

Abstract
We hereby report a case of diffuse pelvic peritoneal involvement by immunoglobulin G4-related disease (IgG4-RD). Numerous pelvic masses and nodules showing delayed enhancement on enhanced abdominal CT were found to congregate in the pelvic organs of a 57-year-old female presenting with intestinal subocclusion. The differentiation between peritoneal IgG4-RD and pelvic peritoneal carcinomatosis was only made by histopathology and immunohistochemistry performed after surgical resection. Autoimmune pancreatitis represents the historical prototype of IgG4-RD, but the spectrum of manifestations involving various organs has expanded during the last decade. In this report, we shortly review this clinical entity.
Keywords: IgG4-related disease; IgG4; Abdomen, CT; Immunohistochemistry; Pathologic correlation
INTRODUCTION
Immunoglobulin G4-related disease (IgG4-RD) is a very recently defined emerging entity characterized by a diffuse or mass forming inflammatory reaction rich in IgG4-positive plasma cells associated with fibrosclerosis and obliterative phlebitis, and it is found to affect an increasing number of organs or systems (1, 2). We here report a case of diffuse pelvic peritoneal involvement by IgG4-RD in a 57-year-old female presenting clinically with intestinal subocclusion. Diffuse pelvic masses and nodules of various sizes were congregated in pelvic organs and showed delayed enhancement on enhanced CT. Some calcifications were also visible but the radiological differentiation from pelvic peritoneal carcinomatosis was not possible. The definitive diagnosis was only made by histopathology and immunohistochemistry performed after surgical resection.
We shortly review this clinical entity and summarize its possible presentations.
CASE REPORT
A 57-year-old female presented with a several-year history of unexplained dyspeptic symptoms. Numerous vague causes were suggested including oesophagitis due to a small hiatal hernia and treated with lansoprazole and dysfunction of the sphincter of Oddi on the basis of a hepato-biliary scintigraphy (data not shown).
During the past year, the patient had experienced three acute episodes characterized by hypogastric colic pain followed colickly pain followed by alimentary-but occasionaly fecaloid-vomiting. These episodes lasted for about 24 to 48 hours and ended by diarrheal debacle. During the last episode, the patient was admitted to the emergency room after the ending diarrheal debacle. Laboratory tests and abdominal plain film were normal at this time. A new episode of abdominal pain compelled the patient to get readmitted to the emergency room. The pain was increasing for a period of 24 hours. The patient remained apyretic. On physical examination, the abdomen was distended. On palpation, there was diffuse tenderness especially in the right iliac fossa. Auscultation revealed a metallic sound. Laboratory test results indicated a mild inflammation with a CRP level of 5.84 mg/L (normal value: < 5 mg/L) and a white blood cell count of 16200 units.
Contrast-enhanced abdominal multidetector-row computed tomography (Fig. 1A-D) revealed a subocclusive intestinal syndrome. The middle and distal small bowel loops were distended along with the presence of hydroaeric levels. The transitional level was found within the pelvic area, and the origin seemed to be related to diffuse peritoneal carcinomatosis including pelvic masses and nodules diffusely covering and encasing the gynecological structures, the terminal ileum and the pelvic folds. A small amount of ascites was present. Numerous smaller nodules were scattered in the hypogastrium and in both iliac fossae. Some nodules were partially calcified and moderately enhanced on delayed contrast-enhanced CT scan. A possible ovarian origin was considered. The option of an emergency surgery was chosen to treat the subocclusive intestinal syndrome. An extensive surgical procedure was performed at one time including total radical hysterectomy, debulking, omentectomy, and resection of the ileo-cecal junction with resection of more than 30 centimeters of the terminal ileum.
Fig. 1.

57-year-old woman presenting with IgG4-related peritoneal disease.
A-D.Four selected contrast-enhanced axial CT views of hypogastric and pelvic areas show diffuse infiltration of pelvic organs by enhancing masses which encase distal ileum (white arrows) causing intestinal subocclusion (grey stars). Diffuse small round nodules are also dispersed within peritoneal spaces (black arrows). White stars show uterus, black stars small amount of fluid in Douglas pouch and grey arrows ovaries. IgG4 = immunoglobulin G4
E. Gross anatomy of resected organs including uterus (white star), ovaries and portion of incarcerated ileum (black arrow). Organs are massively infiltrated by numerous irregular whitish nodules mimicking diffuse carcinomatosis (white arrows). F. Gross anatomy of 30 cm of resected ileum terminale. Numerous white nodules (white arrows) infiltrating mesenteric fat tissue. G. Section through ileum (black star) showing pseudotumoral fibrosing tissue (black arrows) infiltrating full thickness of intestinal wall. IgG4 = immunoglobulin G4
H. Photomicrograph (hematoxylin-eosin [H-E] stain; magnification, × 10) of section of fatty peritoneal tissue showing area of dense storiform fibrosis (white arrow) separated by dense inflammatory lymphoplasmacytic infiltrate (black arrows) with germinal center formation (small black arrows). Black star shows normal fat tissue. I. Photomicrographic (H-E stain; magnification, × 40) details of typical storiform fibrosis intermingled with lymphoplasmacytic inflammatory infiltrate. J. Photomicrographic (orcein stain; magnification, × 40) details of obliterative phlebitis. K. Photomicrograph (IgG4 immunostain; magnification, × 40) showing numerous IgG4-positive plasma cells. IgG4 = immunoglobulin G4
Gross anatomy (Fig. 1E, F) showed numerous whitish nodules which extensively covered and invaded the gynecologic structures, the wall of the ileum and the mesenteric fat tissue. The pattern of invasion was that of diffuse carcinomatosis. On sections, some of these nodules were found to deeply penetrate the entire bowel wall from the serosa to the mucosa (Fig. 1G).
Microscopically (Fig. 1 H-K), these nodules showed areas of dense storiform fibrosis separated by areas of dense lymphoplasmacytic inflammatory infiltrate containing numerous normal plasma cells. Typical mild to moderate eosinophilia, germinal center formation and obliterative phlebitis were also detected. IgG4 immunostaining showed numerous IgG4-positive plasma cells.
The final diagnosis was IgG4-RD with a predominant but a very unusual peritoneal involvement. The serum IgG4 level was not assessed in the preoperative period, but it was found to be normal in the postoperative and follow-up periods.
The patient was treated by methylprednisolone with an initial dose of 32 mg per day. This dose was reduced in a stepwise manner to 12 mg per day. Due to persistent small peritoneal nodules in the right iliac fossa and in the pouch of Douglas after six months (data not shown), azathioprine at a dose of 100 mg daily was added. Concomitantly, the dose of methylprednisolone was gradually lowered to 2 mg per day. At one year of evolution, the remaining small nodules were stable (data not shown).
DISCUSSION
The association of IgG4 with sclerosing diseases was first recognized in 2001 when Hamano et al. (3) reported that patients with autoimmune pancreatitis (AIP) had elevated serum IgG4 levels. Soon thereafter, the examination of pancreatectomy specimens from patients with AIP revealed the infiltration of the pancreas and surrounding tissues by increased numbers of IgG4-positive plasma cells (1).
Immunoglobulin G4-related disease was recognized as a systemic disease since various extrapancreatic lesions were observed in patients with AIP (1, 2, 4-6). The real etiology and pathogenesis of IgG4-RD is still not clearly understood (1, 7). Moreover the exact role of IgG4 or IgG4-positive plasma cells in this disease has not yet been elucidated. Only some inconsistent biological features such as hypergammaglobulinemia or hypocomplementemia support the autoimmune nature of the disease process (7). Various names have been ascribed to this clinicopathological entity including IgG4-related sclerosing disease, IgG4-related systemic sclerosing disease, IgG4-related disease, IgG4-related autoimmune disease, hyper-IgG4 disease and IgG4-related systemic disease (1, 8).
Nevertheless, the current internationally accepted terminology is "IgG4-RD" (9).
The recent increase in the number of reported cases of IgG4-RD and autoimmune pancreatitis probably indicates an increase in the awareness and diagnosis of this entity on the basis of serum markers and IgG4 immunostaining at biopsy rather than a real rise in its incidence (6).
The extrapancreatic lesions of IgG4-RD also exhibit the same characteristic histologic features including dense lymphoplasmacytic infiltrate, massive storiform fibrosis, and obliterative phlebitis as seen in IgG4-related pancreatitis (1, 9).
To date, numerous extrapancreatic organs and/or systems have been found to be affected by IgG4-RD, and they may be divided into various anatomical locations including abdominal organs, thoracic organs, head and neck localization and miscellaneous presentations. Most of these locations are summarized in Table 1 (1, 4, 6, 10-12). All of these organs may be affected either synchronously or metachronously. Several associations with various malignancies have been reported sporadically, but further studies are needed in this area to determine if there is any causal relationship between the malignancies and IgG4-RD (1).
Table 1.
Spectrum of Extrapancreatic IgG4-RD
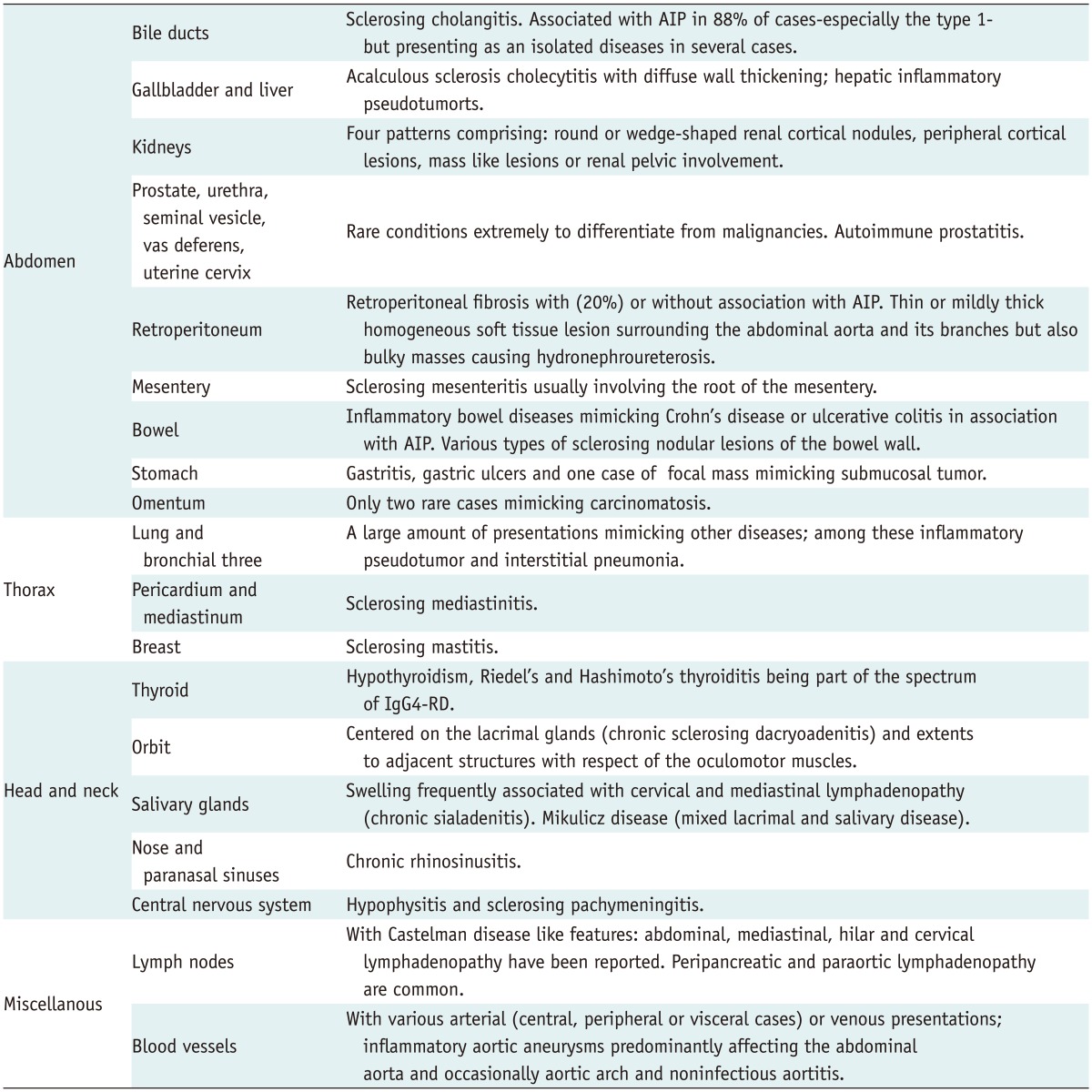
Note.- AIP = autoimmune pancreatitis, IgG4-RD = IgG4-related disease
Currently, there is no published international consensus on the diagnostic criteria for IgG4-RD (2). A raised serum IgG4 level is not mandatory for the diagnosis but may be of valuable assistance (1). Some authors have suggested that a serum IgG4 concentration that is more than twice the upper limit of normal (2.8 g/L) is highly specific for IgG4-RD (2). However, the serum IgG4 level may also be normal. Moreover, not every entity with increased IgG4+ plasma cells and a high IgG4/IgG ratio can be considered to belong to the IgG4-RD spectrum. Most authors agree that a definitive diagnosis of IgG4-RD requires histologic confirmation including the presence of characteristic histopathologic features such as dense lymphoplasmacytic infiltration, storiform fibrosis, obliterative phlebitis, mild to moderate eosinophilia and germinal center formation along with immunohistochemical staining demonstrating an increased number of IgG4+ cells (2, 7, 9).
Because our patient presented with signs of intestinal obstruction, this entity could be confused with IgG4-related sclerosing mesenteritis (4, 13, 14). However, in our patient, intestinal obstruction was caused by encasement of the pelvic organs by numerous peritoneal masses. This feature is significantly different from that in sclerosing mesenteritis, in which fibrosis involves the retroperitoneum along the axis of the mesenteric vessels, a process which may secondarily cause intestinal obstruction by centripetal retraction and/or ischemia of the bowel by incarceration of its vessels.
Patients with IgG4-RD usually show a dramatic response to corticosteroid therapy, often within a few weeks, although spontaneous resolution may also occur. To date, there is no consensus on the steroid therapy regimen or duration of treatment in IgG4-RD. However, disease recurrence is common (occurring in 20-40% of patients) after stopping steroid treatment or during the steroid tapering period. Management of recurrent disease is controversial. Most clinicians administer a repeat course of corticosteroids with an immunomodulator such as azathioprine (1, 7).
To the best of our knowledge, no case of IgG4-RD primarily involving the peritoneum and mimicking peritoneal carcinomatosis has been reported previously. In the reported case, the differentiation of peritoneal IgG4-RD from pelvic peritoneal carcinomatosis was unfortunately not possible with imaging and the final diagnosis was made by histopathology.
References
- 1.Divatia M, Kim SA, Ro JY. IgG4-related sclerosing disease, an emerging entity: a review of a multi-system disease. Yonsei Med J. 2012;53:15–34. doi: 10.3349/ymj.2012.53.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ryu JH, Horie R, Sekiguchi H, Peikert T, Yi ES. Spectrum of Disorders Associated with Elevated Serum IgG4 Levels Encountered in Clinical Practice. Int J Rheumatol. 2012;2012:232960. doi: 10.1155/2012/232960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344:732–738. doi: 10.1056/NEJM200103083441005. [DOI] [PubMed] [Google Scholar]
- 4.Kim JH, Byun JH, Lee SS, Kim HJ, Lee MG. Atypical manifestations of IgG4-related sclerosing disease in the abdomen: imaging findings and pathologic correlations. AJR Am J Roentgenol. 2013;200:102–112. doi: 10.2214/AJR.12.8783. [DOI] [PubMed] [Google Scholar]
- 5.Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. 2003;38:982–984. doi: 10.1007/s00535-003-1175-y. [DOI] [PubMed] [Google Scholar]
- 6.Vlachou PA, Khalili K, Jang HJ, Fischer S, Hirschfield GM, Kim TK. IgG4-related sclerosing disease: autoimmune pancreatitis and extrapancreatic manifestations. Radiographics. 2011;31:1379–1402. doi: 10.1148/rg.315105735. [DOI] [PubMed] [Google Scholar]
- 7.Moh IH, Kim JB, Shin SR, Jung SW, Park SH, Kim JW, et al. A case of intraperitoneal immunoglobulin G4-related inflammatory pseudotumor. Korean J Gastroenterol. 2012;60:258–261. doi: 10.4166/kjg.2012.60.4.258. [DOI] [PubMed] [Google Scholar]
- 8.Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T, et al. A novel clinical entity, IgG4-related disease (IgG4RD): general concept and details. Mod Rheumatol. 2012;22:1–14. doi: 10.1007/s10165-011-0508-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stone JH, Khosroshahi A, Deshpande V, Chan JK, Heathcote JG, Aalberse R, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum. 2012;64:3061–3067. doi: 10.1002/art.34593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamashita H, Takahashi Y, Ishiura H, Kano T, Kaneko H, Mimori A. Hypertrophic pachymeningitis and tracheobronchial stenosis in IgG4-related disease: case presentation and literature review. Intern Med. 2012;51:935–941. doi: 10.2169/internalmedicine.51.6604. [DOI] [PubMed] [Google Scholar]
- 11.Inoue D, Zen Y, Abo H, Gabata T, Demachi H, Yoshikawa J, et al. Immunoglobulin G4-related periaortitis and periarteritis: CT findings in 17 patients. Radiology. 2011;261:625–633. doi: 10.1148/radiol.11102250. [DOI] [PubMed] [Google Scholar]
- 12.Choi JW, Kim SY, Moon KC, Cho JY, Kim SH. Immunoglobulin G4-related sclerosing disease involving the urethra: case report. Korean J Radiol. 2012;13:803–807. doi: 10.3348/kjr.2012.13.6.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen TS, Montgomery EA. Are tumefactive lesions classified as sclerosing mesenteritis a subset of IgG4-related sclerosing disorders? J Clin Pathol. 2008;61:1093–1097. doi: 10.1136/jcp.2008.057869. [DOI] [PubMed] [Google Scholar]
- 14.Minato H, Shimizu J, Arano Y, Saito K, Masunaga T, Sakashita T, et al. IgG4-related sclerosing mesenteritis: a rare mesenteric disease of unknown etiology. Pathol Int. 2012;62:281–286. doi: 10.1111/j.1440-1827.2012.02805.x. [DOI] [PubMed] [Google Scholar]