

Abstract
Uncontrolled fibrosis in organs like heart, kidney, liver and lung is detrimental and may lead to end-stage organ failure. Currently there is no effective treatment for fibrotic disorders. Transforming growth factor (TGF)-β has a fundamental role in orchestrating the process of fibrogenesis; however, interventions directly targeting TGF-β would have undesired systemic side effects due to the multiple physiological functions of TGF-β. Further characterization of the downstream signaling pathway(s) involved in TGF-β-mediated fibrosis may lead to discovery of novel treatment strategies for fibrotic disorders. Accumulating evidence suggests that Nox4 NADPH oxidase may be an important downstream effector in mediating TGF-β-induced fibrosis, while NADPH oxidase-dependent redox signaling may in turn regulate TGF-β/Smad signaling in a feed-forward manner. It is proposed that pharmacological inhibition of the Nox4 function may represent a novel approach in treatment of fibrotic disorders.
Keywords: NADPH oxidase, Nox4, Transforming growth factor-β, Fibrosis, Redox signaling
TGF-β and fibrotic disorders
Fibrosis is caused by uncontrolled wound healing responses during tissue repair in pathological conditions such as myocardial infarction, idiopathic pulmonary fibrosis, hepatitis, and chronic kidney disease [1]. Fibrotic scars are characterized by accumulation of contractile matrix proteins and tend to compromise normal tissue functions. One of the major profibrotic cytokines orchestrating fibrogenesis is transforming growth factor (TGF)-β (see Fig. 1). More than two decades ago it was shown that an exogenous application of TGF-β to incisional wounds accelerated production of collagen and improved the wound healing efficiency [2]. TGF-β is a multifunctional protein, and one of its major biological effects is to promote the recruitment of fibroblasts to wound site and synthesis of structural matrix proteins [3]; both processes are crucial to fibrogenesis (Fig. 1). TGF-β is produced by infiltrating inflammatory cells (for example macrophages), parenchymal cells, and platelets during tissue repair [4], [5]. The profibrotic activity of TGF-β can be further regulated by other endogenous factors such as angiotensin II [6].
Fig. 1.
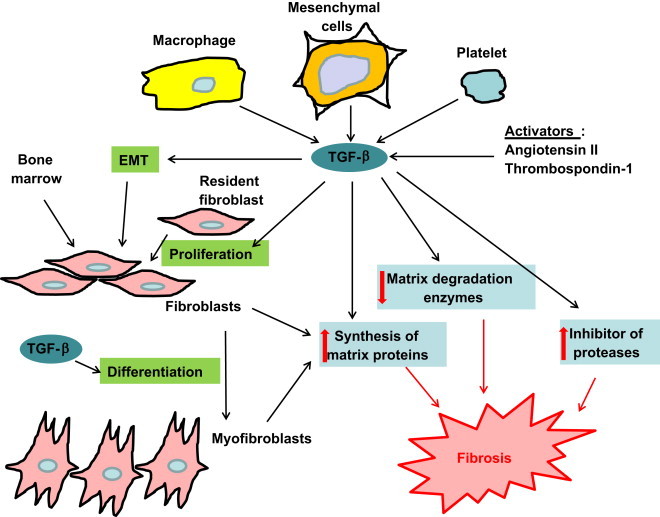
How TGF-β stimulates fibrotic responses during tissue repair. TGF-β is released by platelets, parenchymal cells and macrophages or its activity stimulated by endogenous factors such as angiotensin II. Under the influence of TGF-β, proliferation and migration of resident fibroblasts and the process of epithelial to mesenchymal transition (EMT) occur, while fibroblasts differentiate into myofibroblasts in the wound site. Differentiated myofibroblasts have a higher efficiency in synthesizing extracellular matrix proteins. Accumulation of extracellular matrix is enhanced by the inhibitory and stimulatory effects of TGF-β on expression of matrix metalloproteases and their inhibitors respectively.
A major source of fibroblasts identified in scarred tissues is proliferation and migration of resident fibroblasts to the injured site, partially stimulated by TGF-β [7], [8], while other sources may include epithelial to mesenchymal transition (EMT) and cells derived from the bone marrow [9]. Iwano et al. used bone marrow chimeras and transgenic reporter mice to trace origins of fibroblasts in fibrotic kidneys; two populations of fibroblasts have been identified, with the majority being derived from local EMT and a minor proportion being from the bone marrow [9]. TGF-β mediates EMT in a variety of organs including lung, liver, kidney, heart and eye [10]. Endothelial to mesenchymal transition mediated by TGF-β has also been observed and may play a role in cardiac fibrosis [11]. Hence, TGF-β is a crucial mediator for the generation and mobilization of fibroblasts required for fibrogenesis. Fibroblasts identified in scarred tissues are responsible for the production of matrix proteins [12]. TGF-β promotes accumulation of these proteins by inducing a phenotypic change of fibroblast to a highly efficient matrix-producing type termed myofibroblast [3]. To prevent degradation of the newly synthesized matrix proteins, TGF-β also inhibits expression of matrix catabolizing enzymes such as matrix metalloproteinases, and induces expression of matrix metalloproteinase inhibitors such as the tissue inhibitor of metalloproteinase [4].
Interventions directly targeting TGF-β would have undesired systemic side effects due to the multiple physiological functions of TGF-β [13], [14]. Therefore, further characterization of the downstream signaling pathway(s) involved in TGF-β-induced fibrotic process may provide useful information in the discovery of novel treatment strategies for various fibrotic disorders.
Involvement of NADPH oxidase in TGF-β-mediated profibrotic effects
Accumulative evidence highlights the involvement of NADPH oxidase-dependent redox signaling in the profibrotic responses mediated by TGF-β [15]. Five isoforms of the Nox catalytic subunit, namely Nox1, Nox2, Nox3, Nox4 and Nox5, have been identified [16]. The prototypical NADPH oxidase is composed of the membrane-bound Nox and p22phox, as well as the cytosolic subunits p40phox, p47phox, p67phox and a small GTPase Rac (Fig. 2 insert) [16]. Whereas all of the Nox isoforms except for Nox3 have been shown to be expressed in fibroblasts from different organs (including blood vessels, heart, lung and kidney), current evidence suggests that the Nox4 type NADPH oxidase may have a pivotal role in mediating TGF-β-induced profibrotic responses [15], [17], [18], [19], [20], [21], [22].
Fig. 2.
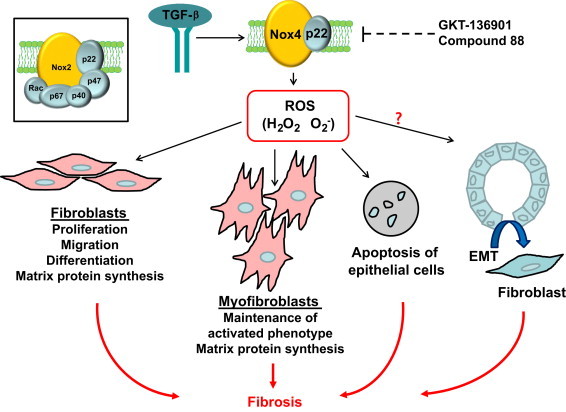
Involvement of Nox4 type NADPH oxidase in TGF-β-induced profibrotic responses. Nox4-derived ROS facilitate TGF-β-mediated fibrosis by inducing differentiation of fibroblasts into myofibroblasts and synthesis of extracellular matrix proteins, while inhibiting their degradation. Nox4-derived ROS also promote apoptosis of epithelial cells. It remains to be confirmed whether Nox4 signaling is also involved in TGF-β-mediated epithelial to mesenchymal transition (EMT). It is proposed that selective inhibitors of Nox4 such as GKT-136901 and compound 88 may represent a novel strategy to combat fibrotic disorders. The inset illustrates the structure of the prototype Nox2.
TGF-β increased Nox4 gene expression without effects on Nox1, Nox2 or Nox5 in human cardiac fibroblasts [18]. Treatment with siRNA against Nox4 suppressed expression of TGF-β target genes including fibronectin, collagen I, α-smooth muscle actin (α-SMA, a marker of myofibroblast), and connective tissue growth factor, indicating that Nox4 was involved in TGF-β-induced differentiation of cardiac fibroblasts to myofibroblasts [18]. In renal tubular epithelial cells, Rhyu et al. showed that inhibition of NADPH oxidase activity with the non-selective Nox inhibitor DPI suppressed EMT and matrix protein production [23], [24]. In a recent study, we showed that in mouse cardiac fibroblasts, TGF-β-stimulated collagen synthesis and myofibroblast differentiation were abrogated by EUK-134 (a scavenger of both superoxide and hydrogen peroxide) or a dominant negative form of Nox4 [17]. Such modulating effects of Nox4 in TGF-β-mediated collagen expression have also been observed in fetal lung mesenchymal cells [19], pulmonary fibroblasts [25], kidney fibroblasts [20] and liver stellate cells [26]; all of these cells are engaged in the development of fibrosis in the affected organs. Moreover, there is evidence showing that Nox4 mRNA and protein are increased in pulmonary fibroblasts isolated from patients with idiopathic pulmonary fibrosis, and the Nox4 expression level correlates with expressions of procollagen I and α-SMA [25].
Amara et al. showed that targeted gene silencing of Nox4 with tracheal administration of siRNA suppressed bleomycin-induced pulmonary fibrosis in mice [25]. Similarly, Carnesecchi et al. reported that bleomycin-induced pulmonary fibrosis was prevented in Nox4 knockout mice, and this protective effect of Nox4 deficiency was attributable to a decrease in epithelial cell apoptosis induced by TGF-β [27]. Moreover, we used a mouse model of subcutaneous sponge implantation and demonstrated that local inhibition of Nox4 expression with RNA interference suppressed the stimulatory effects of TGF-β on collagen accumulation in vivo [17]. These in vitro and in vivo findings support the notion that Nox4 has a pivotal role in mediating the profibrotic actions of TGF-β (Fig. 2).
Currently, a challenge is that we lack isoform-specific NADPH oxidase inhibitors. GKT-136901 is an orally active selective inhibitor of Nox1 and Nox4 [28], which has been shown to be able to suppress liver fibrosis induced by bile duct ligation [29]. Jarman et al. recently showed that oral treatment with a putative Nox4 inhibitor known as compound 88 attenuated bleomycin-induced lung fibrosis in rats and TGF-β-induced induction of procollagen and α-SMA expression in human pulmonary fibroblasts [30]. These studies suggest that pharmacological inhibition of the Nox4 function may represent a novel strategy in treatment of fibrotic disorders.
Redox modulation of TGF-β signaling
In addition to a potential role of NADPH oxidase as downstream mediators in TGF-β-induced profibrotic effects, there is also evidence suggesting that redox pathways may regulate TGF-β/Smad signaling in a feed-forward manner. For example, using cell-free assays people have shown that irradiation or ion-catalyzed reactive oxygen species (ROS) formation elicits conversion of the latent form of TGF-β to its active form, which is one of the most important mechanisms that regulate activation of the TGF-β signaling [31]. ROS may also regulate TGF-β signaling indirectly via certain oxidative intermediate products. In cultured macrophages, treatment with 4-hydroxy nonenal (4-HNE), a major reactive aldehyde formed during lipid peroxidation, upregulated TGF-β expression and release [32], although it was not clear whether the same effect was also present in fibroblasts. Moreover, ROS may have modulating effects on TGF-β-induced Smad2/3 activation. In cardiac fibroblasts, it was observed that TGF-β-induced Smad2/3 phosphorylation was significantly suppressed by various antioxidant agents and by Nox4 gene silencing [18]. In a model of bleomycin-induced pulmonary fibrosis, the level of Smad2 phosphorylation was significantly decreased in Nox4 knockout mice [27]. In a separate study in kidney proximal tubular epithelial cells, it was reported that antioxidant treatment effectively inhibited TGF-β-induced phosphorylation of Smad2, although exogenous H2O2 failed to modulate Smad2 phosphorylation [24]. Nox4 modulation of Smad2/3 phosphorylation was also observed in cultured pulmonary fibroblasts [25]. These findings together indicate that NADPH oxidase and ROS molecules may have critical roles in facilitating TGF-β-induced Smad2/3 activation.
The mechanism of the enhancing effect of ROS on TGF-β-induced Smad phosphorylation is currently unclear. However, several possibilities that may explain this phenomenon are proposed. Activation of TGF-β signaling requires phosphorylation of the type I receptor (ALK5) on serine and threonine residues in the GS domain upon ligand binding, while phosphorylated ALK5 in turn induces phosphorylation of Smad2/3 in their C-terminal SXS motif and thereby initiates downstream signaling events [33]. ALK5 phosphorylation is negatively regulated (dephosphorylated) by protein phosphatase 1 (PP1) and PP2A [33]. There is evidence showing that PP1 and PP2A are redox-sensitive, with ROS molecules such as H2O2 being able to inactivate the activity of these enzymes [34], leading to enhanced ALK5 activation and Smad phosphorylation. Moreover, people have demonstrated that the phosphatase PTEN may induce a decrease in Smad2/3 phosphorylation, although this effect of PTEN appears to require another phosphatase PPM1A [33]. It is well documented that PTEN is sensitive to ROS-induced oxidation and inactivation [35]. In this scenario, ROS may facilitate Smad phosphorylation by inhibiting PTEN/PPM1A-mediated dephosphorylation. Nevertheless, the importance of these potential mechanisms in modulating TGF-β signaling in vivo is currently not understood, which needs to be further corroborated by direct experimental evidence.
In addition to regulating Smad phosphorylation, ROS may also modulate TGF-β signaling via non-canonical, Smad-independent mechanisms. In addition to Smad-transduced signals, TGF-β may activate other signaling pathways including the mitogen-activated protein kinase (MAPK) members c-Jun N-terminal kinase (JNK) and p38 [36]. Of note, it is clear that both JNK and p38 are redox sensitive, which can be activated by ROS in the cytoplasm [35]. Interestingly, experimental evidence has indicated that JNK and p38 may in turn enhance the transcriptional activities of Smad proteins by direct phosphorylation of Smad3 or indirectly by promoting Smad3 association with the transcriptional co-activator p300 [36]. Hence, ROS may facilitate TGF-β-induced signaling by enhancing JNK and p38 activation. These redox mechanisms possibly involved in modulating TGF-β functions are summarized in Fig. 3.
Fig. 3.
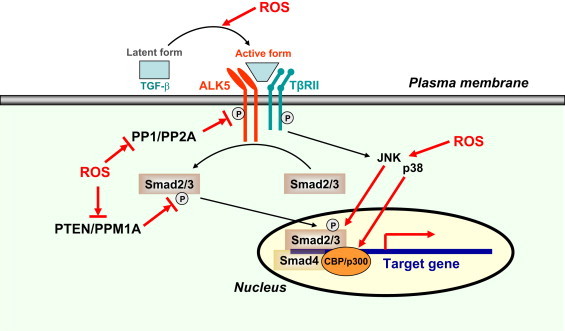
Potential redox-dependent mechanisms that may be involved in modulating TGF-β-induced intracellular signaling. ROS may facilitate TGF-β signaling by (1) promoting conversion of latent TGF-β to its active form; (2) enhancing phosphorylation of the TGF-β receptor I (ALK5); (3) enhancing phosphorylation of Smad2/3; (4) enhancing JNK and p38 activation, leading to increased transcriptional activities of Smad proteins. PP1, protein phosphatase 1.
Mechanisms of TGF-β regulation of Nox4 expression
TGF-β upregulates Nox4 expression in a variety of cells [17], [18], [20], [37], [38], [39], [40]. Two major mechanisms potentially involved in TGF-β-induced Nox4 expression are the classical Smad2/3 pathway and the phosphoinositide 3-kinase (PI3K) pathway (Fig. 4). We and others have demonstrated that TGF-β-induced Nox4 upregulation and ROS generation can be inhibited by inhibitors of ALK5 and Smad3, or dormant negative mutants of Smad2/3 [17], [37], [40], [41], suggesting that Nox4 expression occurs downstream of Smad2/3 activation. Similarly, overexpression of the inhibitory Smad protein Smad7 reversed the stimulating effects of TGF-β on Nox4 expression [41]. In addition to the Smad pathway, there is evidence suggesting that activation of PI3K may also be involved in TGF-β-induced regulation of Nox4 expression. For example, using a pharmacological inhibitor of PI3K, Michaeloudes et al. showed that in airway smooth muscle cells, PI3K inhibition abrogated Nox4 expression in response to TGF-β treatment [41]. Moreover, Sturrock et al. also demonstrated that TGF-β-stimulated Nox4 expression in airway smooth muscle cells was sensitive to PI3K inhibition [42]. However, the role of PI3K in TGF-β-induced modulation of Nox4 expression in fibroblasts is currently unclear, which needs to be further clarified.
Fig. 4.
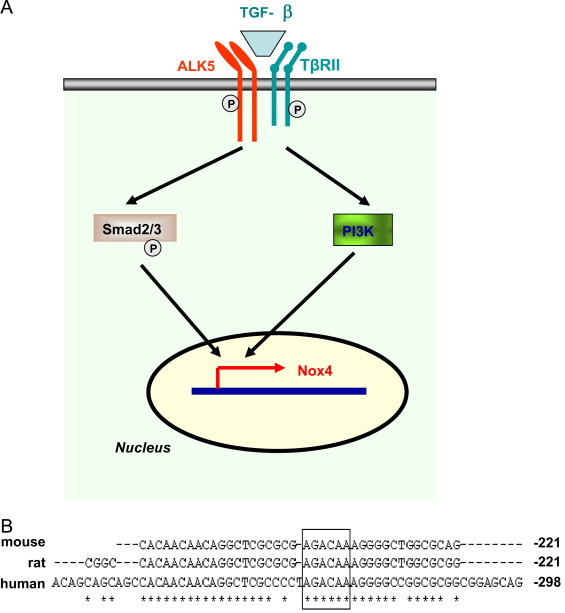
Potential mechanisms of TGF-β-induced upregulation of Nox4 gene expression. (A) TGF-β may stimulate Nox4 expression via the classical Smad2/3 pathway and activation of the PI3K pathway. (B) A putative Smad binding motif (boxed) is present in the Nox4 gene promoter region, which is conserved in human, mouse and rat.
Acknowledgements
This work was supported by the National 973 Basic Research Program (2010CB732605), Natural Science Foundation of China (81271269), Ophthalmic Research Institute of Australia (ORIA), and the Early Career Researcher grant (from University of Melbourne). G.J.D receives a Principal Research Fellowship from NHMRC. G.S.L. and E.C.C. are supported by The Ansell Ophthalmology Foundation. The Centre for Eye Research Australia receives Operational Infrastructure Support from the Victorian Government.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Fan Jiang, Email: fjiang@sdu.edu.cn.
Elsa C. Chan, Email: elsa.chan@unimelb.edu.au.
References
- 1.Ghosh A.K., Quaggin S.E., Vaughan D.E. Molecular basis of organ fibrosis: potential therapeutic approaches. Exp. Biol. Med. (Maywood) 2013;238:461–481. doi: 10.1177/1535370213489441. [DOI] [PubMed] [Google Scholar]
- 2.Mustoe T.A., Pierce G.F., Thomason A., Gramates P., Sporn M.B. Accelerated healing of incisional wounds in rats induced by transforming growth factor-beta. Science. 1987;237:1333–1336. doi: 10.1126/science.2442813. [DOI] [PubMed] [Google Scholar]
- 3.Leask A., Abraham D.J. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 4.Branton M.H., Kopp J.B. TGF-beta and fibrosis. Microb. Infect. 1999;1:1349–1365. doi: 10.1016/s1286-4579(99)00250-6. [DOI] [PubMed] [Google Scholar]
- 5.Meyer A., Wang W., Qu J., Croft L., Degen J.L. Platelet TGF-beta1 contributions to plasma TGF-beta1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood. 2012;119:1064–1074. doi: 10.1182/blood-2011-09-377648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uhal B.D., Kim J.K., Li X., Molina-Molina M. Angiotensin-TGF-beta 1 crosstalk in human idiopathic pulmonary fibrosis: autocrine mechanisms in myofibroblasts and macrophages. Curr. Pharm. Des. 2007;13:1247–1256. doi: 10.2174/138161207780618885. [DOI] [PubMed] [Google Scholar]
- 7.Phan S.H. Biology of fibroblasts and myofibroblasts. Proc. Am. Thorac. Soc. 2008;5:334–337. doi: 10.1513/pats.200708-146DR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalluri R., Neilson E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iwano M., Plieth D., Danoff T.M., Xue C., Okada H. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kovacic J.C., Mercader N., Torres M., Boehm M., Fuster V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: from cardiovascular development to disease. Circulation. 2012;125:1795–1808. doi: 10.1161/CIRCULATIONAHA.111.040352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeisberg E.M., Tarnavski O., Zeisberg M., Dorfman A.L., McMullen J.R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 12.Wynn T.A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Invest. 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pardali E., Goumans M.J., ten Dijke P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol. 2010;20:556–567. doi: 10.1016/j.tcb.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 14.Tian M., Neil J.R., Schiemann W.P. Transforming growth factor-beta and the hallmarks of cancer. Cell Signalling. 2011;23:951–962. doi: 10.1016/j.cellsig.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samarakoon R., Overstreet J.M., Higgins P.J. TGF-beta signaling in tissue fibrosis: redox controls, target genes and therapeutic opportunities. Cell Signalling. 2013;25:264–268. doi: 10.1016/j.cellsig.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan E.C., Jiang F., Peshavariya H.M., Dusting G.J. Regulation of cell proliferation by NADPH oxidase-mediated signaling: potential roles in tissue repair, regenerative medicine and tissue engineering. Pharmacol. Ther. 2009;122:97–108. doi: 10.1016/j.pharmthera.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 17.Chan E.C., Peshavariya H.M., Liu G.S., Jiang F., Lim S.Y. Nox4 modulates collagen production stimulated by transforming growth factor beta1 in vivo and in vitro. Biochem. Biophys. Res. Commun. 2013;430:918–925. doi: 10.1016/j.bbrc.2012.11.138. [DOI] [PubMed] [Google Scholar]
- 18.Cucoranu I., Clempus R., Dikalova A., Phelan P.J., Ariyan S. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 19.Hecker L., Vittal R., Jones T., Jagirdar R., Luckhardt T.R. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009;15:1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bondi C.D., Manickam N., Lee D.Y., Block K., Gorin Y. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J. Am. Soc. Nephrol. 2010;21:93–102. doi: 10.1681/ASN.2009020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sampson N., Berger P., Zenzmaier C. Therapeutic targeting of redox signaling in myofibroblast differentiation and age-related fibrotic disease. Oxid. Med. Cell. Longev. 2012;2012:458276. doi: 10.1155/2012/458276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barnes J.L., Gorin Y. Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int. 2011;79:944–956. doi: 10.1038/ki.2010.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rhyu D.Y., Park J., Sharma B.R., Ha H. Role of reactive oxygen species in transforming growth factor-beta1-induced extracellular matrix accumulation in renal tubular epithelial cells. Transplant. Proc. 2012;44:625–628. doi: 10.1016/j.transproceed.2011.12.054. [DOI] [PubMed] [Google Scholar]
- 24.Rhyu D.Y., Yang Y., Ha H., Lee G.T., Song J.S. Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J. Am Soc. Nephrol. 2005;16:667–675. doi: 10.1681/ASN.2004050425. [DOI] [PubMed] [Google Scholar]
- 25.Amara N., Goven D., Prost F., Muloway R., Crestani B. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax. 2010;65:733–738. doi: 10.1136/thx.2009.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sancho P., Mainez J., Crosas-Molist E., Roncero C., Fernandez-Rodriguez C.M. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS One. 2012;7:e45285. doi: 10.1371/journal.pone.0045285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carnesecchi S., Deffert C., Donati Y., Basset O., Hinz B. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signalling. 2011;15:607–619. doi: 10.1089/ars.2010.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laleu B., Gaggini F., Orchard M., Fioraso-Cartier L., Cagnon L. First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J. Med. Chem. 2010;53:7715–7730. doi: 10.1021/jm100773e. [DOI] [PubMed] [Google Scholar]
- 29.Jiang J.X., Chen X., Serizawa N., Szyndralewiez C., Page P. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic. Biol. Med. 2012;53:289–296. doi: 10.1016/j.freeradbiomed.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jarman E.R., Khambata V.S., Cope C., Jones P., Roger J. An inhibitor of NADPH oxidase-4 attenuates established pulmonary fibrosis in a rodent disease model. Am. J. Respir. Cell Mol. Biol. 2013 doi: 10.1165/rcmb.2013-0174OC. [DOI] [PubMed] [Google Scholar]
- 31.Barcellos-Hoff M.H., Dix T.A. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 1996;10:1077–1083. doi: 10.1210/mend.10.9.8885242. [DOI] [PubMed] [Google Scholar]
- 32.Leonarduzzi G., Scavazza A., Biasi F., Chiarpotto E., Camandola S. The lipid peroxidation end product 4-hydroxy-2,3-nonenal up-regulates transforming growth factor beta1 expression in the macrophage lineage: a link between oxidative injury and fibrosclerosis. FASEB J. 1997;11:851–857. doi: 10.1096/fasebj.11.11.9285483. [DOI] [PubMed] [Google Scholar]
- 33.Wrighton K.H., Lin X., Feng X.H. Phospho-control of TGF-beta superfamily signaling. Cell. Res. 2009;19:8–20. doi: 10.1038/cr.2008.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim H.S., Song M.C., Kwak I.H., Park T.J., Lim I.K. Constitutive induction of p-Erk1/2 accompanied by reduced activities of protein phosphatases 1 and 2A and MKP3 due to reactive oxygen species during cellular senescence. J. Biol. Chem. 2003;278:37497–37510. doi: 10.1074/jbc.M211739200. [DOI] [PubMed] [Google Scholar]
- 35.Jiang F., Zhang Y., Dusting G.J. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011;63:218–242. doi: 10.1124/pr.110.002980. [DOI] [PubMed] [Google Scholar]
- 36.Moustakas A., Heldin C.H. Non-Smad TGF-beta signals. J. Cell Sci. 2005;118:3573–3584. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- 37.Sturrock A., Cahill B., Norman K., Huecksteadt T.P., Hill K. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006;290:L661–L673. doi: 10.1152/ajplung.00269.2005. [DOI] [PubMed] [Google Scholar]
- 38.Carmona-Cuenca I., Roncero C., Sancho P., Caja L., Fausto N. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J. Hepatol. 2008;49:965–976. doi: 10.1016/j.jhep.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 39.Martin-Garrido A., Brown D.I., Lyle A.N., Dikalova A., Seidel-Rogol B. NADPH oxidase 4 mediates TGF-beta-induced smooth muscle alpha-actin via p38MAPK and serum response factor. Free Radic. Biol. Med. 2011;50:354–362. doi: 10.1016/j.freeradbiomed.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boudreau H.E., Casterline B.W., Rada B., Korzeniowska A., Leto T.L. Nox4 involvement in TGF-beta and SMAD3-driven induction of the epithelial-to-mesenchymal transition and migration of breast epithelial cells. Free Radic. Biol. Med. 2012;53:1489–1499. doi: 10.1016/j.freeradbiomed.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Michaeloudes C., Sukkar M.B., Khorasani N.M., Bhavsar P.K., Chung K.F. TGF-beta regulates Nox4, MnSOD and catalase expression, and IL-6 release in airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011;300:L295–L304. doi: 10.1152/ajplung.00134.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sturrock A., Huecksteadt T.P., Norman K., Sanders K., Murphy T.M. Nox4 mediates TGF-beta1-induced retinoblastoma protein phosphorylation, proliferation, and hypertrophy in human airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;292:L1543–L1555. doi: 10.1152/ajplung.00430.2006. [DOI] [PubMed] [Google Scholar]