres D., Molkentin J.D., Duplain H., Wessely R., Chen J., Knowlton K.U. Inducible cardiac-restricted expression of enteroviral protease 2A is sufficient to induce dilated cardiomyopathy. Circulation. 2007;115:94–102. doi: 10.1161/CIRCULATIONAHA.106.631093. [DOI] [PubMed] [Google Scholar]
- Yang D., Yu J., Luo Z., Carthy C.M., Wilson J.E., Liu Z., McManus B.M. Viral myocarditis: identification of five differentially expressed genes in coxsackievirus B3-infected mouse heart. Circ. Res. 1999;84:704–712. doi: 10.1161/01.res.84.6.704. [DOI] [PubMed] [Google Scholar]
- Yuan J., Cheung P.K., Zhang H.M., Chau D., Yang D. Inhibition of coxsackievirus B3 replication by small interfering RNAs requires perfect sequence match in the central region of the viral positive strand. J. Virol. 2005;79:2151–2159. doi: 10.1128/JVI.79.4.2151-2159.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J., Stein D.A., Lim T., Qiu D., Coughlin S., Liu Z., Wang Y., Blouch R., Moulton H.M., Iversen P.L., Yang D. Inhibition of coxsackievirus B3 in cell cultures and in mice by peptide-conjugated morpholino oligomers targeting the internal ribosome entry site. J. Virol. 2006;80:11510–11519. doi: 10.1128/JVI.00900-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Lee C.S., Guo P. The proximate 5′ and 3′ ends of the 120-base viral RNA (pRNA) are crucial for the packaging of bacteriophage phi 29 DNA. Virology. 1994;201:77–85. doi: 10.1006/viro.1994.1267. [DOI] [PubMed] [Google Scholar]
- Zhang C., Tellinghuisen T., Guo P. Confirmation of the helical structure of the 5′/3′ termini of the essential DNA packaging pRNA of phage phi 29. RNA. 1995;1:1041–1050. [PMC free article] [PubMed] [Google Scholar]
- Zhang H.M., Yuan J., Cheung P., Chau D., Wong B.W., McManus B.M., Yang D. Gamma interferon-inducible protein 10 induces HeLa cell apoptosis through a p53-dependent pathway initiated by suppression of human papillomavirus type 18 E6 and E7 expression. Mol. Cell. Biol. 2005;25:6247–6258. doi: 10.1128/MCB.25.14.6247-6258.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]