

Abstract
Huntington’s disease (HD) is an incurable neurodegenerative disease characterized by abnormal motor movements, personality changes, and early death. HD is caused by a mutation in the IT-15 gene that expands abnormally the number of CAG nucleotide repeats. As a result, the translated protein huntingtin contains disease-causing expansions of glutamines (polyQ) that make it prone to misfold and aggregate. While the gene and mutations that cause HD are known, the mechanisms underlying HD pathogenesis are not. Here we will review the state of knowledge of HD, focusing especially on a hallmark pathological feature—intracellular aggregates of mutant Htt called inclusion bodies (IBs). We will describe the role of IBs in the disease. We speculate that IB formation could be just one component of a broader coping response triggered by misfolded Htt whose efficacy may depend on the extent to which it clears toxic forms of mutant Htt. We will describe how IB formation might be regulated and which factors could determine different coping responses in different subsets of neurons. A differential regulation of IB formation as a function of the cellular context could, eventually, explain part of the neuronal vulnerability observed in HD.
Keywords: Huntington’s disease, aggregation, chaperons, ubiquitin proteasome system, autophagy, striatal vulnerability
In Huntington’s disease (HD), other polyQ-dependent disorders, and familial forms of Alzheimer and Parkinson’s disease, symptom onset typically occurs in mid-life, despite the fact that individuals harbor and express the disease-causing mutation from birth. This implies that, early in life, toxicity is buffered by compensatory mechanisms that eventually yield to disease progression. Thus, proteotoxicity and coping responses overlap along the natural history of neurodegeneration. Since neuropathological investigations necessarily focus on the cells that survive—quite likely those that deal best with proteotoxicity—coping responses might be easier to detect than pathogenic ones. Determining whether a particular hallmark is a pathogenic mechanism or a coping response has important therapeutic consequences. In this review, we will consider the role of IBs in HD with these issues in mind.
HD is the most common form of inherited neurodegenerative disease. It is characterized by uncontrolled and excessive motor movements and cognitive and emotional deficits. Unfortunately, there is no treatment for this devastating disease, and death usually occurs 10–15 years after onset. The mutation that causes HD is known: an abnormal expansion of CAG repeats in the IT15 gene results in an autosomal dominant trait (Huntington’s Disease Collaborative Research Group, 1993). The huntingtin (Htt) protein has an abnormal number of glutamine repeats (polyQ). The normal gene contains 6–34 CAG repeats, but a person with a gene exceeding 40 repeats will inevitably develop HD if the person lives long enough. The age of onset correlates inversely with the length of the CAG repeats. Typically, symptoms begin with chorea in mild-life, and other neurological deficits and changes in personality follow. Interestingly, polyQ expansions in other proteins lead to different neurodegenerative diseases, also in a polyQ length–dependent manner. In addition to HD, polyQ-dependent disorders include the spinocerebellar ataxias (SCA1, SCA2, SCA3, SCA7), spinobulbar muscular atrophy (SBMA), and dentatorubropallidoluysian atrophy (DRPLA) (Orr and Zoghbi, 2007).
A deep comprehension of the mechanisms by which polyQ expansions lead to neuronal death in HD is needed to find therapeutic targets to prevent or cure this disease.
Inclusions bodies and Huntington’s disease
Small-animal models are powerful research tools. Soon after discovery of the mutation that causes HD, transgenic lines of mice expressing the first exon of the human HD gene were developed as disease models (Mangiarini, et al., 1996). Of several successful lines with different numbers of disease-associated CAG repeat expansions (115–156), the R6/2 line was the most-extensively characterized and commonly used for HD research. These mice developed a complex and progressive neurological phenotype, with motor abnormalities and premature death, reminiscent of some features of HD.
With the help of the models, a pathological hallmark of HD was soon discovered. Immunostaining with an antibody against abnormal polyQ expansions revealed circular, densely stained intraneuronal inclusions (Davies, et al., 1997). IBs were located in the striatum, cerebral cortex, cerebellum, and the spinal cord. They were specific for mutant Htt and often showed ubiquitin immunoreactivity. Very importantly, immunostaining of HD brains also revealed Htt- and ubiquitin-positive intranuclear inclusions (Becher, et al., 1998, DiFiglia, et al., 1997). Although these initial reports of HD brains described inclusions primarily in the nucleus, subsequent work also found them in the cytoplasm and in neuronal processes (Gutekunst, et al., 1999).
The idea that IBs cause HD was intuitively appealing. They are a pathological hallmark of HD. In initial reports, IBs in transgenic mouse models and human HD brains were closely correlated with HD symptoms. They were found in neurons before the onset of behavioral symptoms and significant neuronal death (Davies, et al., 1997, Ordway, et al., 1997). But if IBs cause HD, how might they do it?
Several hypotheses were proposed. Normal Htt interacts with proteins of the cytoskeleton-based transport, receptor endocytosis and synaptic vesicle recycling (Caviston and Holzbaur, 2009, Harjes and Wanker, 2003, Qin, et al., 2004). Mutant Htt aggregation into IBs might disrupt normal synaptic transmission. Additionally, the aggregation process driven by polyQs might sequester essential proteins, such as transcription factors (McCampbell, et al., 2000, Nucifora, et al., 2001, Steffan, et al., 2000), proteasomes or other ubiquitine proteasome system (UPS) components (Cummings, et al., 1998, Donaldson, et al., 2003) between others (Suhr, et al., 2001). Hence, sequestration of proteins into IBs might trigger different effects, such as transcriptional deregulation or proteasome impairment, affecting neuronal survival. However, several studies found that the extent of protein sequestration (transcription factors and proteasome components) into IBs was not biologically significant (Bennett, et al., 2005, Yu, et al., 2002). Instead, functional sequestration of transcription factors and UPS impairment can occur prior IB formation (Bennett, et al., 2005, Mitra, et al., 2009, Schaffar, et al., 2004).
Interestingly, immunohistochemistry again provided some curious hints. Studies of HD brains revealed a surprising discrepancy between the vulnerability of specific neuronal subsets and IB localization. Early neuropathology reports indicated that the corpus striatum (caudate nucleus, putamen and globus pallidus) was severely affected in HD. In 1985, a system was established for grading the neuropathological severity of the striatum of HD brains. Based upon macroscopic and microscopic criteria, this system assigned patients to one of several grades (0–6, from less to more severe) (Vonsattel, et al., 1985). The best macroscopic indicator of disease severity in the striatum was the atrophy of the caudate-accumbens-putamen and globus pallidus. For microscopic histopathological evaluation, neurons and astrocytes were counted to assess the relative neuronal loss and gliosis. As the grade increased, atrophy and gliosis concomitant with neuronal loss were found to increase in the striatum. The majority of neurons in the striatum are projection neurons, and the enkephalin-positive projection neurons appeared to be mostly affected in HD (Reiner, et al., 1988, Sapp, et al., 1995). At higher grades, cerebral cortex also displayed atrophy and a decrease in neuronal numbers (Vonsattel and DiFiglia, 1998).
Closer examination of the mutant Htt aggregates revealed, however, that they were much more common in the cerebral cortex than in the striatum at a time when cortical neuronal loss was low but striatal neuronal loss was significant (Gutekunst, et al., 1999). Among the neuronal populations in the striatum, aggregates occurred predominantly in interneurons instead of in the more vulnerable medium spiny projection neurons (Kuemmerle, et al., 1999). Therefore, IB localization was not more prevalent in neuronal subpopulations known to die earliest in HD. Furthermore, the presence of IBs and neuronal death did not seem to correlate either. In a primary striatal model of HD, IB formation was recapitulated but IB formation could be dissociated from neuronal death with several experimental manipulations (Saudou, et al., 1998). Additionally, transgenic mice that express mutant ataxin-1 without an essential region for dimerization develop ataxia but do not form IBs (Klement, et al., 1998).
What then is the role of inclusions bodies in Huntington’s disease?
These observations led us to doubt that IB formation has a causative role in HD. Then, what is the role of IB formation in HD neurodegeneration? Are IBs toxic, incidental, or part of a beneficial coping response? And how can we sort out these possibilities?
Inferring causal relationships between a pathogenic feature and neuronal fate is difficult. Conventional approaches provide only “snapshots” of different sets of neurons at different time points. It is tempting to interpret differences observed at later times as an evolution of changes seen at earlier times, but the populations of cells visualized at different time points might be unrelated. New technologies were needed to overcome the limitations of conventional approaches.
We took a different approach: we wanted to follow the same living neurons expressing a particular protein of interest over long times and monitor their survival (“longitudinal survival analysis”). By observing the same neuron repeatedly, we reasoned that we could link the appearance of a particular feature with its ultimate fate. We developed a robotic microscope to do just this. By amassing a very large amount of data on individual neurons, we could also adapt survival analysis to quantitatively determine if a feature predicts neuronal fate (i.e., death). In addition, in the case of multiple factors, survival analysis could quantify the risk associated with each factor, providing a way to rank them in order of importance (Arrasate and Finkbeiner, 2005, Finkbeiner, et al., 2006).
This innovative and powerful approach helped us to resolve the controversial relationship between mutant Htt IBs and death. By applying longitudinal survival analysis to a primary striatal neuronal model of HD, we found that diffuse forms of mutant Htt predicted neuronal death. Unexpectedly, we also found that IB formation reduced the levels of diffuse mutant Htt and the risk of striatal neuronal death. We hypothesized that IB formation reduces levels of toxic misfolded proteins from the diffuse fraction by sequestering them in IBs. Thus, IB formation might be a beneficial coping response, helpful in ameliorating toxicity. In fact, for neurons that form IBs, the risk of death, which is highly correlated to levels of diffuse mutant Htt before IB formation, becomes substantially independent of those levels after an IB forms and for the remainder of that neuron’s lifetime. The finding suggests that IB formation marks a new adapted state for the neuron (Arrasate, et al., 2004, Miller, et al., 2010).
Further research on HD mouse models has confirmed that mutant Htt aggregation and toxicity are not connected. A mouse model expressing an N-terminal human Htt fragment (exons 1 and 2) with 120Qs under the control of the endogenous human promoter showed frequent and widespread IB formation but no evidence of neuronal dysfunction or neurodegeneration (Slow, et al., 2005). Pharmacological promotion of IB formation is starting now to be conceivable as a therapeutic approach for HD (Bodner, et al., 2006). Paradoxically, strategies for reducing aggregation have also been proved beneficial in different mouse models. For example, a small molecule (C2-8) that inhibits aggregation in a yeast-based assay (Zhang, et al., 2005) improves motor performance and reduces neuronal atrophy in the R6/2 mouse model. Interestingly, this molecule reduces the size of the aggregates but not the number (Chopra, et al., 2007). This compound might work by modulating mutant Htt levels and/or the initial steps of Htt misfolding that make it prone to aggregation and hence indirectly affect aggregate formation. Posttranslational modifications could also modulate mutant Htt levels and/or its propensity to misfold and aggregate. In this sense, HD mouse models with mutations on residues susceptible to phosphorylation alter the degradation of mutant Htt and, therefore, the rate of IB formation simultaneously to the survival outcome (Gu, et al., 2009). Interventions or manipulations that prevent Htt misfolding and the generation of toxic species would also be predicted to prevent the need by cells to generate a coping response and could explain why some interventions lead to less IB formation and a better outcome. This fact will be reviewed more extensively in the next section.
Cellular regulation of IB formation
If IB formation is a coping process, might it also be regulated? Other lines of evidence suggest just that. The quantitative relationship between mutant Htt levels and IB formation was recently extended by mining large data sets generated by our automated microscope. The analysis led to the discovery that IB formation saturates with the longest polyQ lengths (Miller, et al., 2010). These findings suggest that IB formation is a regulated process governed by interaction with limited cellular targets and therefore with saturable molecular mechanisms.
Intrinsic factors that influence mutant Htt aggregation capacity
Mutant Htt aggregation is influenced by several intrinsic factors, including the length of the polyQ stretch, the amino acid sequences flanking the polyQ stretch, and the particular conformation of mutant Htt.
The observation that the insertion of a disease-associated polyQ expansion in a heterologous protein could lead to IB formation suggested that polyQ expansions might be sufficient in certain peptide contexts to drive the formation of IBs (Ordway, et al., 1997). A structural model showing that polyQ strands might form a β-sheet structure, held together tightly through hydrogen bonds, suggested a mechanism whereby polyQ expansions could mediate the formation of insoluble aggregates (Perutz, et al., 1994). Of the intrinsic factors contributing to IB formation, the length of the polyQ stretch is most important. Scherzinger and colleagues developed an elegant system based on heterologous chimeric GST-Htt fusion proteins expressed in Escherichia coli to study polyQ-dependent aggregation in vitro. When the GST tag was proteolytically cleaved, aggregation of Htt-containing proteins occurred according to the length of the polyQ stretch (Scherzinger, et al., 1997, Scherzinger, et al., 1999). This principle was recapitulated in the budding yeast (Krobitsch and Lindquist, 2000) and primary cultures of striatal neurons (Arrasate, et al., 2004, Miller, et al., 2010).
Besides the length of the polyQ expansions, the aggregation mechanism involves other intrinsic factors, such as the polyQ-flanking sequences and the particular conformation of mutant Htt. The two domains that flank the polyQ stretch: the prolinerich region downstream of the polyQ domain and the 17–amino acid (N17) peptide at the N-terminal end of Htt greatly influence mutant Htt aggregation capacity. For example, in immortalized striatal cells, the proline-rich region is necessary to form visible IBs of mutant Httex1 (Steffan, et al., 2004). Deleting this region in yeast dramatically influences the morphology of the aggregates, yielding several amorphous dispersed aggregates instead of one or two tightly focused aggregates (Dehay and Bertolotti, 2006, Duennwald, et al., 2006). By comparison, attaching a proline-rich sequence C-terminal to a polyQ peptide decreases the rate of aggregation of the peptide in vitro (Bhattacharyya, et al., 2006). In summary, the proline-rich domain seems to modulate both the rate and mode of aggregation, but the mechanisms by which it contributes to IB formation remain elusive.
Recently, a role in aggregation was proposed for the first 17 amino acids in Htt, N-terminal to the polyQ-stretch (N17). A mutant Httex1 lacking N17 displayed delayed aggregation in vitro and in cellular models. Interestingly, in vitro, the effect on the aggregation kinetics occurs both in cis and trans, since addition of a synthetic N17 peptide promoted aggregation of the deleted construct (Rockabrand, et al., 2007, Tam, et al., 2009, Thakur, et al., 2009). This finding suggests that this portion of Htt has an important role in seeding the aggregation process.
Analysis of purified IBs from HD brains demonstrated the presence of a broad range N-terminal fragments of mutant Htt (DiFiglia, et al., 1997, Hoffner, et al., 2005). Proteolytic cleavage of mutant Htt is thought to lead to the generation of N-terminal fragments and to increase aggregation (Martindale, et al., 1998). Caspases, calpains and aspartic proteases are candidate proteases. Inhibition of cleavage with specific protease inhibitors, as well as mutation of consensus sites for caspases and calpains in Htt, reduces the frequency of aggregate formation (Gafni and Ellerby, 2002, Gafni, et al., 2004, Lunkes, et al., 2002, Wellington, et al., 2000). The specificity of the N-terminal fragments generated by cleavage of mutant Htt might even differentially affect the localization (nuclear versus cytoplasmic) of IB formation (Lunkes, et al., 2002). Interestingly, mice expressing full-length mutant Htt with a mutation of residue 586 (caspase-6 resistant) do not develop striatal degeneration. This result points to proteolytic cleavage of mutant Htt not only as modulator of IB formation but also as an important event in HD progression (Graham, et al., 2006).
The tight correlation among polyQ length, flanking regions, and the temporal pattern of IB formation has been consistently reproduced in genetic mouse models of HD neurodegeneration. The emerging pattern indicates that the longer the polyQ stretch and the shorter the flanking regions, the earlier IBs accumulate (Ferrante, 2009).
The particular monomeric conformation adopted by mutant Htt also influences aggregation. IBs are the culmination of an aggregation process that likely comprises a complex series of intermediate steps. The polyQ-stretch might influence the conformation of normal Htt monomers, resulting in misfolded monomers. These misfolded monomers might even adopt different conformational states before assembling into oligomers, early aggregate precursors that presumably incorporate into IBs. There is in vitro support for this hypothesis: Schaffar and colleagues used intramolecular FRET (Förster resonance energy transfer) to show that soluble monomers of mutant Httex1 displayed a conformational rearrangement before their oligomerization (Schaffar, et al., 2004). From circular dichroism analysis, polyQ expansions undergo a β-sheet conformational transition even as a monomeric protein (Nagai, et al., 2007). Both the proline-rich and N17 flanking regions of the polyQ-stretch may act to stabilize a β-turn, facilitating self-assembly and incorporation into fibrils and, therefore, enhancing aggregation (Lakhani, et al., 2010, Tam, et al., 2009). Importantly, in these studies, the conformational change in Htt preceded its incorporation into aggregates. Finally, experiments involving mutant Htt and atomic force microscopy show that multiple monomeric conformations may coexist, and some might be more prone to aggregation than others (Wacker, et al., 2004).
Modifiers of Htt aggregation
The group of Htt-interacting partners described to date is quite large and includes proteins involved in diverse cellular roles, such as vesicle transport, transcriptional regulation and clathrin-mediated endocytosis between others (Goehler, et al., 2004, Harjes and Wanker, 2003, Kaltenbach, et al., 2007, Li and Li, 2004). A detailed description of the biological meaning of these interactions is beyond the scope of this paper. However, to a large extend, experimental manipulation of the expression of these proteins modulates aggregation of mutant Htt into IB formation. Although the mechanisms are not established in most cases, it is conceivable that expression of huntingtin partners might influence IB formation by diverse mechanisms, for example, changes in subcellular localization or in the ability to bind to another partner among others. In this section we will focus on two general categories of proteins that modulate mutant Htt aggregation. The first group includes proteins that modify mutant Htt posttranslationally and changes the effective concentration of the protein. The second group includes molecular chaperones that will affect mutant Htt conformation.
1) Proteins that modify mutant Htt posttranslationally
Posttranslational modifications that affect mutant Htt degradation would modulate protein levels and consequently IB formation. The two major protein degradation pathways are the ubiquitin-proteasome system (UPS) (non/lysosomal degradative pathway) and autophagy (a lysosome degradative pathway) (Kirkin, et al., 2009). There are three types of autophagy: macroautophagy, microautophagy and chaperone-dependent autophagy. In this review we will refer mainly to macroautophagy, the autophagy type that requires sequestration of substrates in a double membrane vesicle (autophagosome) which then fuses with the lysosome.
Ubiquitin-positive IBs have been found in HD brains and mouse models (DiFiglia, et al., 1997, Sieradzan, et al., 1999) (Davies, et al., 1997). In fact, Htt itself is ubiquitinated (Jana, et al., 2001, Steffan, et al., 2004, Waelter, et al., 2001), and yeast two-hybrid experiments suggest that it interacts with hE2-25K, a specific ubiquitin-conjugating enzyme (Kalchman, et al., 1996). Traditionally ubiquitination has been considered a targeting signal for degradation by the UPS. However, the involving of ubiquitin as a specific factor for selective autophagy is emerging. Selective autophagy requires specific recognition of protein and organelles that are going to be engulfed, in contrast to what is considered non-selective or “bulk” autophagy. Ubiquitin and ubiquitin-binding receptors could be involved in this specific cargo recognition (Kraft, et al., 2010). Therefore, ubiquitinated mutant Htt would be targeted for degradation to the proteasome and/or to the autophagy system, potentially decreasing protein levels and IB formation. The relationship between mutant Htt and the UPS is complex because mutant Htt might cause proteasome impairment leading to IB formation. This issue will be reviewed more extensively in the next section.
The same ubiquitinated lysine residues are targets of sumoylation (Steffan, et al., 2004), a covalent protein modification with small ubiquitin-like modifiers (SUMO). Sumoylation stabilizes mutant Htt and reduces the formation of visible SDS-insoluble aggregates (Steffan, et al., 2004). Rhes, a striatal-specific protein, interacts with mutant Htt (Subramaniam, et al., 2009) and may function as a physiologic regulator to enhance mutant Htt sumoylation (Subramaniam, et al., 2010). Interestingly, Rhes overexpression reduces IB formation, increases levels of soluble mutant Htt, and increases mutant Htt cytotoxicity.
Mutant Htt is also acetylated in HD brains and in HD models (Aiken, et al., 2009, Jeong, et al., 2009). Acetylation at lysine 444 facilitates Htt trafficking into autophagosomes and improves clearance of the protein by macroautophagy. However, mutation of lysine 444 to be resistant to acetylation causes mutant Htt to accumulate without forming visible aggregates in cultured neurons and mouse brains (Jeong, et al., 2009). CREB binding protein (CBP), a histone acetyltransferase, which also interacts with mutant Htt (Steffan, et al., 2001), and HDAC1, a histone deacetylase, strongly increase and decrease acetylation of lysine 444, respectively. Therefore, acetylation may be an interesting mechanism that promotes degradation of mutant Htt by autophagy.
In addition to ubiquitination, sumoylation and acetylation, Htt is subject to palmitoylation at cysteine 214. Palmitoylation is a reversible post-translational modification of cysteine residues by the lipid palmitate that facilitates protein interaction with lipid bilayers, affecting protein sorting and function (Fukata and Fukata, 2010). HIP14, a mammalian palmitoyl transferase, interacts with and palmitoylates Htt (Huang, et al., 2004, Singaraja, et al., 2002). HIP14/Htt interaction is inversely correlated to the polyQ length. Hence, palmitoylation is decreased in mutant Htt vs. the wild-type form in a polyQ-dependent manner. Interestingly, both chemical inhibition of palmitoylation and direct mutation of the cysteine 214 increase the frequency of IBs. Down-regulation of HIP14 with siRNA on primary cultures from mouse models expressing the full-length mutant Htt (YAC128) increases IB formation. Conversely, overexpression of HIP14 substantially reduces IB formation and causes redistribution of endogenous Htt and, to a lesser extent, mutant Htt to the Golgi. These results suggest that palmitoylation regulates the localization of Htt. PolyQ expansions in mutant Htt decrease palmitoylation, and consequently mutant Htt neuronal localization might be affected. Therefore, palmitoylation may regulate IB formation via relocalization of mutant Htt (Yanai, et al., 2006).
Finally, phosphorylation regulates mutant Htt degradation and thus aggregate formation. BAC transgenic mice expressing full-length mutant Htt with serines 13 and 16 mutated to either aspartate (phosphomimetic) or alanine (phosphoresistant) altered the aggregation capacity of mutant Htt (Gu, et al., 2009). In these HD mouse models, phosphorylation of serines 13 and 16 might abrogate HD pathogenesis. Therefore, identifying the kinases involved in Htt phosphorylation might constitute an important step to find therapeutic strategies against HD. Among the candidate kinases, the inflammatory kinase IKK might phosphorylate serine 13 and lead to phosphorylation of serine 16. Phosphorylation of these residues promotes ubiquitination and sumoylation of the adjacent lysines and targets mutant Htt for degradation by the proteasome and lysosomes (Thompson, et al., 2009). That might explain why transgenic mice expressing polyQ-expanded Htt with serines mutated to mimic phosphorylation do not develop aggregates. Recently, high-content screening experiments showed that casein kinase 2 (CK2) inhibitors prevent the phosphorylation of these Htt residues (Atwal, et al., 2011). Paradoxically, inhibitors of IKK enhance Htt phosphorylation. These experiments implicate CK2 and IKK in the modulation of Htt phosphorylation, but it remains to be determined if these inhibitors directly affect Htt phosphorylation or if other Htt-interacting proteins, substrates of these kinases, mediate indirectly the effect. Htt is also phosphorylated at serine 421 by Akt, which reduces its toxicity (Humbert et al., 2002). Phosphorylation of Htt at serine 421 might mitigate toxicity by modulating BDNF vesicle transport (Colin, et al., 2008, Humbert, et al., 2002, Zala, et al., 2008). In addition to the serines, phosphorylation of threonine 3 in Drosophila and mouse HD models increases aggregation and modifies the toxic properties of mutant Htt (Aiken, et al., 2009).
2) Molecular chaperones that affect mutant Htt conformation
Molecular chaperones could also alter the conformational state of mutant Htt and act as potent modulators of aggregation. The effect of chaperones on polyQ-expanded protein aggregation has been extensively studied (see (Muchowski and Wacker, 2005)). Among the most important chaperones are Hsp70 and its co-chaperones Hsp40 and TRiC. Hsp70 and Hsp40 prevent formation of polyQ IBs and modulate the aggregation process in vitro (Muchowski, et al., 2000, Wacker, et al., 2004), in cellular yeast models (Krobitsch and Lindquist, 2000), and Drosophila (Chan, et al., 2000). Interestingly, loss of Hsp70 increases the size but not the number of IBs in a mouse model of HD (Wacker, et al., 2009). In vitro studies suggest that Hsp70/Hsp40 affects the conformation of monomers before oligomerization and, in that way, determines the formation of particular oligomeric species (Schaffar, et al., 2004, Wacker, et al., 2004).
The chaperonin TRiC was identified as a modulator of aggregation in a genome-wide RNA interference screen in Caenorhabditis elegans (Nollen, et al., 2004). It inhibits aggregation. In in vitro experiments, TRiC interfered with polyQ aggregation by blocking or slowing monomer addition to growing fibrils. TRiC may interact directly with mutant Htt monomers or small oligomers to block the N17 amphipathic mutant Htt sequence element that promotes aggregation (Tam, et al., 2006, Tam, et al., 2009). In conditionally TRiC-defective yeast cells, the amount of SDS-insoluble aggregates increases, whereas overexpression of TRiC suppresses polyQ aggregation. Of particular interest is the observation that TRiC might specifically favor formation of ~500-KDa soluble oligomers over a ~200-KDa population of oligomers (Behrends, et al., 2006, Tam, et al., 2006). In agreement with these results, overexpression of TRiC inhibits aggregation of mutant Htt in cellular models (Kitamura, et al., 2006). Interestingly, TRiC may cooperate with Hsp70. In vitro, aggregation experiments with a GST-mutant Htt fusion protein, in the presence of Hsp70, Hsp40, and TRiC, generated ~500-kDa SDS-soluble oligomers. Preincubation of GST-mutant Htt with Hsp70/Hsp40 and later addition of TRiC resulted in formation of the same oligomers, whereas reverse addition did not. These experiments suggest an organized action of chaperones on the early steps of folding of mutant Htt monomers: an initial interaction with Hsp70/Hsp40 is followed by an interaction with TRiC (Behrends, et al., 2006).
In summary, posttranslational modifications that include ubiquitination, sumoylation, and acetylation, influence the targeting of mutant Htt to degradation pathways, such as the proteasome and autophagy, and modulate IB formation. Phosphorylation might determine whether mutant Htt should be ubiquitinated, sumoylated or acetylated and therefore may have a central role in the pathway selected for degradation. Palmitoylation emerges as a posttranslational modification that is impaired by polyQ expansions, which might affect IB formation by determining neuronal trafficking and localization of Htt. Additionally, although further investigation in cellular models is needed, an important cooperative mechanism between chaperones, such as Hsp70/Hsp40 or TRiC, might occur in which the intervention at the very early folding steps of monomeric mutant Htt determine the identity of the oligomers formed and the outcome of the aggregation process.
An important question to be resolved is which monomeric conformers and/or oligomeric forms of mutant Htt are the toxic species. What does the rate of neuronal death in HD suggest about the features of a candidate toxic species? An interesting study demonstrated that in HD the kinetics of neuronal death is better explained by mathematical models in which the risk of neuronal death remains constant (Clarke, et al., 2000). In this study, measurements of 18F-doxyglucose uptake in the caudate nucleus of HD patients were performed as indirect measures of neuronal loss. They found that neurodegeneration in these patients was best described by a constant risk of death. In agreement with these data, the risk of death in cellular HD models is also constant until up to 6 days after transfection with exon-1 mutant Htt (Miller, et al., 2010). Of these studies is suggested that a candidate toxic species would probably be present tonically. Recently, determination of molecular size of mutant Htt species in mouse neuroblastoma cells with sedimentation velocity experiments revealed a population of oligomers that remained constant over time (Olshina, et al., 2010) pointing to this species as potentially toxic. Both monomeric and oligomeric forms have been correlated with toxicity. We reviewed how polyQ expansions undergo a β-sheet conformational transition in a monomeric state. This conformational transition in monomers has been associated with cytotoxic (Nagai, et al., 2007). Additionally, the presence of soluble oligomers of mutant Htt in yeast correlates with a growth defect (Behrends, et al., 2006) and with toxicity in neuroblastoma differentiated cells (Takahashi, et al., 2008). The particular conformation that the polyQs in mutant Htt adopt might determine its toxicity. The latest data support the idea that polyQ might adopt a compact β structure that would confer its toxicity in both monomeric and oligomeric forms of Htt (Poirier, et al., 2005, Zhang, et al., 2011).
Despite this evidence, identifying mutant Htt conformational changes and mutant Htt oligomerization in vivo, as well as assessing their pathological significance, remains technically challenging. However, new tools are becoming available. For example, conformation-specific antibodies (Kayed, et al., 2003, Legleiter, et al., 2009, Peters-Libeu, et al., 2005) are reliable and useful tools for distinguishing mutant Htt monomeric conformations and oligomers. Conformational sensors of mutant Htt that distinguish monomeric proteins from oligomers in live cells have been also developed. These are based on the binding of biarsenical ligands to tetracysteine protein domains (Ramdzan, et al., 2010). These tools should help to assess the role that monomeric and oligomeric mutant Htt species have in striatal neurodegeneration.
What orchestrates the balance between mutant Htt degradation and IB formation?
IB formation is regulated by the gain (production) or loss (degradation) of mutant Htt. The main pathways for mutant Htt degradation are the UPS and autophagy (Sarkar and Rubinsztein, 2008). As explained, ubiquitination can target Htt for degradation by the autophagy and/or the UPS.
The fact that IBs in HD are ubiquitinated led to hypothesize that mutant Htt might impair the ability of the proteasome to degrade mutant Htt itself and other intracellular protein substrates. Experimental support for UPS impairment in HD comes mainly from a mass-spectrometry study that quantified abundance of polyubiquitin chains as a biomarker of UPS function in mouse HD models and human HD brains (Bennett, et al., 2007). IB formation initially appeared the culprit for this impairment. Abnormal accumulation of a fluorescent reporter of proteasomal activity in live cells in presence of IBs led to the idea that IB formation inhibits directly proteasome function (Bence, et al., 2001). However, subsequent studies by the same group and others with techniques that improved greatly the temporal resolution revealed that the accumulation of proteasomal reporter actually preceded IB formation, excluding the possibility that IB formation was the cause of accumulation of the reporter. In fact, IB formation reduces the accumulation of the proteasome reporter (Bennett, et al., 2005, Mitra, et al., 2009).
These results were confirmed in an inducible mouse model that co-expresses mutant Htt and a fluorescent reporter of the UPS (Ortega, et al., 2010). Accumulation of the UPS reporter occurs transiently after acute induction of mutant Htt expression, and restoration of normal proteasome activity correlates with the appearance of IB formation. No accumulation of the UPS reporter is observed later in vivo at symptomatic stages when the presence of IBs is generalized. The transient detectable accumulation of the proteasome reporter may explain why it was not observed in other polyQ disease mouse models with widespread IB formation (Bowman, et al., 2005, Maynard, et al., 2009).
All these results strongly suggest that species of mutant Htt smaller than IBs are causing an accumulation of the proteasomal reporter, phenomenon that it has been generally referred as “proteasomal impairment.” However, two different interpretations equally explain this observation. First, the mutant Htt degradation process is difficult and causes a “jam” in the proteasome. Second, there is a competition between substrates for degradation. This might occur if mutant Htt is preferentially selected, leading to accumulation of other substrates, or if the stress that mutant Htt places on the protein homeostasis system leads to widespread misfolding of other metastable proteins. The increase in the overall load of proteins targeted to the proteasome could therefore lead to the accumulation of proteasome reporters because the capacity of the proteasome is exceeded even if flux through the proteasome was not impaired at all. In any case, IB formation restores normal activity of the UPS probably because it reduces the competition for the UPS by lowering the load of proteins targeted to the UPS (Figure 1).
Figure 1. Model for cellular regulation of coping responses to the presence of misfolded mutant Htt.
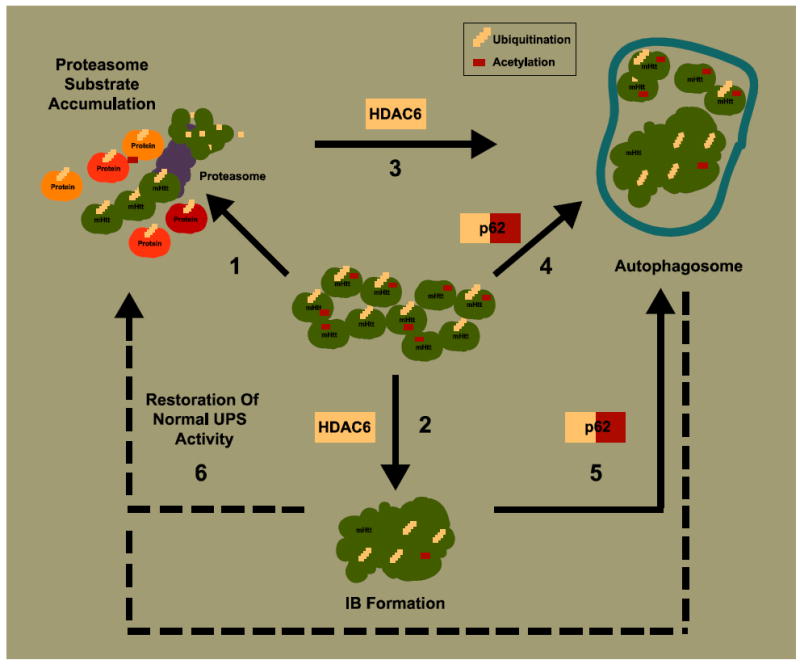
(1) Expression of mutant Htt causes accumulation of substrates requiring proteasome degradation. This might be due to an increase in the amounts of other misfolded proteins that compete for proteasomal degradation or to impairment of proteasome function or both. “Sensor” proteins that recognize ubiquitinated and acetylated forms of mutant Htt, such as HDAC6 and p62, might facilitate IB formation and autophagy activation. (2) HDAC6 binds ubiquitin and favors accumulation of ubiquitinated protein into IBs. (3) HDAC6 also induces autophagy as a response to polyQ-dependent proteasome substrate accumulation. (4) p62 interacts with acetylated mutant Htt potentially targeting the protein to degradation by autophagy. (5) p62 also recognizes ubiquitin and might recruit autophagosomal components to degrade non-aggregated and aggregated forms of ubiquitinated mutant Htt. (6) IB formation and autophagy induction would reduce the load of misfolded mutant Htt protein and potentially restore normal ubiquitin proteasome system (UPS) activity.
Does a neuron “sense” too much ubiquitinated mutant Htt in a way that determines IB formation and eventually restore normal UPS activity? If so, HDAC6, a cytoplasmic deacetylase that interacts with ubiquitin, may be such a sensor (Hook, et al., 2002, Seigneurin-Berny, et al., 2001). HDAC6 binds ubiquitin monomers with high affinity, which mediates its ability to negatively control the cellular polyubiquitin turnover and favors the aggregation of polyubiquitinated proteins (Boyault, et al., 2006). Through its association with the dynein motor complex (Kawaguchi, et al., 2003), it also transports ubiquitinated aggregates to aggresomes (microtubule-dependent inclusion bodies (Kopito, 2000)). Therefore, HDAC6 could be directly controlling the levels of ubiquitinated mutant Htt and the IB formation process (Figure 1).
HDAC6 may also be an essential link between the UPS and autophagy. Mutant Htt accumulates in structures that resemble endosomal-lysosomal organelles in brains from HD patients (Sapp, et al., 1997), and autophagosomes containing mutant Htt have been described in cellular and mouse HD models (Davies and Scherzinger, 1997, Kegel, et al., 2000). Thus, mutant Htt seems to be targeted to autophagosomes for degradation. Further, proteasome impairment by polyQ expansions in Htt has been reported to induce autophagy (Iwata, et al., 2005). In a Drosophila model, expression of the androgen receptor with an abnormal polyQ expansion impairs proteasome function and causes a degenerative phenotype (Pandey, et al., 2007). Interestingly, autophagy was induced in an HDAC6-dependent manner as a compensatory mechanism to counteract the polyQ-dependent proteasome impairment. HDAC6 accelerated the turnover of mutant androgen receptors by autophagy. Upon expression of HDAC6, the degenerative phenotype was suppressed. The deacetylase function of HDAC6 was required for this effect, which may seem paradoxical given that mutant Htt requires acetylation (at least at K444) for targeting to autophagosomes (Jeong, et al., 2009). However, the role of HDAC6 in polyQ-dependent autophagy activation could also involve other mechanisms, such as the recruitment of the autophagy machinery to aggresomes (Iwata, et al., 2005). Therefore, further investigation is required to clarify the mechanisms by which HDAC6 participates in the polyQ induction of autophagy.
p62, a polyubiquitin-binding protein, is another good candidate for sensing ubiquitinated mutant Htt and controlling aggregate formation. Expression of p62 is increased in mutant Htt expressing cells, and p62 co-localizes with IBs (Nagaoka, et al., 2004). Importantly, p62, via interaction with LC3 in autophagosomes, is involved in linking polyubiquitinated protein aggregates to the autophagy machinery (selective autophagy). It might recruit autophagosomal components to polyubiquitinated aggregates, facilitating the clearance of such aggregates and even non-aggregated forms of mutant Htt (Bjorkoy, et al., 2005, Komatsu, et al., 2007, Tung, et al., 2010). Along with recognizing ubiquitinated Htt, p62 interacts with acetylated mutant Htt, which could further contribute to degradation by targeting mutant Htt for autophagy (Jeong, et al., 2009). In considering the role of autophagy, one must consider that, in the context of mutant Htt, autophagosomes form efficiently. However, they may be not fully functional due to a failure in cargo recognition that eventually may lead to inefficient autophagy (Martinez-Vicente, et al., 2010).
Thus, HDAC6 and p62 could be key cellular proteins that govern the fate of ubiquitinated and acetylated polyQ-containing proteins (e.g., mutant Htt) by activating a broad coping response that includes IB formation, activation of autophagy, and other mechanisms. The efficacy of this response to improve neuronal survival would depend on the extent to which misfolded toxic mutant Htt is cleared. IB formation and autophagy would decrease toxic diffuse forms of mutant Htt. This would involve restoration of normal proteasome activity or a better match between the load of misfolded protein to the protein homeostasis system and the available capacity of that system to handle it (Figure 1). Eventually, autophagy may participate in the final clearance of IBs (Ravikumar, et al., 2004, Tsvetkov, et al., 2010).
Could specific neuronal subtypes initiate and mediate IB formation differently? Roles for cell autonomous and non-autonomous mechanisms
We have reviewed two observations about IBs and HD. First, IB formation is dissociated from the vulnerability of different neuronal types and regions of HD brains. Instead, IB formation appears to be part of a broader coping response that acts coordinately with the UPS and autophagy. These findings suggest that specific neuronal subtypes modulate IB formation differently and that this property contributes to survival. What evidence supports this assertion?
In primary neuronal models with mutant Htt expressed by adenoviral vectors, different aggregation patterns were observed among cortical, striatal or cerebellar neurons (Tagawa, et al., 2004). We then measured the risk of IB formation in a longitudinal study of single cortical and striatal primary neurons expressing mutant Htt. Cortical neurons had a higher risk of IB formation than striatal neurons, even with the same dosage of mutant Htt (unpublished results). Thus, distinct subtypes of neurons might regulate IB formation differently and some perhaps in a cell-autonomous manner. Among the candidates for variable modulation of IB formation would be HDAC6, p62, the UPS and autophagy. Variations in the activity of the UPS have been assessed in primary cultures of neurons and astrocytes from rat cerebral cortex upon adenoviral infection with a fluorescent reporter of proteasome function. Cortical neurons accumulated more reporter than astrocytes (Tydlacka, et al., 2008). Whether cortical neurons have lower UPS activity or experience more competition with the fluorescence reporter for the UPS, the result suggests that neurons and glia have cell-specific differences in their protein homeostasis networks. Still, there is no clear evidence of different activities of the UPS and autophagy systems among neuronal subtypes, and the same applies for differential activities of HDAC6 and p62. Differences on other components of the protein homeostasis, such us molecular chaperones, might also play a role in the variable modulation of IB formation. Expression of Hsp70 chaperone could be up-regulated differently in primary granule cells of rat cerebellum than in cortical neurons upon adenoviral expression of mutant Htt (Tagawa, et al., 2007). Further research is warranted to identify key neuron-specific protein players that are involved in the different cell-autonomous mechanisms regulating of IB formation.
When thinking in mechanisms by which mutant Htt cause selective toxicity of striatal neurons, two potential models have to be taken in consideration. Expression of mutant Htt in vulnerable neurons is sufficient to cause dysfunction and neurodegeneration (cell-autonomous mechanisms), or contribution of other cell types (mainly form cortical and nigral afferents) increases the risk of neurodegeneration of the vulnerable ones (non-cell-autonomous mechanisms). Conditional mouse models for HD were created in which the expression of mutant Htt was selectively restricted to specific areas of the brain (Gu, et al., 2007, Gu, et al., 2005). In these models, whereas striatal pathogenesis required non-cell-autonomous mechanisms, IB formation appeared to be a cell-autonomous process.
Other experimental evidence indicates, however, a potential role for non-cell-autonomous mechanisms in IB formation. The striatum receives significant dopaminergic input from the substantia nigra, and dopamine might regulate IB formation since increases aggregation of mutant Htt on primary neuronal and cellular models of HD (Charvin, et al., 2005, Robinson, et al., 2008). Both D1 and D2 receptors could be involved in aggregate modulation. Recently, an interesting non-cell-autonomous mechanism was proposed in which synaptic activity regulates IB formation (Okamoto, et al., 2009). The authors showed that inhibition of NMDA receptors in cortical and striatal primary neurons expressing mutant Htt with the NMDA receptor antagonists APV, memantine or ifenprodil decreased the number of IBs. The balance between synaptic and extra-synaptic NMDA receptor activity seems to critically determine IB formation. A mechanism was proposed in which synaptic activation of NMDA receptors induce expression of the chaperonin TRiC, favoring IB formation. By contrast, extra-synaptic activation of NMDA receptors was proposed to increase Rhes expression and enhance soluble mutant Htt by sumoylation, hence decreasing IB formation. In a previous section, we reviewed how TRiC acts as a potent modulator of mutant Htt aggregation, inhibiting IB formation (Nollen, et al., 2004, Tam, et al., 2009). Interestingly, a cooperative action of Hsp70/Hsp40 with TRiC might modulate the formation of soluble oligomers (Behrends, et al., 2006, Tam, et al., 2006). Although the authors reported that Hsp70 co-localized with IBs, further investigation of the mechanism is required to link synaptic activity–mediated expression of TRiC with oligomer formation and aggregation. In particular, the role of the Hsp70/Hsp40 chaperone system must be examined. Nevertheless, the model proposed by Okamoto et al. provides a plausible mechanistic link between excitotoxicity, striatal vulnerability, and IB formation.
Excitotoxicity mediated by glutamate has been proposed to explain the selective striatal vulnerability in HD (DiFiglia, 1990). YAC transgenic HD mouse models expressing full-length mutant Htt are more susceptible to neuronal death induced by striatal injections of quinolinate and NMDA (Zeron, et al., 2002). This excitotoxic effect could be mediated by NR2B-containing NMDA receptors (Heng, et al., 2009). An exacerbated glutamatergic input to the striatum (mainly from cortical afferents and astrocytes) or an increase in the number and/or activity of NMDA receptors would result in excessive NMDA receptor activity and higher striatal excitotoxicity. In agreement with this idea, alterations in the corticostriatal pathway that suggest abnormal release of glutamate have been reported in mouse models of HD (Cepeda, et al., 2007). Additionally, astrocytes that help to buffer glutamate levels on the striatum may have impaired uptake of glutamate upon mutant Htt expression (Faideau, et al., 2010). Finally, mutant Htt might evoke excitotoxicity by producing changes in NMDA receptors. In fact, a reported increase of NR2B-containing extra-synaptic NMDA receptors in the striatum and extra-synaptic NMDA receptor signaling might contribute to the phenotypic onset in the YAC transgenic HD mouse model (Milnerwood, et al., 2010).
For all these mechanisms, the presence of mutant Htt might actually result in an increase of the glutamatergic input to the striatum, leading to excessive extra-synaptic activation of NMDA receptors and changes in gene expression in the striatum (i.e., Rhes). As a consequence, an altered processing of mutant Htt might favor toxic soluble forms over more aggregated forms (Figure 2). This non-cell-autonomous mechanism might contribute to our understanding of how excitotoxicity and coping responses, such as IB formation, and explain the selective vulnerability of striatal neurons. More importantly, it could have therapeutic implications too. Pharmacological blockage of extra-synaptic NMDA receptors activity improves neuropathological and behavioral deficits of the YAC HD mouse model by eventually favoring activation of pathways that would enhance IB formation (Milnerwood, et al., 2010, Okamoto, et al., 2009).
Figure 2. Potential non-cell-autonomous mechanisms for regulating IB formation in the striatum.
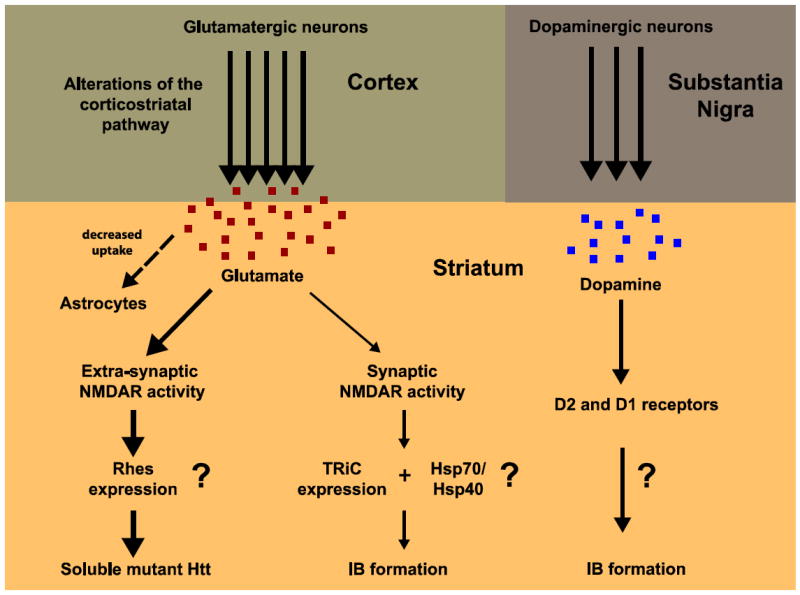
Glutamatergic and dopaminergic neuronal projections are the main afferents to the striatum from the cortex and the substantia nigra, respectively. (Left) Extra-synaptic and synaptic activity mediated by glutamate could modulate IB formation. An excess of glutamatergic stimulation might occur in the striatum of HD brains through different mechanisms: increased release from cortical afferents, decreased uptake by striatal astrocytes, and changes in NMDA receptor (NMDAR) number/subunit composition and localization. Excessive extra-synaptic NMDAR-mediated activity might drive gene expression that stabilizes soluble toxic forms of mutant Htt (thicker arrows), leading to neuronal death. Pharmacological blockage of extra-synaptic NMDAR activity would eventually restore an appropriated balance between extra-synaptic and synaptic activity, favoring gene expression that would increase IB formation and neuroprotection. (Right) Dopamine might also modulate IB formation in the striatum through D2 and/or D1 receptors, via unknown mechanisms.
Summary and final remarks
IB formation, as one of the most important hallmarks of HD, was generally considered to be the main suspect in the pathogenic process of neurodegeneration. Here, we review a growing body of evidence that suggests the IB formation process is part of a broader cellular coping response. Acting together with the UPS and autophagy systems, IB formation might help to reduce the levels of diffuse, toxic mutant Htt.
The incorporation of mutant Htt into IBs depends on the concentration of mutant Htt and on its conformational status. Here, we discussed that cis-acting determinants—the length of the polyQ stretch or the sequences flanking it—and the action of molecular chaperones define specific conformational states of mutant Htt that predispose its aggregation into IBs. On the other hand, a range of post-translational modifications in the mutant Htt molecule (e.g., ubiquitination, acetylation and sumoylation) control the effective concentration of mutant Htt by targeting the protein to degradation via either the UPS or autophagy. In the hierarchy of Htt modifiers, kinases may steer mutant Htt towards a particular post-translational modification and drive the mode of mutant Htt turnover. These findings suggest that IB formation results from the intertwined action of multiple mechanisms controlling mutant Htt folding and turnover.
Finally, mutant Htt displays different toxicity and IB formation in different neuronal subtypes, implying a differential regulation of the mechanisms described here. From that perspective, it is tempting to speculate on the existence of sensing molecules that detect levels of mutant Htt and trigger the clearance of mutant Htt or its accumulation into IBs. Candidates for such a role have been proposed that recognize ubiquitinated polyQ-containing proteins and trigger IB formation, as well as proteasomal and autophagic activity. Further research is needed to identify key neuron-specific protein sensors that regulate IB formation as part of a broader coping response in a cell-autonomous manner. But, interestingly enough, a non-cell-autonomous regulation mechanism was recently proposed in which synaptic activity regulates IB formation. The combination of cell-autonomous and non-cell-autonomous mechanisms regulating the proteastasis of mutant Htt provides a plausible framework to understand the specific striatal vulnerability in HD.
Acknowledgments
We thank Tomás Aragón, Isabel Perez-Otaño and members of the Finkbeiner lab for useful discussions and revision of the manuscript. We thank Gary Howard for editorial assistance, and Kelley Nelson for administrative assistance. Primary support for this work was provided by the Ramón y Cajal Program, Ministry of Science and Innovation (M.A), the FP7-Marie Curie-IRG Program, European Commission (M.A.) and UTE-project / Foundation for Applied Medical Research (M.A.), the National Institute of Neurological Disease and Stroke 2R01 NS039074 and 2R01045091 (S.F.). Additional support was provided by the National Institute on Aging (2P01 AG022074), the Taube-Koret Center for Huntington’s Disease Research, the Hellman Family Foundation for Alzheimer’s Disease Research, and the J. David Gladstone Institutes (S.F.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aiken CT, Steffan JS, Guerrero CM, Khashwji H, Lukacsovich T, Simmons D, Purcell JM, Menhaji K, Zhu YZ, Green K, Laferla F, Huang L, Thompson LM, Marsh JL. Phosphorylation of threonine 3: implications for Huntingtin aggregation and neurotoxicity. J Biol Chem. 2009;284:29427–29436. doi: 10.1074/jbc.M109.013193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arrasate M, Finkbeiner S. Automated microscope system for determining factors that predict neuronal fate. Proc Natl Acad Sci U S A. 2005;102:3840–3845. doi: 10.1073/pnas.0409777102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 4.Atwal RS, Desmond CR, Caron N, Maiuri T, Xia J, Sipione S, Truant R. Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat Chem Biol. 2011;7:453–460. doi: 10.1038/nchembio.582. [DOI] [PubMed] [Google Scholar]
- 5.Becher MW, Kotzuk JA, Sharp AH, Davies SW, Bates GP, Price DL, Ross CA. Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis. 1998;4:387–397. doi: 10.1006/nbdi.1998.0168. [DOI] [PubMed] [Google Scholar]
- 6.Behrends C, Langer CA, Boteva R, Bottcher UM, Stemp MJ, Schaffar G, Rao BV, Giese A, Kretzschmar H, Siegers K, Hartl FU. Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol Cell. 2006;23:887–897. doi: 10.1016/j.molcel.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 7.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 8.Bennett EJ, Bence NF, Jayakumar R, Kopito RR. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell. 2005;17:351–365. doi: 10.1016/j.molcel.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 9.Bennett EJ, Shaler TA, Woodman B, Ryu KY, Zaitseva TS, Becker CH, Bates GP, Schulman H, Kopito RR. Global changes to the ubiquitin system in Huntington’s disease. Nature. 2007;448:704–708. doi: 10.1038/nature06022. [DOI] [PubMed] [Google Scholar]
- 10.Bhattacharyya A, Thakur AK, Chellgren VM, Thiagarajan G, Williams AD, Chellgren BW, Creamer TP, Wetzel R. Oligoproline effects on polyglutamine conformation and aggregation. J Mol Biol. 2006;355:524–535. doi: 10.1016/j.jmb.2005.10.053. [DOI] [PubMed] [Google Scholar]
- 11.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bodner RA, Outeiro TF, Altmann S, Maxwell MM, Cho SH, Hyman BT, McLean PJ, Young AB, Housman DE, Kazantsev AG. Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington’s and Parkinson’s diseases. Proc Natl Acad Sci U S A. 2006;103:4246–4251. doi: 10.1073/pnas.0511256103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowman AB, Yoo SY, Dantuma NP, Zoghbi HY. Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum Mol Genet. 2005;14:679–691. doi: 10.1093/hmg/ddi064. [DOI] [PubMed] [Google Scholar]
- 14.Boyault C, Gilquin B, Zhang Y, Rybin V, Garman E, Meyer-Klaucke W, Matthias P, Muller CW, Khochbin S. HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J. 2006;25:3357–3366. doi: 10.1038/sj.emboj.7601210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caviston JP, Holzbaur EL. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 2009;19:147–155. doi: 10.1016/j.tcb.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cepeda C, Wu N, Andre VM, Cummings DM, Levine MS. The corticostriatal pathway in Huntington’s disease. Prog Neurobiol. 2007;81:253–271. doi: 10.1016/j.pneurobio.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan HY, Warrick JM, Gray-Board GL, Paulson HL, Bonini NM. Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum Mol Genet. 2000;9:2811–2820. doi: 10.1093/hmg/9.19.2811. [DOI] [PubMed] [Google Scholar]
- 18.Charvin D, Vanhoutte P, Pages C, Borrelli E, Caboche J. Unraveling a role for dopamine in Huntington’s disease: the dual role of reactive oxygen species and D2 receptor stimulation. Proc Natl Acad Sci U S A. 2005;102:12218–12223. doi: 10.1073/pnas.0502698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chopra V, Fox JH, Lieberman G, Dorsey K, Matson W, Waldmeier P, Housman DE, Kazantsev A, Young AB, Hersch S. A small-molecule therapeutic lead for Huntington’s disease: preclinical pharmacology and efficacy of C2-8 in the R6/2 transgenic mouse. Proc Natl Acad Sci U S A. 2007;104:16685–16689. doi: 10.1073/pnas.0707842104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarke G, Collins RA, Leavitt BR, Andrews DF, Hayden MR, Lumsden CJ, McInnes RR. A one-hit model of cell death in inherited neuronal degenerations. Nature. 2000;406:195–199. doi: 10.1038/35018098. [DOI] [PubMed] [Google Scholar]
- 21.Colin E, Zala D, Liot G, Rangone H, Borrell-Pages M, Li XJ, Saudou F, Humbert S. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008;27:2124–2134. doi: 10.1038/emboj.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- 23.Davies SW, Scherzinger E. Nuclear inclusions in Huntington’s disease. Trends Cell Biol. 1997;7:422. doi: 10.1016/S0962-8924(97)88136-6. [DOI] [PubMed] [Google Scholar]
- 24.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 25.Dehay B, Bertolotti A. Critical role of the proline-rich region in Huntingtin for aggregation and cytotoxicity in yeast. J Biol Chem. 2006;281:35608–35615. doi: 10.1074/jbc.M605558200. [DOI] [PubMed] [Google Scholar]
- 26.DiFiglia M. Excitotoxic injury of the neostriatum: a model for Huntington’s disease. Trends Neurosci. 1990;13:286–289. doi: 10.1016/0166-2236(90)90111-m. [DOI] [PubMed] [Google Scholar]
- 27.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 28.Donaldson KM, Li W, Ching KA, Batalov S, Tsai CC, Joazeiro CA. Ubiquitin-mediated sequestration of normal cellular proteins into polyglutamine aggregates. Proc Natl Acad Sci U S A. 2003;100:8892–8897. doi: 10.1073/pnas.1530212100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duennwald ML, Jagadish S, Muchowski PJ, Lindquist S. Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc Natl Acad Sci U S A. 2006;103:11045–11050. doi: 10.1073/pnas.0604547103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Faideau M, Kim J, Cormier K, Gilmore R, Welch M, Auregan G, Dufour N, Guillermier M, Brouillet E, Hantraye P, Deglon N, Ferrante RJ, Bonvento G. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington’s disease subjects. Hum Mol Genet. 2010;19:3053–3067. doi: 10.1093/hmg/ddq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferrante RJ. Mouse models of Huntington’s disease and methodological considerations for therapeutic trials. Biochim Biophys Acta. 2009;1792:506–520. doi: 10.1016/j.bbadis.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finkbeiner S, Cuervo AM, Morimoto RI, Muchowski PJ. Disease-modifying pathways in neurodegeneration. J Neurosci. 2006;26:10349–10357. doi: 10.1523/JNEUROSCI.3829-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fukata Y, Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci. 2010;11:161–175. doi: 10.1038/nrn2788. [DOI] [PubMed] [Google Scholar]
- 34.Gafni J, Ellerby LM. Calpain activation in Huntington’s disease. J Neurosci. 2002;22:4842–4849. doi: 10.1523/JNEUROSCI.22-12-04842.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gafni J, Hermel E, Young JE, Wellington CL, Hayden MR, Ellerby LM. Inhibition of calpain cleavage of huntingtin reduces toxicity: accumulation of calpain/caspase fragments in the nucleus. J Biol Chem. 2004;279:20211–20220. doi: 10.1074/jbc.M401267200. [DOI] [PubMed] [Google Scholar]
- 36.Goehler H, Lalowski M, Stelzl U, Waelter S, Stroedicke M, Worm U, Droege A, Lindenberg KS, Knoblich M, Haenig C, Herbst M, Suopanki J, Scherzinger E, Abraham C, Bauer B, Hasenbank R, Fritzsche A, Ludewig AH, Bussow K, Coleman SH, Gutekunst CA, Landwehrmeyer BG, Lehrach H, Wanker EE. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington’s disease. Mol Cell. 2004;15:853–865. doi: 10.1016/j.molcel.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 37.Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, Warby SC, Doty CN, Roy S, Wellington CL, Leavitt BR, Raymond LA, Nicholson DW, Hayden MR. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 38.Gu X, Andre VM, Cepeda C, Li SH, Li XJ, Levine MS, Yang XW. Pathological cell-cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington’s disease. Mol Neurodegener. 2007;2:8. doi: 10.1186/1750-1326-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gu X, Greiner ER, Mishra R, Kodali R, Osmand A, Finkbeiner S, Steffan JS, Thompson LM, Wetzel R, Yang XW. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron. 2009;64:828–840. doi: 10.1016/j.neuron.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu X, Li C, Wei W, Lo V, Gong S, Li SH, Iwasato T, Itohara S, Li XJ, Mody I, Heintz N, Yang XW. Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron. 2005;46:433–444. doi: 10.1016/j.neuron.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 41.Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harjes P, Wanker EE. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci. 2003;28:425–433. doi: 10.1016/S0968-0004(03)00168-3. [DOI] [PubMed] [Google Scholar]
- 43.Heng MY, Detloff PJ, Wang PL, Tsien JZ, Albin RL. In vivo evidence for NMDA receptor-mediated excitotoxicity in a murine genetic model of Huntington disease. J Neurosci. 2009;29:3200–3205. doi: 10.1523/JNEUROSCI.5599-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoffner G, Island ML, Djian P. Purification of neuronal inclusions of patients with Huntington’s disease reveals a broad range of N-terminal fragments of expanded huntingtin and insoluble polymers. J Neurochem. 2005;95:125–136. doi: 10.1111/j.1471-4159.2005.03348.x. [DOI] [PubMed] [Google Scholar]
- 45.Hook SS, Orian A, Cowley SM, Eisenman RN. Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes. Proc Natl Acad Sci U S A. 2002;99:13425–13430. doi: 10.1073/pnas.172511699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang K, Yanai A, Kang R, Arstikaitis P, Singaraja RR, Metzler M, Mullard A, Haigh B, Gauthier-Campbell C, Gutekunst CA, Hayden MR, El-Husseini A. Huntingtin-interacting protein HIP14 is a palmitoyl transferase involved in palmitoylation and trafficking of multiple neuronal proteins. Neuron. 2004;44:977–986. doi: 10.1016/j.neuron.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 47.Humbert S, Bryson EA, Cordelieres FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F. The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves Huntingtin phosphorylation by Akt. Dev Cell. 2002;2:831–837. doi: 10.1016/s1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- 48.Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 49.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 50.Jana NR, Zemskov EA, Wang G, Nukina N. Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum Mol Genet. 2001;10:1049–1059. doi: 10.1093/hmg/10.10.1049. [DOI] [PubMed] [Google Scholar]
- 51.Jeong H, Then F, Melia TJ, Jr, Mazzulli JR, Cui L, Savas JN, Voisine C, Paganetti P, Tanese N, Hart AC, Yamamoto A, Krainc D. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell. 2009;137:60–72. doi: 10.1016/j.cell.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kalchman MA, Graham RK, Xia G, Koide HB, Hodgson JG, Graham KC, Goldberg YP, Gietz RD, Pickart CM, Hayden MR. Huntingtin is ubiquitinated and interacts with a specific ubiquitin-conjugating enzyme. J Biol Chem. 1996;271:19385–19394. doi: 10.1074/jbc.271.32.19385. [DOI] [PubMed] [Google Scholar]
- 53.Kaltenbach LS, Romero E, Becklin RR, Chettier R, Bell R, Phansalkar A, Strand A, Torcassi C, Savage J, Hurlburt A, Cha GH, Ukani L, Chepanoske CL, Zhen Y, Sahasrabudhe S, Olson J, Kurschner C, Ellerby LM, Peltier JM, Botas J, Hughes RE. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007;3:e82. doi: 10.1371/journal.pgen.0030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 55.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 56.Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, DiFiglia M. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci. 2000;20:7268–7278. doi: 10.1523/JNEUROSCI.20-19-07268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34:259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 58.Kitamura A, Kubota H, Pack CG, Matsumoto G, Hirayama S, Takahashi Y, Kimura H, Kinjo M, Morimoto RI, Nagata K. Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat Cell Biol. 2006;8:1163–1170. doi: 10.1038/ncb1478. [DOI] [PubMed] [Google Scholar]
- 59.Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 60.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 61.Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 62.Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12:836–841. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- 63.Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc Natl Acad Sci U S A. 2000;97:1589–1594. doi: 10.1073/pnas.97.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. Huntington aggregates may not predict neuronal death in Huntington’s disease. Ann Neurol. 1999;46:842–849. [PubMed] [Google Scholar]
- 65.Lakhani VV, Ding F, Dokholyan NV. Polyglutamine induced misfolding of huntingtin exon1 is modulated by the flanking sequences. PloS Comput Biol. 2010;6:e1000772. doi: 10.1371/journal.pcbi.1000772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Legleiter J, Lotz GP, Miller J, Ko J, Ng C, Williams GL, Finkbeiner S, Patterson PH, Muchowski PJ. Monoclonal antibodies recognize distinct conformational epitopes formed by polyglutamine in a mutant huntingtin fragment. J Biol Chem. 2009;284:21647–21658. doi: 10.1074/jbc.M109.016923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004;20:146–154. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 68.Lunkes A, Lindenberg KS, Ben-Haiem L, Weber C, Devys D, Landwehrmeyer GB, Mandel JL, Trottier Y. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell. 2002;10:259–269. doi: 10.1016/s1097-2765(02)00602-0. [DOI] [PubMed] [Google Scholar]
- 69.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 70.Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim SU, Bredesen DE, Tufaro F, Hayden MR. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet. 1998;18:150–154. doi: 10.1038/ng0298-150. [DOI] [PubMed] [Google Scholar]
- 71.Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, Cuervo AM. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maynard CJ, Bottcher C, Ortega Z, Smith R, Florea BI, Diaz-Hernandez M, Brundin P, Overkleeft HS, Li JY, Lucas JJ, Dantuma NP. Accumulation of ubiquitin conjugates in a polyglutamine disease model occurs without global ubiquitin/proteasome system impairment. Proc Natl Acad Sci U S A. 2009;106:13986–13991. doi: 10.1073/pnas.0906463106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, Merry D, Chai Y, Paulson H, Sobue G, Fischbeck KH. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet. 2000;9:2197–2202. doi: 10.1093/hmg/9.14.2197. [DOI] [PubMed] [Google Scholar]
- 74.Miller J, Arrasate M, Shaby BA, Mitra S, Masliah E, Finkbeiner S. Quantitative Relationships between Huntingtin Levels, Polyglutamine Length, Inclusion Body Formation, and Neuronal Death Provide Novel Insight into Huntington’s Disease Molecular Pathogenesis. J Neurosci. 2010;30:10541–10550. doi: 10.1523/JNEUROSCI.0146-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM, Hines RM, Boyd JD, Ko RW, Vasuta OC, Graham RK, Hayden MR, Murphy TH, Raymond LA. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron. 2010;65:178–190. doi: 10.1016/j.neuron.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 76.Mitra S, Tsvetkov AS, Finkbeiner S. Single neuron ubiquitin-proteasome dynamics accompanying inclusion body formation in Huntington disease. J Biol Chem. 2009;284:4398–4403. doi: 10.1074/jbc.M806269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU. Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc Natl Acad Sci U S A. 2000;97:7841–7846. doi: 10.1073/pnas.140202897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 79.Nagai Y, Inui T, Popiel HA, Fujikake N, Hasegawa K, Urade Y, Goto Y, Naiki H, Toda T. A toxic monomeric conformer of the polyglutamine protein. Nat Struct Mol Biol. 2007;14:332–340. doi: 10.1038/nsmb1215. [DOI] [PubMed] [Google Scholar]
- 80.Nagaoka U, Kim K, Jana NR, Doi H, Maruyama M, Mitsui K, Oyama F, Nukina N. Increased expression of p62 in expanded polyglutamine-expressing cells and its association with polyglutamine inclusions. J Neurochem. 2004;91:57–68. doi: 10.1111/j.1471-4159.2004.02692.x. [DOI] [PubMed] [Google Scholar]
- 81.Nollen EA, Garcia SM, van Haaften G, Kim S, Chavez A, Morimoto RI, Plasterk RH. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc Natl Acad Sci U S A. 2004;101:6403–6408. doi: 10.1073/pnas.0307697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 83.Okamoto S, Pouladi MA, Talantova M, Yao D, Xia P, Ehrnhoefer DE, Zaidi R, Clemente A, Kaul M, Graham RK, Zhang D, Vincent Chen HS, Tong G, Hayden MR, Lipton SA. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med. 2009;15:1407–1413. doi: 10.1038/nm.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Olshina MA, Angley LM, Ramdzan YM, Tang J, Bailey MF, Hill AF, Hatters DM. Tracking mutant huntingtin aggregation kinetics in cells reveals three major populations that include an invariant oligomer pool. J Biol Chem. 2010;285:21807–21816. doi: 10.1074/jbc.M109.084434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ordway JM, Tallaksen-Greene S, Gutekunst CA, Bernstein EM, Cearley JA, Wiener HW, Dure LSt, Lindsey R, Hersch SM, Jope RS, Albin RL, Detloff PJ. Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell. 1997;91:753–763. doi: 10.1016/s0092-8674(00)80464-x. [DOI] [PubMed] [Google Scholar]
- 86.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 87.Ortega Z, Diaz-Hernandez M, Maynard CJ, Hernandez F, Dantuma NP, Lucas JJ. Acute polyglutamine expression in inducible mouse model unravels ubiquitin/proteasome system impairment and permanent recovery attributable to aggregate formation. J Neurosci. 2010;30:3675–3688. doi: 10.1523/JNEUROSCI.5673-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 89.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci U S A. 1994;91:5355–5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peters-Libeu C, Newhouse Y, Krishnan P, Cheung K, Brooks E, Weisgraber K, Finkbeiner S. Crystallization and diffraction properties of the Fab fragment of 3B5H10, an antibody specific for disease-causing polyglutamine stretches. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61:1065–1068. doi: 10.1107/S1744309105036547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Poirier MA, Jiang H, Ross CA. A structure-based analysis of huntingtin mutant polyglutamine aggregation and toxicity: evidence for a compact beta-sheet structure. Hum Mol Genet. 2005;14:765–774. doi: 10.1093/hmg/ddi071. [DOI] [PubMed] [Google Scholar]
- 92.Qin ZH, Wang Y, Sapp E, Cuiffo B, Wanker E, Hayden MR, Kegel KB, Aronin N, DiFiglia M. Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction. J Neurosci. 2004;24:269–281. doi: 10.1523/JNEUROSCI.1409-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ramdzan YM, Nisbet RM, Miller J, Finkbeiner S, Hill AF, Hatters DM. Conformation sensors that distinguish monomeric proteins from oligomers in live cells. Chem Biol. 2010;17:371–379. doi: 10.1016/j.chembiol.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 95.Reiner A, Albin RL, Anderson KD, D’Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. 1988;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Robinson P, Lebel M, Cyr M. Dopamine D1 receptor-mediated aggregation of N-terminal fragments of mutant huntingtin and cell death in a neuroblastoma cell line. Neuroscience. 2008;153:762–772. doi: 10.1016/j.neuroscience.2008.02.052. [DOI] [PubMed] [Google Scholar]
- 97.Rockabrand E, Slepko N, Pantalone A, Nukala VN, Kazantsev A, Marsh JL, Sullivan PG, Steffan JS, Sensi SL, Thompson LM. The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum Mol Genet. 2007;16:61–77. doi: 10.1093/hmg/ddl440. [DOI] [PubMed] [Google Scholar]
- 98.Sapp E, Ge P, Aizawa H, Bird E, Penney J, Young AB, Vonsattel JP, DiFiglia M. Evidence for a preferential loss of enkephalin immunoreactivity in the external globus pallidus in low grade Huntington’s disease using high resolution image analysis. Neuroscience. 1995;64:397–404. doi: 10.1016/0306-4522(94)00427-7. [DOI] [PubMed] [Google Scholar]
- 99.Sapp E, Schwarz C, Chase K, Bhide PG, Young AB, Penney J, Vonsattel JP, Aronin N, DiFiglia M. Huntingtin localization in brains of normal and Huntington’s disease patients. Ann Neurol. 1997;42:604–612. doi: 10.1002/ana.410420411. [DOI] [PubMed] [Google Scholar]
- 100.Sarkar S, Rubinsztein DC. Huntington’s disease: degradation of mutant huntingtin by autophagy. FEBS J. 2008;275:4263–4270. doi: 10.1111/j.1742-4658.2008.06562.x. [DOI] [PubMed] [Google Scholar]
- 101.Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 102.Schaffar G, Breuer P, Boteva R, Behrends C, Tzvetkov N, Strippel N, Sakahira H, Siegers K, Hayer-Hartl M, Hartl FU. Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol Cell. 2004;15:95–105. doi: 10.1016/j.molcel.2004.06.029. [DOI] [PubMed] [Google Scholar]
- 103.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 104.Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, Bates GP, Lehrach H, Wanker EE. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington’s disease pathology. Proc Natl Acad Sci U S A. 1999;96:4604–4609. doi: 10.1073/pnas.96.8.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Seigneurin-Berny D, Verdel A, Curtet S, Lemercier C, Garin J, Rousseaux S, Khochbin S. Identification of components of the murine histone deacetylase 6 complex: link between acetylation and ubiquitination signaling pathways. Mol Cell Biol. 2001;21:8035–8044. doi: 10.1128/MCB.21.23.8035-8044.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sieradzan KA, Mechan AO, Jones L, Wanker EE, Nukina N, Mann DM. Huntington’s disease intranuclear inclusions contain truncated, ubiquitinated huntingtin protein. Exp Neurol. 1999;156:92–99. doi: 10.1006/exnr.1998.7005. [DOI] [PubMed] [Google Scholar]
- 107.Singaraja RR, Hadano S, Metzler M, Givan S, Wellington CL, Warby S, Yanai A, Gutekunst CA, Leavitt BR, Yi H, Fichter K, Gan L, McCutcheon K, Chopra V, Michel J, Hersch SM, Ikeda JE, Hayden MR. HIP14, a novel ankyrin domain-containing protein, links huntingtin to intracellular trafficking and endocytosis. Hum Mol Genet. 2002;11:2815–2828. doi: 10.1093/hmg/11.23.2815. [DOI] [PubMed] [Google Scholar]
- 108.Slow EJ, Graham RK, Osmand AP, Devon RS, Lu G, Deng Y, Pearson J, Vaid K, Bissada N, Wetzel R, Leavitt BR, Hayden MR. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc Natl Acad Sci U S A. 2005;102:11402–11407. doi: 10.1073/pnas.0503634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, Pandolfi PP, Thompson LM, Marsh JL. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- 110.Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 111.Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Subramaniam S, Mealer RG, Sixt KM, Barrow RK, Usiello A, Snyder SH. Rhes, a physiologic regulator of sumoylation, enhances cross-sumoylation between the basic sumoylation enzymes E1 and Ubc9. J Biol Chem. 2010;285:20428–20432. doi: 10.1074/jbc.C110.127191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Subramaniam S, Sixt KM, Barrow R, Snyder SH. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science. 2009;324:1327–1330. doi: 10.1126/science.1172871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Suhr ST, Senut MC, Whitelegge JP, Faull KF, Cuizon DB, Gage FH. Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J Cell Biol. 2001;153:283–294. doi: 10.1083/jcb.153.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tagawa K, Hoshino M, Okuda T, Ueda H, Hayashi H, Engemann S, Okado H, Ichikawa M, Wanker EE, Okazawa H. Distinct aggregation and cell death patterns among different types of primary neurons induced by mutant huntingtin protein. J Neurochem. 2004;89:974–987. doi: 10.1111/j.1471-4159.2004.02372.x. [DOI] [PubMed] [Google Scholar]
- 116.Tagawa K, Marubuchi S, Qi ML, Enokido Y, Tamura T, Inagaki R, Murata M, Kanazawa I, Wanker EE, Okazawa H. The induction levels of heat shock protein 70 differentiate the vulnerabilities to mutant huntingtin among neuronal subtypes. J Neurosci. 2007;27:868–880. doi: 10.1523/JNEUROSCI.4522-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Takahashi T, Kikuchi S, Katada S, Nagai Y, Nishizawa M, Onodera O. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum Mol Genet. 2008;17:345–356. doi: 10.1093/hmg/ddm311. [DOI] [PubMed] [Google Scholar]
- 118.Tam S, Geller R, Spiess C, Frydman J. The chaperonin TriC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat Cell Biol. 2006;8:1155–1162. doi: 10.1038/ncb1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tam S, Spiess C, Auyeung W, Joachimiak L, Chen B, Poirier MA, Frydman J. The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat Struct Mol Biol. 2009;16:1279–1285. doi: 10.1038/nsmb.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Thakur AK, Jayaraman M, Mishra R, Thakur M, Chellgren VM, Byeon IJ, Anjum DH, Kodali R, Creamer TP, Conway JF, Gronenborn AM, Wetzel R. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat Struct Mol Biol. 2009;16:380–389. doi: 10.1038/nsmb.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Thompson LM, Aiken CT, Kaltenbach LS, Agrawal N, Illes K, Khoshnan A, Martinez-Vincente M, Arrasate M, O’Rourke JG, Khashwji H, Lukacsovich T, Zhu YZ, Lau AL, Massey A, Hayden MR, Zeitlin SO, Finkbeiner S, Green KN, LaFerla FM, Bates G, Huang L, Patterson PH, Lo DC, Cuervo AM, Marsh JL, Steffan JS. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol. 2009;187:1083–1099. doi: 10.1083/jcb.200909067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tsvetkov AS, Miller J, Arrasate M, Wong JS, Pleiss MA, Finkbeiner S. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A. 2010;107:16982–16987. doi: 10.1073/pnas.1004498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tung YT, Hsu WM, Lee H, Huang WP, Liao YF. The evolutionarily conserved interaction between LC3 and p62 selectively mediates autophagy-dependent degradation of mutant huntingtin. Cell Mol Neurobiol. 2010;30:795–806. doi: 10.1007/s10571-010-9507-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tydlacka S, Wang CE, Wang X, Li S, Li XJ. Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J Neurosci. 2008;28:13285–13295. doi: 10.1523/JNEUROSCI.4393-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 126.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 127.Wacker JL, Huang SY, Steele AD, Aron R, Lotz GP, Nguyen Q, Giorgini F, Roberson ED, Lindquist S, Masliah E, Muchowski PJ. Loss of Hsp70 exacerbates pathogenesis but not levels of fibrillar aggregates in a mouse model of Huntington’s disease. J Neurosci. 2009;29:9104–9114. doi: 10.1523/JNEUROSCI.2250-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wacker JL, Zareie MH, Fong H, Sarikaya M, Muchowski PJ. Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat Struct Mol Biol. 2004;11:1215–1222. doi: 10.1038/nsmb860. [DOI] [PubMed] [Google Scholar]
- 129.Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wellington CL, Singaraja R, Ellerby L, Savill J, Roy S, Leavitt B, Cattaneo E, Hackam A, Sharp A, Thornberry N, Nicholson DW, Bredesen DE, Hayden MR. Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J Biol Chem. 2000;275:19831–19838. doi: 10.1074/jbc.M001475200. [DOI] [PubMed] [Google Scholar]
- 131.Yanai A, Huang K, Kang R, Singaraja RR, Arstikaitis P, Gan L, Orban PC, Mullard A, Cowan CM, Raymond LA, Drisdel RC, Green WN, Ravikumar B, Rubinsztein DC, El-Husseini A, Hayden MR. Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function. Nat Neurosci. 2006;9:824–831. doi: 10.1038/nn1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yu ZX, Li SH, Nguyen HP, Li XJ. Huntingtin inclusions do not deplete polyglutamine-containing transcription factors in HD mice. Hum Mol Genet. 2002;11:905–914. doi: 10.1093/hmg/11.8.905. [DOI] [PubMed] [Google Scholar]
- 133.Zala D, Colin E, Rangone H, Liot G, Humbert S, Saudou F. Phosphorylation of mutant huntingtin at S421 restores anterograde and retrograde transport in neurons. Hum Mol Genet. 2008;17:3837–3846. doi: 10.1093/hmg/ddn281. [DOI] [PubMed] [Google Scholar]
- 134.Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 135.Zhang QC, Yeh TL, Leyva A, Frank LG, Miller J, Kim YE, Langen R, Finkbeiner S, Amzel ML, Ross CA, Poirier MA. A compact beta model of huntingtin toxicity. J Biol Chem. 2011;286:8188–8196. doi: 10.1074/jbc.M110.192013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang X, Smith DL, Meriin AB, Engemann S, Russel DE, Roark M, Washington SL, Maxwell MM, Marsh JL, Thompson LM, Wanker EE, Young AB, Housman DE, Bates GP, Sherman MY, Kazantsev AG. A potent small molecule inhibits polyglutamine aggregation in Huntington’s disease neurons and suppresses neurodegeneration in vivo. Proc Natl Acad Sci U S A. 2005;102:892–897. doi: 10.1073/pnas.0408936102. [DOI] [PMC free article] [PubMed] [Google Scholar]