

Abstract
Nickel nanotubes have been synthesized by the popular and versatile method of template-assisted electrodeposition, and a surface-directed growth mechanism based on the adsorption of the nickel-borate complex has been proposed.
The synthesis of metal nanotubes (NTs) is a rapidly growing area of study due to their unique physical, chemical, and magnetic properties. Compared to their nanowire (NW) counterparts, NTs possess higher specific surface area due to the accessibility of their inner cavity, which makes them ideal candidates for applications in nanoelectronic devices,1,2 high-density magnetic storage,3 catalysis,4 and solar cells.5 The fabrication of metal NTs has been achieved by various methods,2,6 with template-assisted electrodeposition being the most popular method.7 Two major growth mechanisms have been proposed for the electrodeposition of metal NTs within porous templates: current-directed and surface-directed. In the current-directed growth mechanism the morphology of the deposited structure is determined by the electrode shape and the value of the applied current. Under high current density, the annular base electrode employed for deposition induces tubular growth due to the ‘tip effect’ in which the rate of metal deposition is greater at the sharp pore edge compared to the transverse area.8 However, under low current density, the diffusion rate of metal ions overcomes the tip effect resulting in the formation of solid NWs. For the surface-directed growth mechanism the structure of the deposited material is based on the affinity of the metal ion species for the surface of the template, which results in an increased rate of metal reduction at the pore wall. Various methods of surface modification have been investigated to increase the metal ion concentration along the pore wall surface, including nanoparticle deposition,9 and adsorption or silanization of a chelating group.10,11
Both of these proposed mechanisms rely on a physical effect in order to generate tubular nanostructures, however it is important to fully understand the chemical effect that allows for NT growth. Therefore, in the presented work we demonstrate the synthesis of nickel NTs in the absence of annular electrodes and prior surface modification. Furthermore, we explore the NT growth mechanism within an anodized aluminium oxide (AAO) template in aqueous electrolyte solution, similar to the well-known Watts plating solution, containing nickel sulfate (NiSO4) and boric acid (H3BO3). Our standard electrolyte solution contained 0.5 M NiSO4 and 0.4 M H3BO3 at pH 4.2. Under these conditions, we propose a surface-directed NT growth mechanism based on the presence of boric acid.
Boric acid has long been viewed as a buffering agent for electrodeposition solutions.14,16 In addition, recent reports have cited its catalytic effect on the electrodeposition of metals.12,14,15 It has been suggested that boric acid enhances metal deposition due to its ability to lower the over-potential and increase current efficiency, but to the best of the authors’ knowledge a detailed mechanism of these effects has not been thoroughly described yet. We clearly demonstrate herein that boric acid plays a key role in the electrodeposition of nickel NTs by forming a surface-bound nickel-borate complex on the template wall.
In this work, NT growth was observed within a commercial AAO template in aqueous solutions with various pH and potential values. First, gold was sputtered on the bottom of the porous AAO membrane. Then gold NWs were gently electrodeposited from a commercial gold plating solution to form short, flat NWs at the bottom of the AAO in order to eliminate the tip effect on nickel deposition (Fig. S1). Figure 1 presents the electrodeposited Ni nanostructures from aqueous NiSO4 in the presence (Fig. 1A) and absence (Fig. 1B) of H3BO3. As was demonstrated by SEM and TEM imaging, deposition within the AAO nanochannel in our standard electrolyte solution produced nickel NTs with uniform wall thickness (Fig. 1A). However, NT formation was not observed in the absence of boric acid; instead only nickel NWs with NT ends were formed (Fig. 1B). Based on these results and previous reports from the literature,12,13 we attributed the NT growth to the presence of boric acid.
Fig. 1.

TEM images of nickel nanostructures synthesized by electrodeposition at −1V in electrolyte containing (a) 0.5 M NiSO4 and 0.4 M H3BO3 and (b) 0.5 M NiSO4. The scale bar shown in the inset represents 200 nm.
In order to test this hypothesis and to explore the mechanism behind it, we electrodeposited Ni in various concentrations of boric acid under a constant potential of −1V in 0.5 M NiSO4. Based on the morphology of the resultant nanostructures, we determined that the concentration of boric acid in the electrolyte controlled the thickness of the deposited NT wall. In the standard electrolyte solution (0.4 M H3BO3), NTs with thin, flexible walls were formed. The lack of rigidity caused the NTs to collapse onto one another, forming a film-like coverage over the flat-top electrodes (Fig. S2A). As the boric acid concentration was reduced the NT walls became thicker and more rigid until NW structures eventually formed in the absence of boric acid (Fig. S2C).
It is clear from these results that boric acid plays a significant role in the growth of nickel NTs. Boric acid is known to complex with metal ion species in aqueous solution, such as the nickel-borate complex shown in Scheme 1.14,16 Since boric acid can also complex with diol compounds, it can readily bind to the hydroxyl moieties on the alumina surface of the AAO template.17,18 We propose that the formation of these complexes dictate the shape of the deposited nickel nanostructures in the AAO template by modifying the local concentration of the Ni2+ ions in the proximity of the pore walls. The high concentration of boric acid in the electrolyte solution suggests that it will primarily be found in the bound state, hence a layer of surface-bound borate molecules are formed around the template pore wall. Given that the surface-bound borate molecules can complex with Ni2+ ions, the nickel concentration at the pore wall can increase nearly 60 times that of the bulk solution. As a result, preferential deposition of nickel along the pore wall will occur when a negative potential is applied, leading to the formation of nickel NTs (Scheme 2A).
Scheme 1.

Formation of the nickel-borate complex.
Scheme 2.
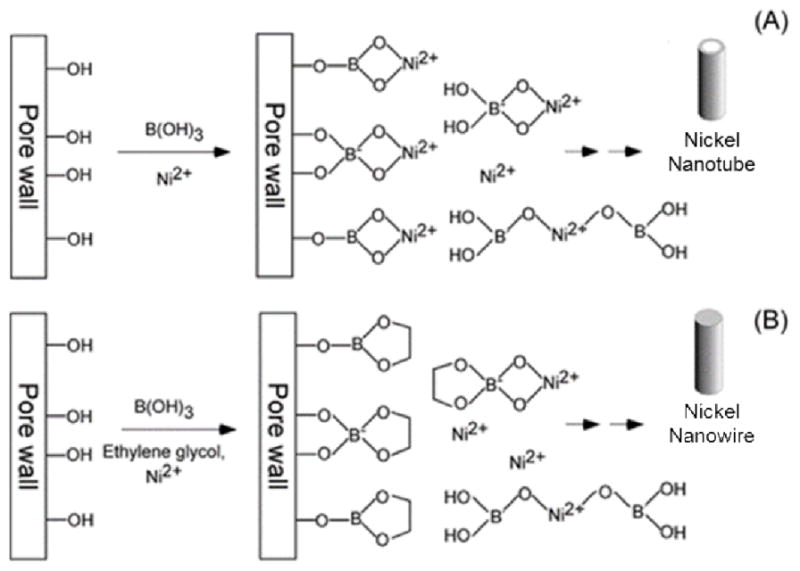
Schematic illustration of the formation of (a) nickel NTs in the presence of boric acid, and (b) nickel NWs in the presence of boric acid and ethylene glycol.
In order to test our hypothesis, unbound boric acid and nickel ions were removed from the template prior to deposition. First, the AAO membrane, with embedded Au NWs, was equilibrated in the standard electrolyte for several minutes. Then, the template was rinsed with acetonitrile and the deposition was performed in 1 M NaSO4. Figure S3 illustrates that despite the lack of bulk Ni ions, NT formation was achieved due to the presence of the surface-bound layer of nickel-borate on the template pore wall surface.
Further support of our hypothesis was obtained by inhibiting the formation of the nickel-borate ion complex with the addition of a competitive binder to the standard electrolyte. A hydroxyl-containing compound, such as ethylene glycol, can compete with the Ni2+ ions for complexation to boric acid. Since boric acid has a higher affinity for the competitive binder, adsorption of the Ni2+ ions to the surface-bound borate molecules was impeded. Scheme 2B illustrates that under these conditions, the Ni2+ concentration at the pore wall is not significantly increased thus deposition occurs more uniformly along the template pore to form NWs. Indeed, as presented in Figure 2, the addition of 0.4 M ethylene glycol to the standard electrolyte solution resulted in the formation of nickel NWs. Given that ethylene glycol does not readily react with Ni2+ ions,19 we concluded that NT formation was inhibited by its interaction with boric acid.
Fig. 2.

(a) SEM and (b) TEM images of nickel NWs electrodeposited from a 0.5M NiSO4 solution containing 0.4M H3BO3 and 0.4M ethylene glycol.
Following this proposed mechanism, NT formation can also be inhibited by preventing the adsorption of the nickel-borate complex to the surface of the template pore. For instance, when a polycarbonate (PC) membrane was used as the template in place of AAO, Ni NWs were formed due to the lack of hydroxyl moieties on the surface of the PC membrane. Since the nickel-borate complex could not adsorb to the PC surface, there was no significant increase in concentration or preferential deposition of nickel along the pore wall, which led to more uniform deposition across the nanochannel.
We further investigated the effect of boric acid on the deposition of nickel as NTs rather than NWs with the electrodeposition of nickel in the presence of alternative buffering agents. Figure S4 shows that the addition of 0.4 M acetic acid to the nickel plating solution resulted in the electrodeposition of nickel NWs. Similar NWs were also synthesized in the presence of 0.2 M citric acid (Fig. S5), indicating that nickel NT growth is mediated specifically by boric acid. It is important to note that nickel NTs were synthesized in the standard electrolyte over a pH range of 1.9 to 5.2 (Fig. S6). Therefore, slight pH changes accompanied by the different electrolyte conditions did not inhibit NT formation.
From these results, we conclude that boric acid plays a key role in the growth of nickel NTs, which is strongly dependent on the chemistry of the pore wall. Figure 4 presents further evidence for a growth mechanism regulated by the template surface chemistry. The branched NT structure is a clear indication that deposition of Ni2+ ions followed the template surface morphology. Commercial AAO membranes possess two sides; one with well-ordered, uniform pores and the other, referred to as the branched side, with nanochannels that are randomly interconnected. It is clear from Figure 4 that the branched morphology was maintained during NT synthesis. This demonstrates the influence of the template surface structure on the deposition process and supports our understanding of a surface-directed growth mechanism in which NT growth occurs due to the adsorption of the nickel-borate complex on the alumina surface of the template.
Figure 4.

TEM image of a nickel NT displaying branched structures formed from the branched AAO membrane.
Finally, to confirm that a current-directed mechanism was not a factor in NT growth under the specified conditions, we deposited nickel at a range of various current densities. As described above, in the current-directed mechanism NT growth is observed as a result of the tip effect. Under this mechanism, NT formation requires high current density to produce significantly faster growth along the pore wall. However, our results demonstrate that decreasing the current density from −3 to −0.3 mA/cm2 did not inhibit NT growth (Fig. S8). The lack of NW formation at low current density indicates that the current-directed mechanism plays a less significant role in nickel NT synthesis when flat-top electrodes are employed. Therefore, we conclude that the borate effect is responsible for the surface-directed growth mechanism of electrochemically synthesized nickel NTs.
In summary, we have provided a systematic investigation into the mechanism of template-assisted electrodeposition of nickel NTs in aqueous solution. It was demonstrated that under the specified conditions synthesis proceeded through a surface-directed growth mechanism in which borate acts as a dynamic surface modifier that binds Ni2+ ions to the AAO walls, thereby significantly increasing the nickel concentration along the template surface compared to the bulk solution. This phenomenon, which appears to be specific to borate, induced preferential deposition along the template wall and led to the formation of nickel NTs. Modifications to pH and applied potential did not inhibit NT formation when flat-top electrodes were employed. However, the addition of competitive borate binders to the electrolyte solution prevented the adsorption of the nickel-borate complex and resulted in the formation of NWs. Understanding the growth mechanism and the role of boric acid in NT formation more completely allows for further improvement of the electrodeposition process. Additionally, the borate effect may prove to be a general mechanism applicable to the synthesis of a variety of metal NTs. Therefore, we plan to investigate the borate effect on the synthesis of other metal and alloy nanostructures.
Supplementary Material
Acknowledgments
This work was supported by a grant from the NIH (GM 021248). SKK, MN and SBL were supported (electrochemical mechanism) as part of the Nanostructures for Electrical Energy Storage (NEES), an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DESC0001160. We also acknowledge the Maryland NanoCenter and its NISP Laboratory for the use of TEM, and Tim Maugel for expert SEM assistance.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental details; SEM and TEM images; current density profile. See DOI: 10.1039/b000000x/
Notes and references
- 1.Li L, Yang YW, Huang XH, Li GH, Ang R, Zhang LD. Appl Phys Lett. 2006;88:3. [Google Scholar]
- 2.Sander MS, Cote MJ, Gu W, Kile BM, Tripp CP. Adv Mater. 2004;16:2052. [Google Scholar]
- 3.Liu HR, Lu QF, Han XF, Liu XG, Xu BS, Jia HS. Appl Surf Sci. 2012;258:7401. [Google Scholar]
- 4.Luo Y, Lee S, Hofmeister H, Steinhart M, Gosele U. Nano Lett. 2004;4:143. [Google Scholar]; Niwa S, Eswaramoorthy M, Nair J, Raj A, Itoh N, Shoji H, Namba T, Mizukami F. Science. 2002;295:105. doi: 10.1126/science.1066527. [DOI] [PubMed] [Google Scholar]
- 5.Martinson ABF, Elam JW, Hupp JT, Pellin MJ. Nano Lett. 2007;7:2183. doi: 10.1021/nl070160+. [DOI] [PubMed] [Google Scholar]
- 6.Remskar M. Adv Mater. 2004;16:1497. [Google Scholar]; Martin CR, Nishizawa M, Jirage K, Kang MS, Lee SB. Adv Mater. 2001;13:1351. [Google Scholar]; Chu SZ, Kawamura H, Mori M. J Electrochem Soc. 2008;155:D414. [Google Scholar]; Sun YG, Xia YN. Adv Mater. 2004;16:264. [Google Scholar]
- 7.Han XF, Shamaila S, Sharif R, Chen JY, Liu HR, Liu DP. Adv Mater. 2009;21:4619. [Google Scholar]; Li XR, Wang YQ, Song GJ, Peng Z, Yu YM, She XL, Sun J, Li JJ, Li PD, Wang ZF, Duan XF. J Phys Chem C. 2010;114:6914. [Google Scholar]; Nicewarner-Pena SR, Freeman RG, Reiss BD, He L, Pena DJ, Walton ID, Cromer R, Keating CD, Natan MJ. Science. 2001;294:137. doi: 10.1126/science.294.5540.137. [DOI] [PubMed] [Google Scholar]
- 8.Yoo WC, Lee JK. Adv Mater. 2004;16:1097. [Google Scholar]; Cao H, Wang L, Qiu Y, Wu Q, Wang G, Zhang L, Liu X. Chemphyschem. 2006:7. doi: 10.1002/cphc.200500690. [DOI] [PubMed] [Google Scholar]; Guo YY, Wand M, Mao XB, Jiang YX, Wang C, Yang YL. Acta Phys-Chim Sin. 2010;26:2037. [Google Scholar]; Liu L, Zhou W, Xie S, Song L, Luo S, Liu D, Shen J, Zhang Z, Xiang Y, Ma W, Ren Y, Wang C, Wang G. J Phys Chem C. 2008;112 [Google Scholar]; Xiao R, Il Cho S, Liu R, Lee SB. J Am Chem Soc. 2007;129:4483. doi: 10.1021/ja068924v. [DOI] [PubMed] [Google Scholar]
- 9.Lee W, Scholz R, Niesch K, Gosele U. Angew Chem Int Edit. 2005:44. doi: 10.1002/anie.200501341. [DOI] [PubMed] [Google Scholar]
- 10.Bao JC, Tie CY, Xu Z, Zhou QF, Shen D, Ma Q. Adv Mater. 2001:13. [Google Scholar]; Liu L, Park S. Chem Mater. 2011;23:1456. [Google Scholar]; Brumlik CJ, Martin CR. J Am Chem Soc. 1991:113. [Google Scholar]
- 11.Chowdhury T, Casey DP, Rohan JF. Electrochem Commun. 2009;11:1203. [Google Scholar]
- 12.Hoare JP. J Electrochem Soc. 1987;134:3102. [Google Scholar]; Supicova M, Rozik R, Trnkova L, Orinakova R, Galova M. J Solid State Electrochem. 2005;10:61. [Google Scholar]
- 13.Lu J, Yang QH, Zhang Z. Trans Nonferrous Met Soc China. 2010;20:S97. [Google Scholar]
- 14.Ji J, Cooper WC, Dreisinger DB, Peters E. J Appl Electrochem. 1995;25:642. [Google Scholar]
- 15.Horkans J. J Electrochem Soc. 1979;126:1861. [Google Scholar]
- 16.Mukherjee G, Das A. J Indian Chem Soc. 2002;79:45. [Google Scholar]; Tilak B, Gendront A, Mosoiu M. J Appl Electrochem. 1977;7:495. [Google Scholar]
- 17.Springsteen G, Wang BH. Tetrahedron. 2002;58:5291. [Google Scholar]
- 18.Bouguerra W, Mnif A, Hamrouni B, Dhahbi M. Desalination. 2008;223:31. [Google Scholar]; Bouguerra W, Marzouk I, Hamrouni B. Water Environ Res. 2009;81:2455. [PubMed] [Google Scholar]
- 19.Furia TE. In: Stability constants of metal-ion complexes. Sillen LG, Martell AE, Bjerrum J, editors. Chemical Society; London: 1964. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.