

Abstract
Background
Activation of the platelet-specific collagen receptor, glycoprotein (GP) VI, induces intracellular reactive oxygen species (ROS) production; however the relevance of ROS to GPVI-mediated platelet responses remains unclear.
Objective
The objective of this study was to explore the role of the ROS-producing NADPH oxidase (Nox)1 and 2 complexes in GPVI-dependent platelet activation and collagen-induced thrombus formation.
Methods and results
ROS production was measured by quantitating changes in the oxidation-sensitive dye, H2DCF-DA, following platelet activation with the GPVI-specific agonist, collagen related peptide (CRP). Using a pharmacological inhibitor specific for Nox1, 2-acetylphenothiazine (ML171), and Nox2 deficient mice, we show that Nox1 is the key Nox homolog regulating GPVI-dependent ROS production. Nox1, but not Nox2, was essential for CRP-dependent thromboxane (Tx)A2 production, which was mediated in part through p38 MAPK signaling; while neither Nox1 nor Nox2 was significantly involved in regulating CRP-induced platelet aggregation/integrin αIIbβ3 activation, platelet spreading, or dense granule and α-granule release (ATP release and P-selectin surface expression, respectively). Ex-vivo perfusion analysis of mouse whole blood revealed that both Nox1 and Nox2 were involved in collagen-mediated thrombus formation at arterial shear.
Conclusion
Together these results demonstrate a novel role for Nox1 in regulating GPVI-induced ROS production, which is essential for optimal p38 activation and subsequent TxA2 production, providing an explanation for reduced thrombus formation following Nox1 inhibition.
Keywords: Platelets, Glycoprotein VI, Reactive oxygen species, NAPDH oxidase, Thrombus
Graphical abstract
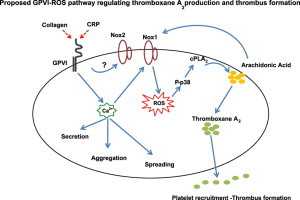
Highlights
-
•
Nox1, but not Nox2 mediates GPVI-induced ROS production.
-
•
GPVI-specific, CRP-activated platelet aggregation, spreading, secretion and αIIbβ3 activation is Nox1/2-independent.
-
•
GPVI-induced thromboxane A2 production is ROS-dependent, which is mediated by p38 signaling.
-
•
Collagen-induced ROS production and aggregation is Nox1-dependent.
-
•
Both Nox1 and Nox2 regulate collagen-induced thrombus formation at arterial shear.
Introduction
Upon vascular damage, platelets rapidly adhere to the exposed extracellular matrix (ECM). Collagen, the most abundant and thrombogenic constituent of the ECM, signals mainly through the glycoprotein (GP) VI transmembrane receptor on platelets, initiating a series of events including platelet activation and spreading, secretion of storage granules, thromboxane A2 (TxA2) production, and activation of the VWF- and fibrinogen-binding integrin αIIbβ3, which results in platelet aggregation. Another consequence of platelet activation through GPVI is the production of reactive oxygen species (ROS). A significant function of ROS is their participation in the body's host-defence mechanism. However, in non-innate immune cells, ROS are more often associated with oxidative stress, as chronic imbalance in the cellular reduction–oxidation (redox) state is implicated in various diseases, including vascular thrombosis [1], [2]. ROS also represent important secondary messengers in signal transduction cascades through their ability to oxidatively modify protein thiols, such as catalytic cysteinyl residues in protein tyrosine phosphatases, or interact with other reactive compounds [3], [4]. As activated platelets produce predominantly intracellular ROS; platelet-derived ROS are believed to have a role in regulating cellular function rather than providing a physiological response to pathogen invasion [5], [6]. Although much is already known about the GPVI signaling pathway, the precise mechanism linking GPVI-dependent ROS production to changes in platelet function remains to be elucidated.
Platelets activated by collagen generate a variety of ROS including superoxide anions, hydrogen peroxide, hydroxyl radicals and peroxynitrite, which appear necessary for maximal platelet activation [7], [8], [9]. The source of these reactive species is suggested to be primarily generated from NADPH oxidases (Nox), as the presence of antioxidants or Nox inhibitors have been shown to significantly reduce the ability of platelets to fully aggregate, recruit additional platelets or to form normal thrombus [7], [10], [11], [12], [13].
Seven Nox family members have so far been discovered in mammalian cells (Nox1-5 and Duox1-2) [13], [14], [15]. There are conflicting data in regard to which Nox homolog is responsible for ROS production in platelets. Nox2, first discovered in phagocytes, is suggested to be the main catalytic unit present as platelets derived from X-linked chronic granulomatous disease (CGD) patients with Nox2 deficiency showed significantly less ROS after collagen or arachidonic acid challenge as compared to normal platelets [16]. Immunodetection of Nox2 (gp91phox) and its associated subunits p22phox, p47phox and p67phox in human platelets, and RNA transcriptome analysis of both mouse and human platelets further support the existence of Nox2 in platelets [7], [17], [18], [19], [20]. Additionally, ROS-dependent antibody lysis of platelets is reduced compared to wildtype in Nox2 knockout mouse platelets [21]. However, mRNA and protein expression of Nox1, and weak mRNA expression of Nox4, have been reported in mouse bone marrow and megakaryocytes [22]. Others have confirmed both Nox1 and Nox2 in human platelets by western blot analysis [11], [23].
In this study, we have investigated the role of Nox1 and Nox2 in GPVI-dependent platelet activation using a pharmacological inhibitor specific for Nox1 (ML171) and a Nox2-deficient mouse model. Our findings indicate that Nox1, but not Nox2, is the key Nox homolog responsible for GPVI-dependent ROS production, which subsequently regulates TxA2 generation via a p38 MAP kinase-dependent signaling pathway. Both Nox homologs are required for collagen-mediated thrombus formation at arterial shear, but the mechanism contributing to the defective thrombus phenotype in the Nox2-deficient model remains unclear.
Materials and methods
Reagents and antibodies
The Nox1 inhibitor, ML171 (2-acetylphenothiazine), was purchased from Sigma Aldrich (St. Louis, MO, USA) and dissolved in DMSO. Apocynin, aspirin and phorbol 12-myristate 13-acetate (PMA) were purchased from Tocris (R&D systems, Inc.) and reconstituted in DMSO. The final concentration of the vehicle DMSO in all experiments was 0.1% (except for studies with aspirin which contained 0.5% DMSO). Anti-ERK 1/2, anti-pERK 1/2Thr 202/Tyr 204, anti-Akt, anti-pAktSer 473, anti-p38 and anti-P-p38Thr 180/Tyr 182 antibodies were from Cell Signaling Technology (Boston, MA, USA). HRP-conjugated anti-rabbit light chain-specific IgG was from Millipore (Lake Placid, NJ, USA). Crosslinked collagen related peptide (CRP) was purchased from the Department of Biochemistry, University of Cambridge, UK. Fibrillar Horm collagen was from Nycomed (Munich, Germany).
Preparation of washed mouse platelets
Wildtype and Nox2-deficient mice, both on a C57BL6/J background, were originally from Jax® Mice and Services. Blood was drawn from the inferior vena cava of CO2 terminally-asphyxiated mice using a 25-gauge needle containing acid citrate dextrose (ACD-15% v/v) as anticoagulant (140 µL of ACD per 1 mL final volume). Blood was further diluted with 200 µL of ACD and 800 µL of CGS buffer (123 mM NaCl, 33.3 mM glucose, 14.7 mM trisodium citrate, pH 7.0) and centrifuged at 190g for 10 min without centrifugal braking at room temperature. Platelet-rich plasma (PRP) was removed into a new tube and the concentrated blood was diluted up to 2 mL with CGS buffer and re-centrifuged to obtain more PRP. Platelets were isolated from PRP by centrifugation for 10 min at 650g, resuspended in modified HEPES-Tyrode's buffer (5 mM HEPES, 5.5 mM glucose, 138 mM NaCl, 12 mM NaHCO3, 0.49 mM MgCl2, 2.6 mM KCl, 0.36 mM NaH2PO4, pH 7.4) and maintained at 37 °C before use.
Preparation of mouse neutrophils
Following platelet isolation, the remaining blood cell population was incubated for 3 min with a red blood cell lysis buffer (Sigma). Leukocytes were then isolated by centrifugation at 500g for 5 min, washed (two times) in Tris-buffered saline (TBS)/0.1% bovine serum albumin (BSA) and resuspended at a concentration of 1×106/mL. The neutrophil population, distinguished from other cell types by its light-scattering properties, was analyzed on a Becton Dickinson FACSCanto™ (San Jose, CA, USA) using the FACSDiva software.
Platelet aggregation
Platelet aggregation was performed in a PAP 4-C aggregometer (Bio/Data Corporation) using washed platelets (2×108/mL, final concentration) with constant stirring at 1100 rpm. For all studies with pharmacological inhibitors, platelets were incubated with either vehicle control or inhibitors for 10 min at 37 °C before the addition of CRP.
Mouse platelet integrin αIIbβ3 activation
Platelet suspension at 2.5×108/mL was preincubated with 5 µM ML171 or vehicle control. The platelets (50 µL reactions) were stimulated with 0, 0.25 or 1 µg/mL CRP and labeled with a PE-conjugated antibody specific for the functionally active form of integrin αIIbβ3 (1/10 dilutions, clone JON/A, EMFRET Analytics GmbH & Co. KG, Germany) for 10 min at 37 °C under non-stirring conditions. The reactions were stopped by adding 450 µL phosphate-buffered saline and the samples were analyzed immediately by flow cytometry.
Granule secretion
ATP release from dense granules was measured using Chrono-Lume luciferin–luciferase reagent as previously described [24]. P-selectin surface expression from platelet α-granules was measured by flow cytometry.
Measurement of intracellular ROS
Intracellular ROS was measured as previously described with some minor modifications [25]. Briefly, washed platelets (2.5×108/mL) were incubated for 15 min at 37 °C with 10 µM 2',7'-dihydrodichlorofluorescein diacetate, H2DCFDA (Cambridge Bioscience, UK), pretreated with inhibitors and then stimulated with the relevant agonist for up to 10 min at 37 °C. Samples were diluted 10-fold in modified HEPES-Tyrode's buffer containing 10 µM H2DCFDA and analyzed immediately by flow cytometry. For all ROS experiments, geo-mean values were expressed as fold change, relative to unstimulated levels set as 1.
Ex vivo thrombus formation
Analysis of thrombus formation was performed using a microvolume parallel-plate flow chamber apparatus as previously described [26]. In brief, glass coverslips were coated with 200 µg/mL acid-soluble type I collagen (Sigma) overnight at 4 °C and blocked in 1% fatty acid free BSA (Sigma) for 1 h. Mouse blood was collected via the vena cava, as described above, using recombinant hirudin (70 µg/mL final concentration, Allphar Services, Ireland) as an anticoagulant. Hirudinated whole blood was diluted with an equal volume of modified HEPES-Tyrode's buffer and blood cells were fluorescently labeled with 1 µM DiOC6(3) (Molecular Probes, Eugene, OR, USA) for 10 min at 37 °C in the presence of vehicle control or inhibitors. Labeled, diluted blood was perfused at a constant shear rate of 1500 s−1 for 6 min over the collagen-coated surface, at which point samples were washed at a similar shear rate for 5 min in modified HEPES-Tyrode's buffer and subsequently fixed with 3.7% formaldehyde. Images of platelet thrombi were captured using a Zeiss FLUAR 20× objective on a Zeiss Axiovert 200M microscope, with 10 randomly selected fields of view acquired in each sample for representative purposes.
Measurement of thrombus surface coverage and volume
Quantitative assessment of various indices of thrombus formation was based on previous studies [10], [27]. Image analysis was performed using the object extraction tool within JMicroVision (http://www.jmicrovision.com/), establishing an unbiased, constant gray intensity threshold between images to differentiate between signal (platelet thrombi) and image background. Computed values allowed calculation of % surface coverage by dividing the total thrombus area by the image area and thrombus volume by dividing total gray intensity levels (which is proportional to the number of platelets) by thrombi number. Blind experimental analysis between genotypes was applied throughout.
Immunoblot analysis
Western blot analysis of mouse platelets was performed essentially as previously described (PMID 11606185). Quantitative band densitometry was performed using ImageJ.
Measurement of TxA2 production
Activated platelet samples (from aggregation reactions) were stopped at 3 min with 5 mM EDTA and 200 µM indomethacin to prevent further TxA2 formation. TxB2 levels (as stable metabolite) were analyzed with a commercial ELISA kit (Enzo Life Sciences, UK) according to the manufacturer's instructions.
Statistical analysis
All analysis was performed using GraphPad Prism 5. Results are shown as mean±SEM. Statistical significance of difference between means was determined using one- and two-way ANOVA, with Bonferonni post-hoc analysis. A value of ⁎P≤0.05 was considered to be statistically significant.
Results
Detection of GPVI-induced reactive oxygen species in platelets isolated from wildtype and Nox2-knockout (KO) mice
As no previous data existed in regards to the generation of ROS from Nox2-KO mouse platelets, we first evaluated the level of ROS from platelets prepared from age- and sex-matched wildtype (Wt) and Nox2-KO mice. Fig. 1A shows that platelets from both genotypes produced comparable levels of ROS when stimulated with CRP (1 µg/mL). These values represent 2 min time points, as temporal monitoring (0–10 min) of the GPVI-ROS response in both WT and Nox2-KO platelets identified peak ROS levels 2 min post-CRP-stimulation with equally comparable levels of ROS between genotypes at all other time points (data not shown). As a proof that Nox2-KO mice lack the ability to generate ROS in a manner similar to X-linked CGD patients (i.e. Nox2 defects), we stimulated white blood cells with 5 µM phorbol-12-myristate-13-acetate (PMA) and showed that neutrophils derived from Nox2-KO mice failed to generate ROS (Fig. 1B). Additionally, we confirmed that the Nox1 inhibitor, ML171, did not block Nox2-dependent activity as the compound did not significantly affect ROS generation by Wt neutrophils (Supplement Fig. S1).
Fig. 1.
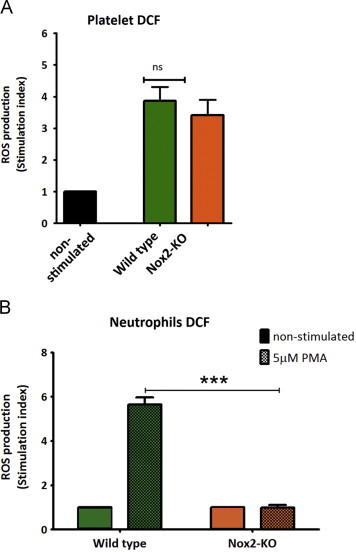
GPVI-specific ROS production does not involve Nox2. (A) Washed platelets from wildtype (Wt) or Nox2-knockout (KO) mice were pre-loaded with 10 µM H2DCFDA, then stimulated with 1 µg/mL CRP for 2 min and monitored for ROS production by flow cytometry. Data are mean±SEM, n=8, non-significant (ns). (B) H2DCFDA loaded neutrophils from Wt or Nox2-KO mice were stimulated with 5 µM PMA for 5 min and analyzed by flow cytometer to assess ROS production. Data are mean±SEM, n=4, ⁎⁎⁎P<0.001. Two-way ANOVA with Bonferroni post-hoc analysis were performed for A and B.
Effect of Nox1 inhibition and Nox2 deficiency on GPVI-dependent platelet activation
Production of ROS has been suggested by some studies to be essential for optimal platelet activation, affecting functional responses such as platelet aggregation and recruitment. Here we examined the effect of Nox1 inhibition and Nox2 deficiency in several in vitro platelet function tests. Firstly, ML171 significantly suppressed ROS production from both wildtype (Wt) and Nox2-deficient platelets activated with threshold (0.25 µg/mL) and high (1 µg/mL) doses of CRP without significantly reducing platelet aggregation response or the surface level expression of active αIIbβ3 (Fig. 2A and B, Supplement Fig. S2). Nox2-deficient platelets stimulated with low dose CRP showed a mildly reduced, though not statistically significant aggregation response compared to Wt. Similarly, ML171 treatment of Nox2-deficient platelets did not cause a statistically-significant reduction in the aggregation response.
Fig. 2.
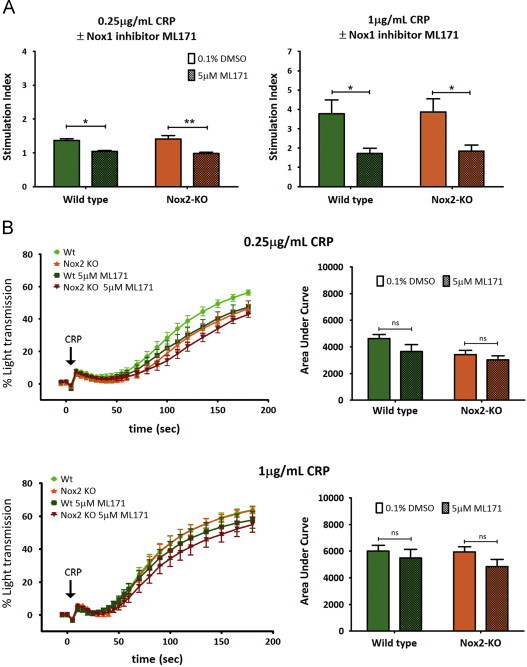
Nox1 is the key Nox homolog regulating GPVI-dependent ROS production. Platelet aggregation is independent of Nox1/2. (A) Wildtype (Wt) and Nox2-KO H2DCFDA-loaded platelets were pretreated with vehicle control (0.1% DMSO) or the Nox1 specific inhibitor (5 µM ML171) and monitored for ROS production using 0.25 µg/mL (left panel) and 1 µg/mL (right panel) CRP with 2 min stimulation. Data are mean±SEM, n=3 (0.25 µg/mL), n=4 (1 µg/mL); ⁎P<0.05, ⁎⁎P<0.01. (B) Washed platelets from Wt or Nox2-KO platelets containing 0.1% DMSO or 5 µM ML171 were monitored for CRP-dependent platelet aggregation (left panels) with area under the curve analysis (right panels) using 0.25 µg/mL and 1 µg/mL CRP. Data are mean±SEM, n=7, non-significant (ns). Two-way ANOVA with Bonferroni analysis were performed for A and B.
As ROS generation in mouse platelets appeared to be independent of Nox2, we extended our Nox inhibitory studies to other indices of GPVI-mediated platelet function, including secretion, spreading and TxA2 production using 1 µg/mL CRP. The ability of CRP-activated platelets to secrete ATP (dense granule), express surface P-selectin (α-granule) or to spread on a fibrinogen-coated surface were not affected by Nox1 inhibition or Nox2 deficiency (Fig. 3A and B, Supplement Fig. S3). Interestingly, the production of TxA2 by activated platelets was completely abrogated in the presence of ML171 (Fig. 3C). This reduction was also observed with CRP-activated platelets using a non-homolog-specific Nox inhibitor and antioxidant, apocynin (data not shown), suggesting a role for Nox1-derived ROS in the GPVI-mediated TxA2 production pathway.
Fig. 3.
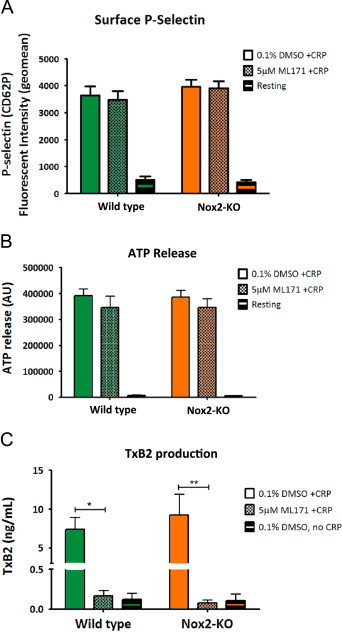
GPVI-mediated platelet α and dense granule release is independent of Nox1/2, but thromboxane A2 generation requires Nox1-derived ROS. Washed platelets from wildtype or Nox2-KO mice were pre-incubated with vehicle control (0.1% DMSO) or Nox1 inhibitor (5 µM ML171) and assessed following stimulation with 1 µg/mL CRP for (A) α-granule release by measuring P-selectin (CD62P) surface expression, (B) dense body secretion by ATP release and (C) thromboxane A2 production using a TxB2 ELISA. Data are mean±SEM, n=4 (n=3 for TxA2 assay), ⁎P<0.05, ⁎⁎P<0.01 by two-way ANOVA with Bonferroni correction.
Effect of Nox1 and Nox2 on collagen-mediated platelet activation
Previous studies have shown that specific stimulation of GPVI with CRP elicits a rapid and powerful platelet aggregation response, whereas collagen induces a delayed aggregation response that is primarily dependent on the secondary wave agonists, ADP and TxA2 [28], [29]. Since our data indicated that Nox1-derived ROS was essential for GPVI-dependent TxA2 formation, we hypothesized that Nox1 inhibition would affect collagen-induced response in platelets. Indeed, ML171 significantly attenuated collagen-mediated aggregation and ROS production, an effect independent of Nox2 (Fig. 4A and B).
Fig. 4.
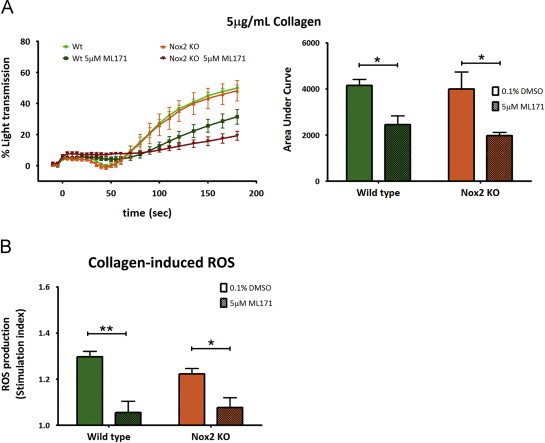
Collagen-induced platelet aggregation and ROS production requires Nox1. Wildtype and Nox2-KO washed platelets were pretreated with vehicle control (0.1% DMSO) or the Nox1 specific inhibitor (5 µM ML171), then stimulated with 5 µg/mL collagen and monitored for (A) platelet aggregation (left panel) with area under the curve analysis (right panel) and (B) ROS production. Data are mean±SEM, n=4, ⁎P<0.05, ⁎⁎P<0.01 by two-way ANOVA with Bonferroni analysis.
Effect of Nox1 inhibition on GPVI-dependent signaling
In an attempt to better understand the role of Nox1 in GPVI-dependent mouse platelet activation we examined several proteins in the GPVI signaling pathway that could potentially regulate TxA2 production. As preliminary studies demonstrated that the intracellular Ca2+ chelator, BAPTA, completely suppressed GPVI-dependent ROS and aggregation (data not shown), we focused our attention on signaling molecules known to be downstream of or distinct from Ca2+ release, and proximal to TxA2 production. The Akt, ERK and p38 phosphorylation profiles showed that ML171, and the antioxidant apocynin, reproducibly reduced phosphorylation of p38(Thr180/Tyr182) 1 min post-CRP stimulation without significantly affecting the phosphorylation of Akt or ERK. At 3 min, ML171, although less effective than apocynin, still decreased the level of phospho-p38, although this was not statistically significant (Fig. 5A and B). To confirm a potential causal link between p38 and TxA2 generation, we found that the p38 inhibitor, SB202190 (10 µM), significantly blocked CRP-induced TxA2 (Fig. 5C).
Fig. 5.
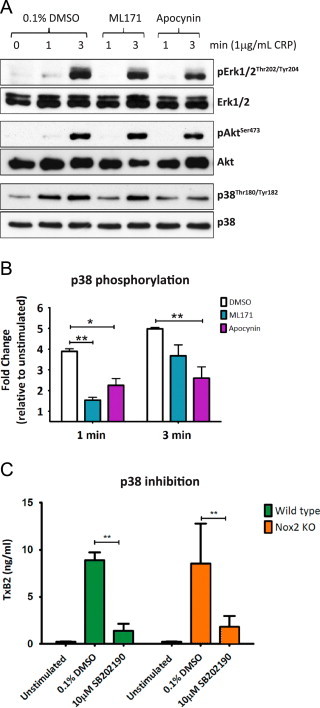
CRP-mediated p38 activation requires Nox1-derived ROS. (A) Washed platelets from wildtype (WT) mice were pre-treated with vehicle control (0.1% DMSO), Nox1 inhibitor (5 µM ML171) or ROS scavenger apocynin (250 µM) and stimulated for up to 3 min with 1 µg/mL CRP under non-stirring conditions. Platelets were lysed, separated by SDS-PAGE and immunoblotted using phospho-specific antibodies for active ERK1/2 (upper panel), Akt (middle panel) and p38 (lower panel), with subsequent loading controls for each protein. Blots are representative of three independent experiments. (B) Densitometric analysis of p38 phosphorylation expressed as fold change relative to unstimulated control. Data are mean±SEM, n=3, ⁎P<0.05, ⁎⁎P<0.01 by two-way ANOVA with Bonferroni correction. (C) Washed platelets from WT mice were pre-incubated with 0.1% DMSO or p38 inhibitor (10 µM SB202190), stimulated with 1 µg/mL CRP and monitored for thromboxane A2 production using a TxB2 ELISA. Data are mean±SEM, n=3 ⁎⁎P<0.01 by two-way ANOVA with Bonferroni correction.
Effect of Nox2 deficiency and Nox1 inhibition on thrombus formation in mouse blood at arterial shear
To examine the role of Nox1 and Nox2 on thrombus formation, ML171- or vehicle-treated blood isolated from Wt or Nox2-KO mice were perfused over collagen at an arterial shear rate of 1500 s−1. Representative end-point images of the different conditions are shown in Fig. 6A. Interestingly, Nox2 deficiency significantly reduced thrombus volume without affecting the overall surface coverage (Fig. 6B). This effect was not due to differential blood profiles between Wt and Nox2-KO mice. The number of platelets, leukocytes and hematocrit level in whole blood was not significantly different between the genotypes (Table 1). Additionally, GPVI and GPIbα, the subunit of the receptor complex GPIb-V-IX required under high shear conditions for platelet adhesion, were detected at similar expression levels.
Fig. 6.
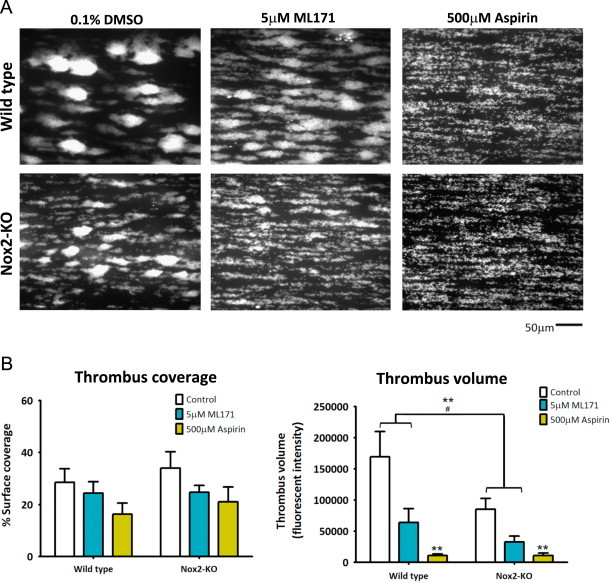
Collagen-dependent thrombus formation at arterial shear requires both Nox1 and Nox2. Hirudin-anticoagulated whole blood from wildtype (Wt) or Nox2 knockout (KO) mice was diluted 1:2 with modified HEPES-Tyrode's buffer, then pre-incubated with vehicle control (0.5% DMSO), Nox1 inhibitor (5 µM ML171) or aspirin (500 µM). Samples were perfused over a collagen-coated microfluidic channel at a constant shear of 1500 s−1 for 6 min, followed by washing in modified HEPES-Tyrode's buffer for 5 min with subsequent fixation in 3.7% formaldehyde. (A) Representative fluorescent, end point images of platelet thrombi from Wt (upper panel) and Nox2 KO (lower panel) mice following vehicle control/inhibitor treatment. (B) Platelet surface coverage (left panel) and thrombus volume (right panel) are expressed as mean±SEM, n=5 (n=3 for aspirin), ⁎P<0.05, ⁎⁎P<0.01 by two-way ANOVA with Bonferroni analysis.
Table 1.
Key blood components and expression levels of platelet receptors GPVI and GPIb.
| Mouse | WBC count (103/μl) | Plt count (103/μl) | Hct (%) | GPVI Exp* | GPIb Exp* |
|---|---|---|---|---|---|
| Wildtype | 6.4±0.4 (n=13) | 886.6±47.3 (n=13) | 39.0±1.2 (n=13) | 3904±125 (n=4) | 2505±209 (n=4) |
| Nox2-KO | 5.5±0.2 (n=13) | 830.9±44.4 (n=13) | 42.2±1.0 (n=13) | 3589±75 (n=4) | 2098±146 (n=4) |
Female and male mice 8–16 weeks of age were used. Values are means±standard errors of the means. Unpaired t-test analysis indicated no significance difference between the two genotypes in the parameters shown above.
Abbreviations: WBC, white blood cell; Plt, platelet; Hct, hematocrit; Exp*, expression level as determined by flow cytometry.
The effect on thrombus formation by Nox2 deficiency was further compounded by the presence of ML171. Nox1 inhibition alone was also effective at significantly reducing thrombus volume. Using higher doses of ML171, at 50 µM, the effect of Nox1 inhibition was similar to aspirin, an irreversible cyclooxygenase inhibitor and an enzyme essential in the TxA2 production pathway (data not shown). Blood treated with ML171 or aspirin showed no significant reduction in the percentage surface area covered by platelets.
Effect of Nox1 inhibition on human platelets
To establish whether ML171 asserted a similar effect on human platelets, we performed several in vitro functional assays using washed human platelets from healthy donors. Overall, ML171 had no significant impact on the ability of human platelets to aggregate, spread on fibrinogen, secrete ATP, or to expose membrane phosphatidylserine in response to GPVI activation by CRP (data not shown). As for mouse platelets, ROS and TxA2 productions from GPVI-activated human platelets were significantly suppressed by 5 µM ML171 (Supplement Fig. S4).
Discussion
In this study we have examined the relative roles of Nox1 and Nox2 in GPVI-dependent platelet function. Our findings demonstrate that Nox2 plays a minimal role in GPVI-dependent ROS production and other platelet function assays in vitro. On the contrary, suppression of Nox1 activity by the pharmacological inhibitor, ML171, significantly decreased CRP-induced ROS and TxA2 generation in platelets. The effect of ML171 was not limited to CRP-activated platelets as ML171 also suppressed thrombin- and arachidonic acid-induced ROS generation in both mouse and human platelets (data not shown). Interestingly, the ex vivo experiments revealed that both Nox homologs were required for optimal collagen-induced thrombus formation at arterial shear flow rates.
Previous studies have implicated Nox2 as the key Nox homolog regulating ROS-mediated platelet responses [16], [30]. Our initial experiments with GPVI-stimulated human platelets did not substantiate these findings as apocynin and the Nox2 blocking peptide (gp91ds-tat), which are both commonly described as specific Nox2 inhibitors, proved to have off target effects (data not shown). A recent report, as well as our own data, found apocynin to be a potent antioxidant. We therefore investigated the ability of platelets from Nox2 knockout (KO) mice to generate ROS. Activated platelets from both Nox2-KO and wildtype (Wt) mice were equally capable of generating ROS. However, only Wt and not Nox2-KO neutrophils produced ROS, suggesting a role for other Nox homologs in platelets. ML171 has been recently identified as a specific Nox1 inhibitor, with no ROS scavenging ability and minimal activity toward other Nox isoforms. Gianni et al. demonstrated IC50 of >10 µM for ML171 against Nox2 in a cell-free ROS production system and in isolated human neutrophils [31]. Our initial experiments confirmed that ML171 blocked ROS production by all platelet agonists tested (collagen, CRP, thrombin, arachidonic acid, Par1 and Par4 activating peptides). We confirmed that ML171 does not inhibit Nox2 as Wt neutrophils stimulated with PMA were unaffected by ML171 pre-treatment. Regrettably, we were not able to test ML171 on platelets derived from CGD patients or Nox1-KO mouse models to ascertain the specificity of ML171 against the Nox1 isoform. As with all genetic deficiency models, there is an expected compensatory effect afforded by related proteins and knockout models do not always provide a definitive answer. Although it is possible that there are other yet to be identified ROS-producing proteins in platelets that can be inhibited by ML171, the research in this area is beyond the scope of this study. Nevertheless, we showed here that ML171, the most specific Nox1 inhibitor commercially available to date, abrogated ROS production in activated platelets without overtly affecting other platelet functions.
As there are contradicting studies in regards to the role of ROS in platelet aggregation, integrin αIIbβ3 (GPIIb/IIIa) activation, and α and dense granule release [5], [6], [7], [32], we investigated the effect of Nox1 inhibition and Nox2 deficiency on these responses following platelet stimulation with CRP. Platelet aggregation and the expression level of activated αIIbβ3 on CRP-stimulated Wt and Nox2-deficient platelets treated with either ML171 or vehicle control were comparable. Furthermore, the secretion of ATP (from platelet dense granules) and P-selectin surface expression (translocated from α-granules) after CRP stimulation were not significantly different between the two genotypes or treatments, indicating that these functions were independent of Nox2 and Nox1-derived ROS.
It has been previously shown that ROS scavenging blocks collagen-mediated TxA2 production [12]. In this paper we confirmed this finding and show for the first time that the specific inhibition of Nox1, but not the absence of Nox2, completely suppresses TxA2 production in CRP-activated platelets. Although TxA2 is known to contribute and enhance platelet response to various stimuli, inhibition of Nox1 did not significantly affect platelet aggregation in response to CRP. This may be explained by the observation that normal aggregation can still be elicited by GPVI-specific agonists in the presence of cyclooxygenase inhibitors or the absence of the TxA2 receptor [28], [29], [33]. However, in in vitro assays utilizing suspensions of fibrillar collagen, the platelet aggregation response is believed to be primarily mediated by the secondary agonists, TxA2 and ADP [34], [35]. Our aggregation data using fibrillar type I Horm collagen supported this finding as similar decreases in the aggregation responses from both Wt and Nox2-KO platelets were observed in the presence of ML171, confirming that TxA2 was an important factor in secondary platelet responses, and that this effect was mediated by Nox1-derived ROS. In comparison to CRP, Horm collagen induced a very low level of ROS, which may reflect the fact that platelets stimulated by fibrillar collagen and CRP are markedly different. GPVI activation is initiated by its interaction with repeated glycine–proline–hydroxyproline (GPO) motifs, which in the case of fibrillar collagen are scarce and scattered heterogeneously along the elongated fibrils, whereas in synthetic crossed-linked CRP, the GPO repeats are much more abundant and contiguous [36], [37]. Thus in a physiological environment, an abrogation of Nox1-dependent TxA2 production may exert a significant impact on platelet function.
In an effort to understand the regulatory link between GPVI-derived ROS (via Nox1) and TxA2 production, we explored the potential involvement of a number of signaling kinases. As chelation of intracellular Ca2+ with BAPTA completely blocked multiple GPVI-dependent platelet responses including aggregation and ROS formation (data not shown), we focussed our attention on signaling components distal to Ca2+ mobilization. Of significance, the MAP kinases, ERK1/2 and p38 have been implicated in the regulation of cytosolic phospholipase A2 (cPLA2), a key enzyme involved in the formation of TxA2 by liberating arachidonic acid from the plasma membrane [39], [40], while the serine/threonine kinase, Akt, has been shown to be a key downstream effector of PI3 kinase-dependent thrombosis [41]. Our findings demonstrated that p38, but not ERK1/2 or Akt activation required Nox1-dervied ROS. This was consistent with the observation that the generic ROS inhibitor, apocynin, elicited similar effects. Interestingly both p38 and ERK1/2 have been designated as redox sensitive kinases in different cell types [42], [43]; however in platelets, our data suggest p38 as the protein target for Nox1-derived ROS. Furthermore, our observation that Nox1 did not significantly inhibit late (3 min) p38 activation suggests a Nox1-independent mechanism of p38 regulation. In this regard, ADP release, which we show to be independent of Nox1-derived ROS with GPVI activation, is known to be an important mediator of p38 activation in platelets [44], We subsequently confirmed a potential link between CRP-mediated ROS and TxA2, via p38, by demonstrating that inhibition of p38 by the selective inhibitor, SB202190, significantly blocked TxA2 production following GPVI activation.
To examine the importance of Nox1 and Nox2 under more physiological conditions, we performed whole blood perfusion assays over a collagen-coated surface at arterial shear flow rates. There was no obvious defect in initial platelet adhesion or thrombus surface coverage as both ML171- and vehicle control-treated blood from Wt or Nox2 KO mice showed no statistically significant difference in the percentage of surface area covered by platelets. However, subsequent thrombus build-up was inhibited in the presence of the Nox1 inhibitor ML171. This suggests that thrombus growth requires Nox1-derived ROS and/or TxA2 to recruit and activate more platelets and that this growth is independent of further platelet activation by collagen, presumably because the first layer of platelets already mask the GPVI-activating GPO motifs. Whole blood treated with 50 µM ML171 gave a much more pronounced defect in thrombus volume (data not shown), which very closely resembled the thrombus phenotype observed with aspirin, consistent with our in vitro findings that Nox1 is essential for TxA2 production. Importantly, our findings that GPVI-dependent ROS production and TxA2 generation are Nox1-dependent have been replicated in human platelets. Furthermore, we confirm that collagen-induced thrombus volume in human blood is Nox1-dependent (data not shown). Interestingly however, a recent study by Vara et al. [11] demonstrated a substantial defect in thrombus coverage with Nox1 inhibition using ML171 and ROS scavenging via Apocynin. These findings contradict our observations with ML171 and also a study by Pignatelli et al. [10] using Apocynin. However, it must be noted that all the collagen-activated thrombus phenotype (including DMSO-treated control sample) observed by Vara et al. is localized to the edges of the flow channel, whereas our control blood shows consistent thrombi distribution. In addition to the disparity between our control results, the differences in experimental design and equipment further make direct comparison between our studies difficult. For instance, after venepuncture our blood samples were swiftly labeled whole and perfused while Vara et al. used reconstituted Calcein™-labeled PRP. It may be that the ~1 h labeling and reconstitution steps, absent in ours and the Pignatelli et al. studies, altered platelet responses, e.g. through receptor desensitization.
Our observation that Nox2 was also important for thrombus volume is consistent with a previous report [10], however the mechanism for this is presently unclear based on our present functional studies. Studies on CGD patients have shown that surface expression of CD40L is Nox2-dependent [16], however we could not reliably detect substantial differences in surface expression or soluble forms of CD40L following platelet activation (data not shown). To date the only clear function of NAPDH oxidases is to generate ROS and considering our data, it would be interesting to speculate on a ROS-independent function of Nox2 in platelets [14], [38]. However, it is possible that the presence of Nox2 in blood neutrophils, monocytes or vascular endothelial cells contributes to thrombus formation ex vivo and in vivo. Therefore, future studies comparing thrombus formation using blood of Nox2-KO mice with either Wt or Nox2-deficient platelets, and bone marrow transplant chimeric studies, may highlight a potential role of leukocyte-derived ROS during thrombosis.
In summary, our study demonstrates Nox1 as an important regulator of platelet function in thrombosis. Furthermore, our data suggest that targeting Nox1 may be an effective substitute for the use of cyclooxygenase inhibitors, such as aspirin, to reduce TxA2 release and thereby suppress thrombotic events. Using anti-Nox1 as an anti-platelet strategy may thus have significant therapeutic implications for individuals with aspirin allergy or sensitivity.
Contribution of authors
PM and MCB designed the study. TW, NC and PM performed experiments and acquired data. TW and PM interpreted the results. PM supervised data analysis. TW composed the majority of the manuscript. PM prepared the structure of the manuscript and the final figures. PM and MCB revised and edited the manuscript. JC assisted with blood perfusion experiment. DK provided the key proprietary instrumentation.
Acknowledgment
We would like to acknowledge Science Foundation Ireland, Biomedical Diagnostics Institute, Royal College of Surgeons in Ireland and Curtin University for their financial, infrastructure and technical support.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2013.12.023.
Appendix. Supplementary materials
Supplementary material
References
- 1.Freedman J.E. Oxidative stress and platelets. Arterioscler. Thromb. Vasc. Biol. 2008;28(3):s11–s16. doi: 10.1161/ATVBAHA.107.159178. [DOI] [PubMed] [Google Scholar]
- 2.Touyz R.M. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension. 2004;44(3):248–252. doi: 10.1161/01.HYP.0000138070.47616.9d. [DOI] [PubMed] [Google Scholar]
- 3.Wu R.F., Xu Y.C., Ma Z., Nwariaku F.E., Sarosi G.A., Terada L.S. Subcellular targeting of oxidants during endothelial cell migration. J. Cell Biol. 2005;171(5):893–904. doi: 10.1083/jcb.200507004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen K., Craige S.E., Keaney J.F., Jr. Downstream targets and intracellular compartmentalization in Nox signaling. Antioxid. Redox Signal. 2009;11(10):2467–2480. doi: 10.1089/ars.2009.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Begonja A.J., Gambaryan S., Geiger J., Aktas B., Pozgajova M., Nieswandt B. Platelet NAD(P)H-oxidase-generated ROS production regulates aIIbβ3-integrin activation independent of the NO/cGMP pathway. Blood. 2005;106(8):2757–2760. doi: 10.1182/blood-2005-03-1047. [DOI] [PubMed] [Google Scholar]
- 6.Bakdash N., Williams M.S. Spatially distinct production of reactive oxygen species regulates platelet activation. Free Radic. Biol. Med. 2008;45(2):158–166. doi: 10.1016/j.freeradbiomed.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 7.Krotz F., Sohn H.Y., Gloe T., Zahler S., Riexinger T., Schiele T.M. NAD(P)H oxidase-dependent platelet superoxide anion release increases platelet recruitment. Blood. 2002;100(3):917–924. doi: 10.1182/blood.v100.3.917. [DOI] [PubMed] [Google Scholar]
- 8.Pignatelli P., Pulcinelli F.M., Lenti L., Paolo Gazzaniga P., Violi F. Hydrogen peroxide is involved in collagen-induced platelet activation. Blood. 1998;91(2):484–490. [PubMed] [Google Scholar]
- 9.Praticò D., Pasin M., Barry O.P., Ghiselli A., Sabatino G., Iuliano L. Iron-dependent human platelet activation and hydroxyl radical formation: involvement of protein kinase C. Circulation. 1999;99(24):3118–3124. doi: 10.1161/01.cir.99.24.3118. [DOI] [PubMed] [Google Scholar]
- 10.Pignatelli P., Carnevale R., Di Santo S., Bartimoccia S., Sanguigni V., Lenti L. Inherited human gp91phox deficiency is associated with impaired isoprostane formation and platelet dysfunction. Arterioscler. Thromb. Vasc. Biol. 2011;31(2):423–434. doi: 10.1161/ATVBAHA.110.217885. [DOI] [PubMed] [Google Scholar]
- 11.Vara D., Campanella M., Pula G. The novel NOX inhibitor 2-acetylphenothiazine impairs collagen-dependent thrombus formation in a GPVI-dependent manner. Br. J. Pharmacol. 2013;168(1):212–224. doi: 10.1111/j.1476-5381.2012.02130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chlopicki S., Olszanecki R., Janiszewski M., Laurindo F.R., Panz T., Miedzobrodzki J. Functional role of NADPH oxidase in activation of platelets. Antioxid. Redox Signal. 2004;6(4):691–698. doi: 10.1089/1523086041361640. [DOI] [PubMed] [Google Scholar]
- 13.Drummond G.R., Selemidis S., Griendling K.K., Sobey C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011;10(6):453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nauseef W.M. Biological roles for the NOX family NADPH oxidases. J. Biol. Chem. 2008;283(25):16961–16965. doi: 10.1074/jbc.R700045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leto T.L., Morand S., Hurt D., Ueyama T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid. Redox Signal. 2009;11(10):2607–2619. doi: 10.1089/ars.2009.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pignatelli P., Sanguigni V., Lenti L., Ferro D., Finocchi A., Rossi P. gp91phox-Dependent expression of platelet CD40 ligand. Circulation. 2004;110(10):1326–1329. doi: 10.1161/01.CIR.0000134963.77201.55. [DOI] [PubMed] [Google Scholar]
- 17.Pleines I., Elvers M., Strehl A., Pozgajova M., Varga-Szabo D., May F. Rac1 is essential for phospholipase Cγ2 activation in platelets. Pflügers Arch. Eur. J. Physiol. 2009;457(5):1173–1185. doi: 10.1007/s00424-008-0573-7. [DOI] [PubMed] [Google Scholar]
- 18.Rowley J.W., Oler A.J., Tolley N.D., Hunter B.N., Low E.N., Nix D.A. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood. 2011;118(14):e101–e111. doi: 10.1182/blood-2011-03-339705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dharmarajah J., Arthur J., Sobey C., Drummond G. The anti-platelet effects of apocynin in mice are not mediated by inhibition of NADPH oxidase activity. Naunyn-Schmiedeberg's Arch. Pharmacol. 2010;382(4):377–384. doi: 10.1007/s00210-010-0552-3. [DOI] [PubMed] [Google Scholar]
- 20.Arthur J.F., Shen Y., Gardiner E.E., Coleman L., Kenny D., Andrews R.K. TNF receptor-associated factor 4 (TRAF4) is a novel binding partner of glycoprotein Ib and glycoprotein VI in human platelets. J. Thromb. Haemost. 2011;9(1):163–172. doi: 10.1111/j.1538-7836.2010.04091.x. [DOI] [PubMed] [Google Scholar]
- 21.Nardi M., Feinmark S.J., Hu L., Li Z., Karpatkin S. Complement-independent Ab-induced peroxide lysis of platelets requires 12-lipoxygenase and a platelet NADPH oxidase pathway. J. Clin. Invest. 2004;113(7):973–980. doi: 10.1172/JCI20726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCrann D.J., Eliades A., Makitalo M., Matsuno K., Ravid K. Differential expression of NADPH oxidases in megakaryocytes and their role in polyploidy. Blood. 2009;114(6):1243–1249. doi: 10.1182/blood-2008-12-195883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gambim M.H., de Oliveira do Carmo A., Marti L., Verissimo-Filho S., Lopes L.R., Janiszewski M. Platelet-derived exosomes induce endothelial cell apoptosis through peroxynitrite generation: experimental evidence for a novel mechanism of septic vascular dysfunction. Crit. Care. 2007;11:R107. doi: 10.1186/cc6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lombardi F., De Chaumont C., Shields D.C., Moran N. Platelet signalling networks: pathway perturbation demonstrates differential sensitivity of ADP secretion and fibrinogen binding. Platelets. 2011;23(1):17–25. doi: 10.3109/09537104.2011.594190. [DOI] [PubMed] [Google Scholar]
- 25.Arthur J.F., Qiao J., Shen Y., Davis A.K., Dunne E., Berndt M.C. ITAM receptor-mediated generation of reactive oxygen species in human platelets occurs via Syk-dependent and Syk-independent pathways. J. Thromb. Haemost. 2012;10(6):1133–1141. doi: 10.1111/j.1538-7836.2012.04734.x. [DOI] [PubMed] [Google Scholar]
- 26.Kent N.J., Basabe-Desmonts L., Meade G., MacCraith B.D., Corcoran B.G., KENNY D. Microfluidic device to study arterial shear-mediated platelet-surface interactions in whole blood: reduced sample volumes and well-characterised protein surfaces. Biomed. Microdev. 2010 Dec;12(6):987–1000. doi: 10.1007/s10544-010-9453-y. [DOI] [PubMed] [Google Scholar]
- 27.Gurbel P.A., Bliden K.P., Antonino M.J., Stephens G., Gretler D.D., Jurek M.M. The effect of elinogrel on high platelet reactivity during dual antiplatelet therapy and the relation to CYP2C19⁎2 genotype: first experience in patients. J. Thromb. Haemost. 2010;8(1):43–53. doi: 10.1111/j.1538-7836.2009.03648.x. [DOI] [PubMed] [Google Scholar]
- 28.Nieswandt B., Bergmeier W., Eckly A., Schulte V., Ohlmann P., Cazenave J.P. Evidence for cross-talk between glycoprotein VI and Gi-coupled receptors during collagen-induced platelet aggregation. Blood. 2001;97(12):3829–3835. doi: 10.1182/blood.v97.12.3829. [DOI] [PubMed] [Google Scholar]
- 29.Atkinson B.T., Stafford M.J., Pears C.J., Watson S.P. Signalling events underlying platelet aggregation induced by the glycoprotein VI agonist convulxin. Eur. J. Biochem. 2001;268(20):5242–5248. doi: 10.1046/j.0014-2956.2001.02448.x. [DOI] [PubMed] [Google Scholar]
- 30.Carnevale R., Pignatelli P., Lenti L., Buchetti B., Sanguigni V., Di Santo S. LDL are oxidatively modified by platelets via GP91phox and accumulate in human monocytes. FASEB J. 2007;21(3):927–934. doi: 10.1096/fj.06-6908com. [DOI] [PubMed] [Google Scholar]
- 31.Gianni D., Taulet N., Zhang H., DerMardirossian C., Kister J., Martinez L. A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem. Biol. 2010;5(10):981–993. doi: 10.1021/cb100219n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sill J.C., Proper J.A., Johnson M.E., Uhl C.B., Katusic Z.S. Reactive oxygen species and human platelet GP IIb/IIIa receptor activation. Platelets. 2007;18(8):613–619. doi: 10.1080/09537100701481385. [DOI] [PubMed] [Google Scholar]
- 33.Quinton T.M., Ozdener F., Dangelmaier C., Daniel J.L., Kunapuli S.P. Glycoprotein VI-mediated platelet fibrinogen receptor activation occurs through calcium-sensitive and PKC-sensitive pathways without a requirement for secreted ADP. Blood. 2002;99(9):3228–3234. doi: 10.1182/blood.v99.9.3228. [DOI] [PubMed] [Google Scholar]
- 34.Nieswandt B., Watson S.P. Platelet-collagen interaction: is GPVI the central receptor? Blood. 2003;102(2):449–461. doi: 10.1182/blood-2002-12-3882. [DOI] [PubMed] [Google Scholar]
- 35.Cho M.J., Liu J., Pestina T.I., Steward S.A., Thomas D.W., Coffman T.M. The roles of αIIbβ3-mediated outside-in signal transduction, thromboxane A2, and adenosine diphosphate in collagen-induced platelet aggregation. Blood. 2003;101(7):2646–2651. doi: 10.1182/blood-2002-05-1363. [DOI] [PubMed] [Google Scholar]
- 36.Herr A.B., Farndale R.W. Structural insights into the interactions between platelet receptors and fibrillar collagen. J. Biol. Chem. 2009;284(30):19781–19785. doi: 10.1074/jbc.R109.013219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kehrel B., Wierwille S., Clemetson K.J., Anders O., Steiner M., Graham Knight C. Glycoprotein VI is a major collagen receptor for platelet activation: it recognizes the platelet-activating quaternary structure of collagen, whereas CD36, glycoprotein IIb/IIIa, and von willebrand factor do not. Blood. 1998;91(2):491–499. [PubMed] [Google Scholar]
- 38.Paravicini T.M., Nadph Oxidases Touyz RM. Reactive oxygen species, and hypertension. Diabetes Care. 2008;31(Suppl. 2):S170–S180. doi: 10.2337/dc08-s247. [DOI] [PubMed] [Google Scholar]
- 39.Zhou H., Das S., Murthy K.S. Erk1/2- and p38 MAP kinase-dependent phosphorylation and activation of cPLA2 by m3 and m2 receptors. Am. J. Physiol.—Gastrointest. Liver Physiol. 2003;284(3):G472–G480. doi: 10.1152/ajpgi.00345.2002. [DOI] [PubMed] [Google Scholar]
- 40.Lin L.L., Wartmann M., Lin A.Y., Knopf J.L., Seth A., Davis R.J. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72(2):269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- 41.Woulfe D.S. Akt signaling in platelets and thrombosis. Expert Rev. Hematol. 2010;3(1):81–91. doi: 10.1586/ehm.09.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Catarzi S., Romagnoli C., Marcucci G., Favilli F., Iantomasi T., Vincenzini M.T. Redox regulation of ERK1/2 activation induced by sphingosine 1-phosphate in fibroblasts: Involvement of NADPH oxidase and platelet-derived growth factor receptor. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2011;1810(4):446–456. doi: 10.1016/j.bbagen.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 43.Robinson K.A., Stewart C.A., Pye Q.N., Nguyen X., Kenney L., Salzman S. Redox-sensitive protein phosphatase activity regulates the phosphorylation state of p38 protein kinase in primary astrocyte culture. J. Neurosci. Res. 1999;55(6):724–732. doi: 10.1002/(SICI)1097-4547(19990315)55:6<724::AID-JNR7>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 44.Begonja A.J., Geiger J.+, Rukoyatkina N., Rauchfuss S., Gambaryan S., Walter U. Thrombin stimulation of p38 MAP kinase in human platelets is mediated by ADP and thromboxane A2 and inhibited by cGMP/cGMP-dependent protein kinase. Blood. 2007;109(2):616–618. doi: 10.1182/blood-2006-07-038158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material