

Abstract
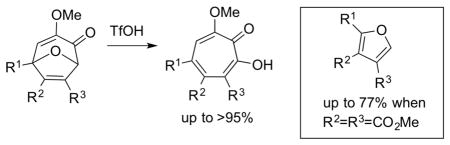
Methoxytropolones are useful scaffolds for therapeutic development due to their known biological activity and established value in the synthesis of α-hydroxytropolones. Upon treatment with triflic acid, a series of 3-methoxy-8-oxabicyclo[3.2.1]octa-3,6-dien-2-ones rearrange rapidly and cleanly to form methoxytropolones. Interestingly, bicycles that are derived from dimethyl acetylenedicarboxylate (R2=R3=CO2Me) instead form furans as the major product.
Introduction
MY3-469 (Figure 1) is a methoxytropolone inhibitor of platelet-type 12-lipoxygenase, an enzyme that has been implicated in cardiovascular and renal diseases, as well as cancer and inflammatory responses.1 In addition, the methoxytropolone-containing natural product pareirubrine A has demonstrated potent antileukemic properties.2 Our lab’s interest in methoxytropolones stem from their demonstrated value as intermediates in the synthesis of α-hydroxytropolones, which have a broad range of bio-activity.3 For example, α-hydroxytropolones are the most potent known inhibitors of ANT(2″),4 a major enzyme associated with bacterial resistance to aminoglycoside antibiotics,5 and are among the most potent inhibitors of HIV RT RNase H,6 which is a promising target for HIV treatment.7 Crystal structures of α-hydroxytropolones bound to the binuclear active site of RNase H reveal an ordered three oxygen, two metal binding pattern that likely provides this potency.8 Similar binding is thought to be responsible for α-hydroxytropolone inhibition of other binuclear metalloenzymes such as inositol mono-phosphatase, alkaline phosphatase,9 and phospholipase C,10 and it is possible that this binding mode may lead to the pharmacophore having privilege for many other similar metalloenzymes.
Figure 1.

Methoxytropolones, hydroxytropolones and examples of their bioactivity
Despite the potential of hydroxytropolones and methoxytropolones as therapeutic leads, very few synthetic chemistry-driven structure-function studies have been conducted on them,11 perhaps due to the scarcity of synthetic methods available to access them.12 Early strategies to access this class of molecules focused on tropone oxidation (scheme 1A), which can be an efficient method for generating the parent α-hydroxytropolone but can make introduction of functionality with control challenging.13 This has led to efforts to generate the α-hydroxytropolones through other, more controlled, strategies. One method that could be particularly useful in structure-function studies is a cyclopropanation/ring-opening strategy used extensively by the Banwell group for tropone and tro-polone synthesis.14 In one representive example of this work, the group leveraged a bromine handle to perform cross-coupling chemistry to synthesize different α-hydroxytropolones (scheme 1B).14a More recently, Föhlisch and coworkers showed a very efficient route to α-hydroxytropolones starting with furans that they then converted to dialkoxy-8-oxabicyclo[3.2.1]oct-6-en-3-ones (scheme 1C).15 These bicyclic substrates were then opened with the aid of base and heat to produce methoxytropolones, which could be converted to α-hydroxytropolones through standard demethylation reaction conditions.
Scheme 1.
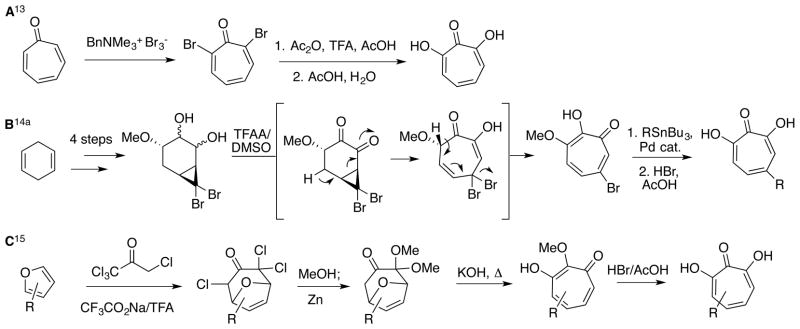
Representative examples illustrating established strategies for the synthesis of α-hydroxytropolones
Inspired by literature examples where oxidopyrylium cycloaddition chemistry and ring-opening methods were used in tropolone synthesis (scheme 2A), we have been studying similar strategies toward α-hydroxytropolones.16 Adapting this approach required an α-hydroxy-γ-pyrone oxidopyrylium cycloaddition reaction17 modified for intermolecular reactions by Wender and Mascareñas (scheme 2B).18 By using an optimized version of the Wender-Mascareñas oxidopyrylium cycloaddition procedure along with a demethylative boron trichloride ring-opening, we demonstrated that α-hydroxytropolones can be synthesized from kojic acid through a two-step sequence (scheme 2C).19 Among the benefits of this route are the low cost of the kojic acid starting material (10kg can be purchased for $850 through Chem Impex), the scalability of the synthesis of 2, which can be made on a gram scale within a few days without any need for chromatography, and the ability to quickly generate di- and polysubstituted α-hydroxytropolones.20 Among the limitations to the process are the need for electronically stabilizing groups in conjugation with the alkynes (ie aryl acetylenes, propiolates or ynones). The synthesis of various hydroxytropolones can be achieved in two to three steps from a common, scalable intermediate, making the route very appealing for SAR studies. Moreover, the boron trichloride method that we have reported led directly to α-hydroxytropolones for several substrates tested. However, in some instances methoxytropolones were also generated, either as byproducts or, in once instance, the only product formed. In addition, a phenylacetylene-derived bicycle led to a mixture of at least three compounds that were inseparable. During the course of medicinal chemistry-driven pursuits on these molecules, the unpredictable nature of the boron trichloride chemistry became an apparent barrier, and thus we became interested in developing a more robust and predictable method for this ring-opening. The following outlines our studies on triflic acid-mediated ring-openings of 3-methoxy-8-oxabicyclo[3.2.1]octa-3,6-dien-2ones to generate methoxytropolones.
Scheme 2.
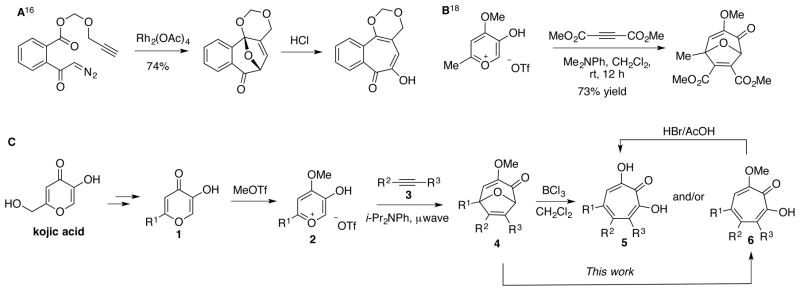
Precedence (A and B) for oxidopyrylium cycloaddition/ring-opening strategy toward substituted α-hydroxytropolones (C).
Results and Discussion
Early studies focused on phenyl acetylene-derived 3-methoxy-8-oxabicyclo[3.2.1]octa-3,6-dien-2-one 4a due to its problematic nature under the previously described conditions,19 and deuterated chloroform was used to readily monitor reaction progress by 1H NMR (Scheme 3). While milder acids, including trifluoroacetic and toluenesulfonic acid, were not capable of mediating the ring-opening, sulfuric acid and triflic acid both led to methoxytropolone 6a within a half an hour under ambient temperature and atmosphere, with triflic acid promoting a considerably cleaner reaction. During these studies, sub-stoichiometric amounts of triflic acid (0.5 equivalents) were found capable of leading to quantitative yields of 6a (Scheme 3, Conditions B).21 When similar conditions were attempted on nitroaryl 4b, incomplete conversion and other by-products were observed. Fortunately, increasing the amount of acid to 4 equivalents led to a near quantitative reaction (Conditions C). This was in contrast to our previous studies with BCl3 in which case a 1:1 mixture of hydroxytropolone and methoxytropolone was obtained (Conditions A).19 Higher concentrations of triflic acid were also found to work well on 4a. Thus, 4 equivalents were identified as an optimal amount to use for our substrate scope studies.
Scheme 3.
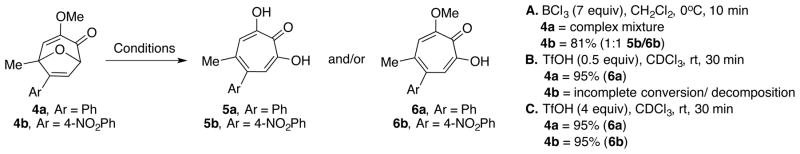
Prior results from aryl-substituted bicycles 4a and 4b with boron trichloride (A) along with triflic acid conditions (B and C)
Our attention was next turned toward cycloadducts arising from alkynyl caboxylates, as some of these provided yield and selectivity issues under our previous conditions (Scheme 4, Conditions A).19 Ethyl ester bicycle 4c had previously been found to lead to low yields, which we suspect may be due to hydrolysis of an exposed ester during the longer quench used in the boron trichloride reaction workup. Gratifyingly, we found that using the new triflic acid conditions (Conditions C), methoxytropolone 6c was formed in excellent yields. Another substrate, phenyl ketone bicycle 4d had previously led to a completely unselective 1:1 ratio of hydroxy and methoxytropolone. Under the new conditions, methoxytropolone are formed exclusively in near quantitative yields. Finally, the compound bearing a methyl substituent at R2 had previously led to 6e in 77% yield, which was satisfactory. However, under the new conditions this reaction was again near quantitative, representing a significant improvement.
Scheme 4.
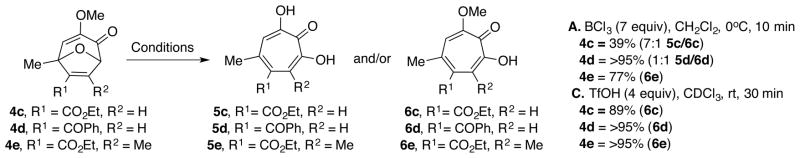
Prior results from bicycles derived from alkynyl carboxylates (4c–e) with boron trichloride (A) along with new and methoxytropolone selective triflic acid conditions (C)
Cross-coupling of methoxytropolones have been widely precedented by the work of Banwell (Scheme 1B), and thus we also wanted to synthesize methoxytropolones with halogen handles that may be useful for structure-function studies. Two halogen-containing bicycles were synthesized through the optimized oxidopyrylium cycloaddition reaction previously described. One of these, 4f, contained a bromide on the phenyl appendage, while the other, 4g, had an alkyl chloride. Of note was the sluggish reaction toward 4g that employed a chloromethyl triflate salt 2b and phenylacetylene (3a). In this instance, four hours under microwave irradiation at 100°C was necessary to obtain respectable yields of the bicycle. Gratifyingly, both bicycles rearranged cleanly to afford the anticipated products in excellent yields.
An example of the utility can be seen in the synthesis of biphenyl methoxytropolone 7 from bromophenyl 6f (Scheme 3). While the (PPh3)2Pd(II)Cl2/dioxane Stille conditions previously described by Banwell did not work in this case, we found that the use of Pd(PPh3)4 in refluxing toluene provided the anticipated cross-coupling product in yields ranging from 44–55%. These compounds can also be demethylated under known conditions to provide α-hydroxytropolone 8.
Attempts at similar cross-coupling reactions with chloromethyl-containing compound 6g were unsuccessful. However, 6g does react with nucleophiles such as sodium acetate and sodium azide to generate new methoxytropolones 9 and 10, the latter of which will undergo copper-catalyzed Huisgen [3+2] dipolar cycloaddition coupling with phenyl acetylene to generate triazole 11.22
When bicycle 4h was subjected to the triflate reaction conditions, we noticed a surprising change in reactivity. In this instance, the anticipated methoxytropolone was only a minor component (<20%), and the major isolated compound was instead furan 12h,23 as confirmed by comparison to known 1H NMR spectral data (Scheme 8A). Given the surprising nature of this discovery, and our initial skepticism, we synthesized 4i, which would lead to another known furan that had symmetry and would thus confirm this discovery. The oxidopyrylium cycloaddition chemistry with triflate salt 2c did not react at all at 100°C, and had to be heated to 150°C to promote the conversion to 4i. With 4i in hand, treatment of the molecule to the reaction conditions led to the known, symmetric furan 12i,24 which helped confirm this rearrangement.
Scheme 8.
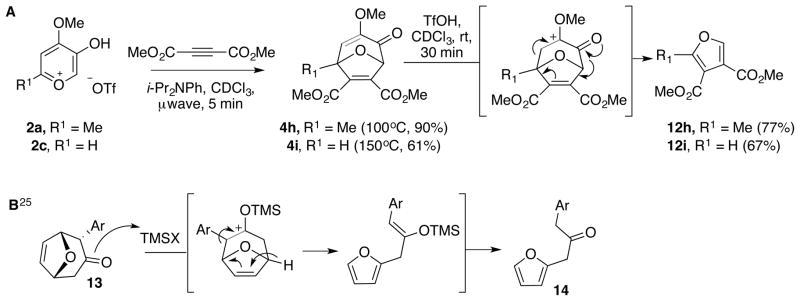
(A) Unanticipated furan rearrangment of dimethylacetylene dicarboxylate-derived bicycles 4h and 4i and (B) Mechanistically similar rearrangment reported by Mann and coworkers.
Our current hypothesis is that furan formation is favored when the two ethyl esters destabilize the allylic carbocation character necessary for ring-opening. This may allow for protonation of the enol ether, which may initiate a cation-mediated cycloreversion reaction (as is illustrated in Scheme 8A). Mann and coworkers disclosed a mechanistically related fragmentation that supports this proposal (Scheme 8B).25 This could happen either through sp hybridized oxonium formation (as shown), or through a hemiacetal or hydrate intermediate. However, no noticeable differences in reactivity were observed when the reaction was run over molecular sieves. Another possibility is that protonation of one or multiple carbonyls may promote a cycloreversion reaction, generating cyclo-propenone byproducts. Unfortunately, we have been unable to identify any byproducts that could provide insight into the reaction, which may be the result of polymerization of acrylate-like byproducts. Mechanistic studies are currently underway.
While syntheses of furan and furan-containing compounds had been described as early as the 19th century,26 polysubstituted furan synthesis remains a significant challenge and one of high interest to chemists.27 This serendipitous finding reveals a two step oxi-dopyrylium cycloaddition/fragmentation strategy to furans that is reminiscent of widely used thermal processes involving oxazole or 1,3,4 oxadiazole-based Diels-Alder/retro-Diels-Alder processes (Scheme 9),28 and could find complementary use. The development of conditions for this fragmentation that would work for a broader range of substrates is clearly needed in order to meet this potential.
Scheme 9.
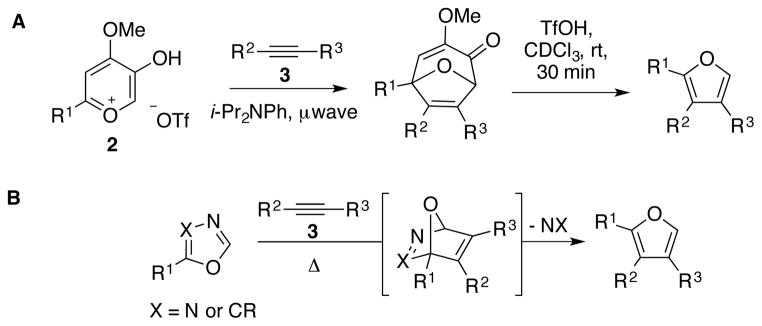
Oxidopyrylium cycloaddition/ring-opening (A) and oxazole/oxadiazole Diels-Alder/retro-Diels-Alder (B) strategy for furan synthesis. R1, R2, R3 are labeled as such for comparative purposes.
Conclusion
In conclusion, we have demonstrated that several methoxytropolones can be readily synthesized through acid-mediated ring-openings of 3-methoxy-8-oxabicyclo[3.2.1]octa-3,6-dien-2-ones. The high yielding nature of the chemistry, along with the ease of synthesis (ambient atmosphere and temperature) and rapid conversion (30 min) makes it an excellent method for quickly generating methoxytropolones, and should have value in structure-function studies on methoxy and hydroxytropolones.
Experimental
General Experiments Methods
All starting materials and reagents were purchased from commercially available sources and used without further purification, with the exception of dichloromethane, which was purified on a solvent purification system prior to the reaction. Microwave reactions were carried out in a Biotage® Initiator (External IR Temperature Sensor). 1H NMR shifts are measured using the solvent residual peak as the internal standard (CDCl3 δ 7.26), and reported as follows: chemical shift, multiplicity (s = singlet, bs = broad singlet, d = doublet, t = triplet, dd = doublet of doublet, q = quartet, m = multiplet), coupling constant (Hz), integration. 13C NMR shifts are measured using the solvent residual peak as the internal standard (CDCl3 δ 77.20), and reported as chemical shifts. Infrared (FTIR) spectral bands are characterized as broad (br), strong (s), medium (m), and weak (w).
General procedure for oxidopyrylium cycloaddition
Known 3-methoxy-8-oxabicyclo[3.2.1]octa-3,6-dien-2-ones 4a–e and 4h were synthesized as previously described. New 3-methoxy-8-oxabicyclo[3.2.1]octa-3,6-dien-2-ones 4f, 4g, and 4i were synthesized using previously described conditions.19 In short, into a microwave reactor vial was added triflate salt (2a–c), alkyne (20 equiv) and CHCl3 (0.2 M). N,N-Diisopropylaniline (1.2 or 2.0 equiv) was added, and the reaction vessel was sealed, and heated under microwave irradiation at 100°C (controlled temperature). The reaction was monitored periodically by 1H NMR, and deemed complete when the oxidopyrylium dimer was no longer observable.
6-(4-bromophenyl)-3-methoxy-5-methyl-8-oxabicyclo[3.2.1]octa-3,6-dien-2-one (4f)
5-hydroxy-4-methoxy-2-methylpyrylium 2a (100 mg, 0.34 mmol), and 1-ethynyl-4-bromobenzene (1.0 g, 6.9 mmol) were suspended in CHCl3 (2 mL). N,N-Diisopropylaniline (81 μL, 0.41 mmol) was added to the reaction, the reaction vessel was sealed, and the reaction mixture was heated under microwave irradiation at 100ºC (controlled temperature) for 30 min. The reaction mixture was then concentrated and purified by chromatography (silica gel, 18 × 1.8 cm, 50 mL Hexanes, 200 mL 2% EtOAc in Hexanes, 100 mL 10% EtOAc in Hexanes, 200 mL 15% EtOAc in Hexanes) to lead to 4f as a light yellow solid (68 mg, 61% yield). MP = 192–200ºC. Rf = 0.17 in 16% EtOAc/Hexanes. FTIR (KBr, thin film) 525 (m), 714 (m), 1057 (w), 1132(m), 1606(s), 1708(s), 1905 (w), 2978(m) cm−1. 1H NMR (400 MHz, CDCl3) δ 7.49 (d, J = 8.5 Hz, 2H), 7.14 (d, J = 8.5 Hz, 2H), 6.29 (d, J = 2.5 Hz, 1H), 6.14 (s, 1H), 4.96 (d, J = 2.5 Hz, 1H), 3.58 (s, 3H), 1.64 (s, J = 3H). 13C NMR (100 MHz, CD3CN/CDCl3) δ 189.43, 157.75, 145.66, 132.19, 131.73, 127.99, 124.04, 122.10, 119.73, 86.16, 85.77, 54.47, 21.35. HRMS (ESI+): calc’d for C15H14BrO3 (M+H): 321.0121; Found: 321.0114.
5-(chloromethyl)-3-methoxy-6-phenyl-8-oxabicyclo[3.2.1]octa-3,6-dien-2-one (4g)
2-(chloromethyl)-5-hydroxy-4-methoxypyrylium 2b (200 mg, 0.616 mmol), and phenylacetylene (1.35 mL, 12.3 mmol) were dissolved in CHCl3 (3.08 mL). N,N-Diisopropylaniline (240 μL, 1.23 mmol) was added to the reaction, the reaction vessel was sealed, and the reaction mixture was heated under microwave irradiation at 100ºC (controlled temperature) for 4 h. The reaction mixture was then concentrated and purified by chromatography (silica gel, 18 × 1.8 cm, 50 mL Hexanes, 200 mL 2% EtOAc in Hexanes, 100 mL 10% EtOAc in Hexanes, 200 mL 15% EtOAc in Hexanes) to lead to 4g as a highly viscous yellow oil (114 mg, 67% yield). Rf = 0.14 in 20% EtOAc in Hexanes. FTIR (KBr, thin film) 656(w), 798(s), 1078(m), 1261(s), 1607(b), 1709(b) 1960(w), 2254(w), 2838(w), 2962(s) cm−1. 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.09 (m, 5H), 6.22 (d, J = 2.4 Hz, 1H), 6.11 (s, 1H), 4.98 (d, J = 2.4 Hz, 1H), 3.94 (d, J = 12.4 Hz, 1H), 3.77 (d, J = 12.4 Hz, 1H), 3.52 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 189.2, 156.3, 146.9, 132.7, 129.4, 129.2, 125.9, 124.9, 114.8, 88.7, 86.4, 55.2, 45.9. HRMS (ESI+): calc’d for C15H13ClO3 : 276.0553; Found: 276.0552.
dimethyl 3-methoxy-2-oxo-8-oxabicyclo[3.2.1]octa-3,6-diene-6,7-dicarboxylate (4i)
3-hydroxy-4-methoxypyrylium29 2c (108 mg, 0.391 mmol), and dimethylacetylene dicarboxylate (1.11 mg, 7.82 mmol) were dissolved in CHCl3 (780 μL). N,N-Diisopropylaniline (91 μL, 0.47 mmol) was added to the reaction, the reaction vessel was sealed, and the reaction mixture was heated under microwave irradiation at 150ºC (controlled temperature) for 5 min. The reaction mixture was then concentrated and purified by chromatography (silica gel, 18 × 1.8cm, 50 mL of Hexanes, 75 mL 10% EtOAc in Hexanes, 100 mL 20% EtOAc in Hexanes, 200 mL 30% EtOAc in Hexanes) to lead to 4i as a white solid (64 mg, 61% yield). MP = 113–116ºC. Rf = 0.15 in 25% EtOAc/Hexanes. FTIR (KBr, thin film) 688 (m), 794 (w), 1000 (b), 1129 (s), 1611 (s), 1654 (m), 1716 (b), 2838 (m), 2956 (s), 3019 (w) cm−1; 1H NMR (400 MHz, CDCl3) δ 6.17 (d, J = 4.8 Hz, 1H), 5.46 (d, J = 4.8 Hz, 1H), 5.28 (s, 1H), 3.86 (s, 3H), 3.81 (s, 3H), 3.60 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 186.8, 162.7, 161.9, 149.5, 146.9, 138.5, 114.2, 88.9, 80.9, 55.2, 53.2, 53.1. HRMS (ESI+): calc’d for C12H13O7 (M+H): 269.0656; Found: 269.0652.
General procedure for methoxytropolone synthesis
3-methoxy-8-oxabicyclo[3.2.1]octa-3,6-dien-2-ones (4a–g) were dissolved in CHCl3 (0.1M) and trifluoromethanesulfonic acid (4 equiv) was added to the reaction. The reaction was stirred for 30 minutes, at which time it was quenched with phosphate buffer (1.6 M, pH 7), extracted with CH2Cl2, dried over Na2SO4, filtered and concentrated to yield methoxytropolones (6a–g).
2-hydroxy-7-methoxy-5-methyl-4-phenylcyclohepta-2,4,6-trienone (6a)
3-methoxy-5-methyl-6-phenyl-8-oxabicyclo[3.2.1]octa-3,6-dien-2-one 4a (10 mg, 0.041 mmol) was dissolved in CDCl3 (410 μL) and trifluoromethanesulfonic acid (15 μL, 0.165 mmol) was added to the reaction. The reaction was stirred for 30 min and was quenched with phosphate buffer (1.6 M, pH 7, 20 mL), extracted with CH2Cl2 (3 × 20 mL), dried over Na2SO4, filtered and concentrated to yield 6a as a yellow solid (9.5 mg, 95% yield) that decomposes at 175°C. FTIR (KBr, thin film) 799 (s), 1095 (s), 1260 (s), 1451 (w), 1726 (b), 2923 (w), 2962 (m) cm−1; 1H NMR (400 MHz, CDCl3) δ 7.45 – 7.24 (m, 5H), 7.23 (s, 1H), 7.18 (s, 1H), 4.04 (s, 3H), 2.27 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.9, 158.9, 158.4, 143.9, 143.6, 135.4, 128.9, 128.6, 128.0, 122.7, 122.4, 56.8, 27.0. HRMS (ESI+): calc’d for C15H15O3 (M+H): 243.1016; Found: 243.1019.
2-hydroxy-7-methoxy-5-methyl-4-(4-nitrophenyl)cyclohepta-2,4,6-trienone (6b)
3-methoxy-5-methyl-6-(4-nitrophenyl)-8-oxabicyclo[3.2.1]octa-3,6-dien-2-one 4b (29 mg, 0.10 mmol) was dissolved in CDCl3 (1.0 mL) and trifluoromethanesulfonic acid (61 μL, 0.41 mmol) was added to the reaction. The reaction was stirred for 30 min and was quenched with phosphate buffer (1.6 M, pH = 7, 20 mL), extracted with CH2Cl2 (3 × 20 mL), dried over Na2SO4, filtered and concentrated to yield 6b as a fine, powdery yellow solid (30 mg, >95% yield). 1H NMR matched previously reported data.20 1H NMR (400 MHz, CDCl3) δ 8.32 (d, J = 8.6 Hz, 2H), 7.45 (d, J = 8.6 Hz, 2H), 7.29 (s, 1H), 7.14 (s, 1H), 4.05 (s, 3H), 2.26 (s, 3H).
ethyl 6-hydroxy-4-methoxy-2-methyl-5-oxocyclohepta-1,3,6-trienecarboxylate (6c)
Ethyl 3-methoxy-5-methyl-2-oxo-8-oxabicyclo[3.2.1]octa-3,6-diene-6-carboxylate 4c (40 mg, 0.168 mmol) was dissolved in CDCl3 (1.68 mL) and trifluoromethanesulfonic acid (59 μL, 0.672 mmol) was added to the reaction. The reaction was stirred for 30 min and was quenched with phosphate buffer (1.6 M, pH = 7, 50 mL), extracted with CH2Cl2 (3 × 50 mL), dried over Na2SO4, filtered and concentrated to yield 6c as an orange solid (35.7 mg, 89% yield). MP = 99–106ºC. FTIR (KBr, thin film) 750 (s), 1056 (w), 1457 (s), 1561 (s), 1717 (s), 2927 (w), 3252 (b) cm−1; 1H NMR (400 MHz, CDCl3) δ 7.60 (s, 1H), 7.03 (s, 1H), 4.46 – 4.29 (q, J = 7.1 Hz, 2H), 4.02 (s, 3H), 2.56 (s, 3H), 1.39 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 171.2, 168.9, 159.8, 137.4, 132.4, 121.9, 117.8, 62.3, 56.8, 26.2, 14.5. HRMS (ESI+): calc’d for C12H15O5 (M+H): 239.0914; Found: 239.0913.
4-benzoyl-2-hydroxy-7-methoxy-5-methylcyclohepta-2,4,6-trienone (6d)
6-benzoyl-3-methoxy-5-methyl-8-oxabicyclo[3.2.1]octa-3,6-dien-2-one 4d (20 mg, 0.074 mmol) was dissolved in CDCl3 (0.74 mL) and trifluoromethanesulfonic acid (26.2 uL, 0.30 mmol) was added to the reaction. The reaction was stirred for 30 min and was quenched with phosphate buffer (1.6 M, pH = 7, 25 mL), extracted with CH2Cl2 (3 × 25 mL), dried over Na2SO4, filtered and concentrated to yield 6d as a dark yellow solid (19.7 mg, >95% yield). 1H NMR matched previously reported.20 1H NMR (400 MHz, CDCl3) δ 7.82 (d, J = 7.4 Hz, 2H), 7.63 (t, J = 7.0 Hz, 1H), 7.49 (t, J = 7.3 Hz, 2H), 7.19 (d, J = 29.1 Hz, 1H), 7.10 (s, 1H), 4.05 (s, 3H), 2.33 (s, 3H).
ethyl 6-hydroxy-4-methoxy-2,7-dimethyl-5-oxocyclohepta-1,3,6 trienecarboxylate (6e)
Ethyl 3-methoxy-5,7-dimethyl-2-oxo-8-oxabicyclo[3.2.1]octa-3,6-diene-6-carboxylate 4e (19 mg, 0.075 mmol) was dissolved in CDCl3 (750 μL) and trifluoro-methanesulfonic acid (26 μL, 0.30 mmol) was added to the reaction. The reaction was stirred for 30 min and was quenched with phosphate buffer (1.6 M, pH = 7, 30 mL), extracted with CH2Cl2 (3 × 30 mL), dried over Na2SO4, filtered and concentrated to yield 6e as a red solid (18.5 mg, >95% yield). 1H NMR matched previously reported data.20 1H NMR (400 MHz, CDCl3) δ 6.97 (s, 1H), 4.45 (q, J = 7.1 Hz, 2H), 3.97 (s, 3H), 2.43 (s, 3H), 2.40 (s, 3H), 1.42 (t, J = 7.1 Hz, 3H).
4-(4-bromophenyl)-2-hydroxy-7-methoxy-5-methylcyclohepta-2,4,6-trienone (6f)
6-(4-bromophenyl)-3-methoxy-5-methyl-8-oxabicyclo[3.2.1]octa-3,6-dien-2-one 4f (70.1 mg, 0.22 mmol) was dissolved in CDCl3 (2.2 mL) and trifluoromethanesulfonic acid (77 μL, 0.87 mmol) was added to the reaction. The reaction was stirred for 30 min and was quenched with phosphate buffer (1.6 M, pH = 7, 50 mL), extracted with CH2Cl2 (3 × 50 mL), dried over Na2SO4, filtered and concentrated to yield 6f as a yellow solid (70 mg, >95% yield). MP = 110–115ºC. FTIR (KBr, thin film) 815.4 (w), 1211.4 (s), 1264.2 (s), 1713.9 (w), 2939.4 (w), 3259.2 (b) cm−1. 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 8.3 Hz, 2H), 7.32 (s, 1H), 7.14 (s, 1H), 7.12 (d, J = 8.3 Hz, 2H), 4.02 (s, 3H), 2.25 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.1, 158.8, 158.5, 142.7, 141.8, 135.1, 132.1, 130.4, 122.4, 122.2, 121.7, 56.7, 26.9. HRMS (ESI+): calc’d for C15H14BrO3 (M+H): 321.0121; Found: 321.0128.
4-(chloromethyl)-7-hydroxy-2-methoxy-5-phenylcyclohepta-2,4,6-trienone (6g)
5-(chloromethyl)-3-methoxy-6-phenyl-8-oxabicyclo[3.2.1]octa-3,6-dien-2-one 4g (37 mg, 0.13 mmol) was dissolved in CDCl3 (1.34 mL) and trifluoromethanesulfonic acid (47 μL, 0.54 mmol) was added to the reaction. The reaction was stirred for 20 min and was quenched with phosphate buffer (1.6 M, pH = 7, 25 mL), extracted with CH2Cl2 (3 × 25 mL), dried over Na2SO4, filtered and concentrated to yield 6g as a brownish solid (34 mg, 92% yield). MP = 159–167ºC. FTIR (KBr, thin film) 702 (s), 735 (m), 762 (w), 1157 (w), 1261 (s), 1559 (b), 1713 (b), 2927 (b), 3247 (b) cm− 1. 1H NMR (400 MHz, CDCl3) δ 7.39 – 7.14 (m, 7H), 4.31 (s, 2H), 3.99 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.49, 159.48, 158.96, 144.55, 141.92, 134.06, 129.04, 128.72, 128.57, 122.05, 121.37, 56.97, 49.01. HRMS (ESI+): calc’d for C15H14ClO3 (M+H): 277.0626; Found: 277.0627.
4-([1,1′-biphenyl]-4-yl)-2-hydroxy-7-methoxy-5-methylcyclohepta-2,4,6-trienone (7)
4-(4-bromophenyl)-2-hydroxy-7-methoxy-5-methylcyclohepta-2,4,6-trienone (6f) (28 mg, 0.087 mmol) was suspended in toluene (1.75 mL) and trimethyl(phenyl)tin (47.6 uL, 62 mg, 0.25 mmol) and tetrakis(triphenylphosphine)palladium(0) (10.02 mg, 0.0088 mmol) were added. The reaction was stirred vigorously for 4 days at reflux under argon. The solution was cooled to room temperature and the solvent was removed under pressure. The resulting residue was dissolved in CH2Cl2 (2 mL). The solution was then treated with aqueous NaOH (1 M, 25 mL), aqueous KF (saturated, 25 mL) and stirred vigorously for 30 minutes. The solution was acidified with aqueous HCl (2M, 25 mL) to a pH of 3. The solution was extracted with CH2Cl2 (3 × 25 mL), dried over Na2SO4, filtered and concentrated. Chromatography (C18 column [10 g], 35% MeCN/H2O [0.05% TFA] to 100% MeCN [0.05% TFA] gradient over 20 column volumes) followed by concentration of product peaks yielded 7 as a light yellow solid (15 mg, 55% yield). MP = 174–180ºC. FTIR (thin film, KBr) 1113 (s), 1132 (w), 1211 (s), 1554 (s), 1774 (w), 2933 (w), 2986 (w), 3018 (w), 3257 (s) cm−1; 1H NMR (400 MHz, CDCl3) δ 7.70 – 7.21 (m, 11H), 4.04 (s, 3H), 2.35 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.79, 158.77, 158.24, 143.03, 142.66, 140.69, 140.57, 135.29, 129.06, 128.97, 127.77, 127.40, 127.26, 122.54, 122.18, 56.61, 26.89. HRMS (ESI+): calc’d for C21H18O3 mass: 318.1251; Found: 318.1256.
4-([1,1′-biphenyl]-4-yl)-2,7-dihydroxy-5-methylcyclohepta-2,4,6-trienone (8)
4-([1,1′-biphenyl]-4-yl)-2-hydroxy-7-methoxy-5-methylcyclohepta-2,4,6-trienone (7) (16.5 mg, .052 mmol) was dissolved in 33%HBr/AcOH (0.52 mL). The reaction was heated to reflux for 3 h with constant stirring. The solution was cooled to room temperature, quenched with phosphate buffer (pH 7), extracted with CH2Cl2 (3 × 20 mL), dried over Na2SO4, filtered and concentrated to yield 8 as a light yellow solid (16.1 mg, >95% yield). MP = 170–175ºC. FTIR (KBr, thin film) 1525 (s), 2923 (w), 3246 (br), cm−1; 1H NMR (400 MHz, CDCl3) δ 7.79 – 7.20 (m, 11H), 2.32 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 167.35, 158.12, 156.87, 143.80, 142.84, 140.92, 140.70, 139.41, 129.24, 129.13, 127.96, 127.58, 127.44, 126.51, 124.79, 26.86. HRMS (ESI+): calc’d for C20H16O3 mass: 304.1099; Found: 304.1105.
(4-hydroxy-6-methoxy-5-oxo-2-phenylcyclohepta-1,3,6-trien-1-yl)methyl acetate (9)
To a solution of chloromethoxytropolone 6g (33 mg, 0.1191 mmol) in acetonitrile (11.9 ml) was added sodium acetate (195 mg, 2.38 mmol). The reaction was stirred for 12 h at ambient temperature and atmosphere, and phosphate buffer (pH 6) was added. The aqueous layer was extracted with CH2Cl2, and the combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to yield 7 as a light yellow solid (20 mg, 56% yield). MP = 159–161ºC. FTIR (KBr, thin film) 3243(s), 1743(s), 1579(s), 1478(s), 1264(s), 1103(s), 704(m) cm−1. 1H NMR (200 MHz, CDCl3) δ 7.72 – 6.96 (m, 7H), 4.88 (s, 2H), 4.04 (s, 3H), 2.08 (s, 3H). 13C NMR (50 MHz, CDCl3) δ 170.70, 159.53, 158.55, 144.72, 141.82, 131.75, 128.74, 128.57, 128.39, 121.45, 120.33, 67.72, 56.63, 21.16. 13C NMR (50 MHz, CDCl3/CD3CN) δ 169.82, 169.73, 158.64, 157.85, 143.49, 141.20, 131.33, 127.95, 127.86, 127.54, 120.40, 119.76, 66.76, 55.89, 20.08. HRMS (ESI+) m/z calc’d for C17H17O5 (M+H): 301.1071. Found: 301.1071.
4-(azidomethyl)-7-hydroxy-2-methoxy-5-phenylcyclohepta-2,4,6-trien-1-one (10)
To a solution of chloromethoxytropolone 6g (62 mg, 0.22 mmol) in acetonitrile (22 ml), was added sodium azide (285 mg, 4.39 mmol). The reaction was stirred for 12 h at ambient temperature and atmosphere, and phosphate buffer (pH 6) was added. The aqueous layer was extracted with CH2Cl2 and the combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to yield 10 as a brown solid (40mg, 64% yield). MP = 115–119ºC. FTIR (KBr, thin film) 3228(s), 2102(s), 1524(s), 1463(s), 1228(s), 1128(s), 703(s) cm−1. 1H NMR (400 MHz, CDCl3) δ 7.71 – 7.37 (m, 7H), 4.22 (s, 2H), 4.08 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.57, 159.18, 158.56, 143.64, 141.67, 131.40, 128.81, 128.42, 128.30, 121.13, 119.56, 56.54, 56.16. HRMS (ESI+) m/z calc’d for C15H14N3O3 (M+H): 284.1030. Found: 284.1024.
2-hydroxy-7-methoxy-4-phenyl-5-((4-phenyl-1H-1,2,3-triazol-1-yl)methyl)cyclohepta -2,4,6-trien-1-one (11)
To a suspension of azidomethoxytropolone 10 (21 mg, 0.074 mmol) in water (800 mL) and tert-butanol (800 mL) was added phenylacetylene (20 mL, 0.18 mmol), copper sulfate pentahydrate (2 mg, 0.008 mmol), and sodium ascorbate (3 mg, 0.015 mmol). The reaction mixture was heated under microwave irradiation at 110ºC (controlled temperature) for 30 min. The t-BuOH/H2O was evaporated and the reaction was then re-suspended in NaCl(aq) (1 mL), extracted with CH2Cl2 (3 × 1 mL), dried over Na2SO4, filtered, and concentrated to yield 11 as a brown solid (14 mg, 50% yield). MP = 171–173ºC. FTIR (KBr, thin film) 3221(s), 2939(s), 1568(s), 1467(s), 1229(s), 1121(s) cm−1. 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 7.5 Hz, 2H), 7.63 – 7.29 (m, 10H), 7.23 (s, 1H), 5.42 (s, 2H), 3.91 (s, 3H). 13C NMR (100 MHz, CDCl3/CD3OD) δ 170.33, 159.35, 158.48, 147.62, 143.59, 141.20, 130.17, 129.42, 128.56, 128.40, 128.02, 128.00, 127.87, 125.16, 121.44, 120.40, 118.95, 55.62, 54.72. HRMS (ESI+) m/z calc’d for C23H20N3O3 (M+H): 386.1499. Found: 386.1492.
General procedure for furan synthesis
3-methoxy-8-oxabicyclo[3.2.1]octa-3,6-dien-2-ones (4h, 4i) were dissolved in CHCl3 (0.1M) and trifluoromethanesulfonic acid (4 equiv) was added to the reaction. The reaction stirred for 30 min, at which time the reaction was quenched with triethylamine, concentrated, and purified by silica gel chromatography.
dimethyl 2-methylfuran-3,4-dicarboxylate (12h)
dimethyl 3-methoxy-1-methyl-4-oxo-8-oxabicyclo[3.2.1]octa-2,6-diene-6,7-dicarboxylate 4h (20 mg, 0.071 mmol) was dissolved in CHCl3 (0.71 mL) and trifluoromethanesulfonic acid (25 μL, 0.283 mmol) was added to the reaction. The reaction stirred for 30 min and was quenched with triethylamine (50 μL), The reaction mixture was then concentrated and purified by chromatography (silica gel, 18 × 1.8 cm, 50 mL Hexanes, 100 mL 5% EtOAc in Hexanes, 100 mL 10% EtOAc in Hexanes, 200 mL 20% EtOAc in Hexanes) to lead to 12h as a light yellow solid (10.9 mg, 78% yield). 1H NMR matched previously reported data.22 1H NMR (400 MHz, CDCl3) δ 7.73 (s, 1H), 3.85 (s, 3H), 3.82 (s, 3H), 2.50 (s, 3H).
dimethyl furan-3,4-dicarboxylate (12i)
dimethyl 3-methoxy-2-oxo-8-oxabicyclo[3.2.1]octa-3,6-diene-6,7-dicarboxylate 4i (20 mg, 0.074 mmol) was dissolved in CHCl3 (0.746 mL) and trifluoromethanesulfonic acid (0.026 mL, 0.289 mmol) was added to the reaction. The reaction stirred for 30 min and was quenched with triethylamine (50 μL), The reaction mixture was then concentrated and purified by chromatography (silica gel, 18 × 1.8 cm, 50 mL Hexanes, 100 mL 2% EtOAc in Hexanes, 100 mL 5% EtOAc in Hexanes, 100 mL 8% EtOAc in Hexanes) to lead to 12i as a light yellow solid (8.6 mg, 63% yield). 1H NMR matched that previously reported.23 1H NMR (400 MHz, CDCl3) d 7.94 (s, 2H), 3.86 (s, 6H).
Supplementary Material
Scheme 5.
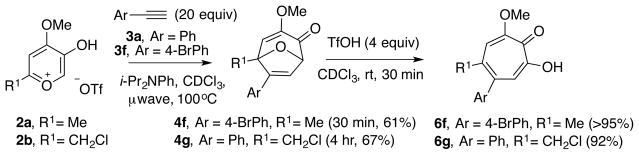
Synthesis of halogen containing bicycles 4f and 4g and ring-expansion using triflic acid
Scheme 6.
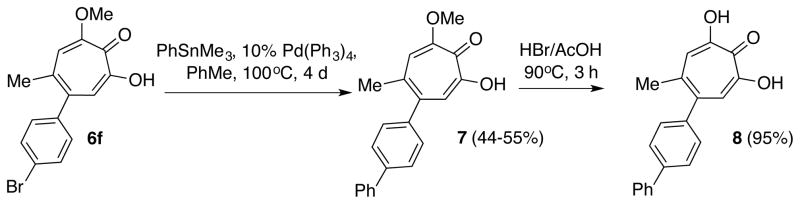
Stille cross-coupling of 6f to afford 7 and demethylation to afford 8
Scheme 7.

Modifications of methoxytropolone 6g
Acknowledgments
The authors are grateful to Brooklyn College and the National Institutes of Health (SC2GM099596) for generous funding. We also thank Connie M. David (Louisiana State University) for Mass Spectroscopy studies.
Footnotes
Supporting Information. 1H and 13C NMR spectra of all new compounds and 1H NMR data of all known compounds with new experimental procedures; HMBC and HSQC spectra of 6f with assignments. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kitamura S, Iida T, Shirahara K, Kase H. J Antiobiot. 1986;39:589–93. doi: 10.7164/antibiotics.39.589. [DOI] [PubMed] [Google Scholar]
- 2.Morita H, Matsumoto K, Takeya K, Itokawa H, Iitaka Y. Chem Pharm Bull. 1993;41:1418. doi: 10.1248/cpb.41.1418. [DOI] [PubMed] [Google Scholar]
- 3.For examples, see: Iwatsuki M, Takada S, Mori M, Ishiyama A, Namatame M, Otoguro K, Nishihara-Tsukashima A, Nonaka K, Masuma R, Otoguro O, Shiomi K, Omura S. J Antibiot. 2011;64:183. doi: 10.1038/ja.2010.124.Semenova EA, Johnson AA, Marchand C, Davis DA, Yarchoan R, Pommier Y. Mol Pharmacol. 2006;69:1454. doi: 10.1124/mol.105.020321.Tavis JE, Cheng X, Hu Y, Totten M, Cao F, Michailidis E, Aurora R, Meyers MJ, Jacobsen EJ, Parniak MA, Sarafianos SG. PLoS Pathog. 2013;9:e1003125. doi: 10.1371/journal.ppat.1003125.
- 4.Allen NE, Alborn WE, Jr, Hobbs JN, Jr, Kirst JHA. Antimicrob Agents Chemother. 1982;22:824. doi: 10.1128/aac.22.5.824.For a mini-review on drugs targeting resistance pathways, see: Wright G. Chem Biol. 2000;7:R127. doi: 10.1016/s1074-5521(00)00126-5.
- 5.Mayer KH. Am J Med. 1986;80:56. doi: 10.1016/0002-9343(86)90480-8. [DOI] [PubMed] [Google Scholar]
- 6.Budihas SR, Gorshkova I, Gaidamakov S, Wamiru A, Bona MK, Parniak MA, Crouch RJ, McMahon JB, Beutler JA, Le Grice SFJ. Nucleic Acids Res. 2005;33:1249. doi: 10.1093/nar/gki268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ilina T, LaBarge K, Sarafianos SG, Ishima R, Parniak MA. Biology. 2012;1:521. doi: 10.3390/biology1030521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Himmel DM, Maegley KA, Pauly TA, Bauman JD, Das K, Dharia C, Clark AD, Jr, Ryan K, Hickey MJ, Love RA, Hughes SH, Bergqvist S, Arnold E. Structure. 2009;17:1625. doi: 10.1016/j.str.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piettre SR, Ganzhorn A, Hoflack J, Islam K, Hornsperger JM. J Am Chem Soc. 1997;119:3201. [Google Scholar]
- 10.Martin SF, Follows BC, Hergenrother PJ, Franklin CL. J Org Chem. 2000;65:4509–14. doi: 10.1021/jo9915731. [DOI] [PubMed] [Google Scholar]
- 11.(a) Chung S, Himmel DM, Jiang JK, Wojtak K, Bauman JD, Rausch JW, Wilson JA, Beutler JA, Thomas CJ, Arnold E, Le Grice SFJ. J Med Chem. 2011;54:4462. doi: 10.1021/jm2000757. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Didierjean J, Isel C, Querré F, Mouscadet JF, Aubertin AM, Valnot JV, Piettre SR, Marquest R. Antimicrob Agents Chemother. 2005;49:4884. doi: 10.1128/AAC.49.12.4884-4894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For some relevant examples, see: Brechy R, Buettner F, Boehm M, Seitz M, Frenzen G, Pilz A, Massa W. J Org Chem. 2001;66:2911. doi: 10.1021/jo991171t.Yamatani T, Masafumi Y, Kahei T. Tetrahedron Lett. 1970;11:1725–8.Davies HML, Clark TJ. Tetrahedron. 1994;50:9883–9892.For a review, see: Banwell MG. Aust J Chem. 1991;44:1–36.
- 13.Takeshita H, Mori A. Synthesis. 1986:578. [Google Scholar]
- 14.(a) Banwell MG, Collis MP. J Chem Soc, Chem Commun. 1991:1343–5. [Google Scholar]; (b) Banwell MG, Crisp GT, Reum JN, Scoble ME, Scoble JA. J Chem Soc, Chem Commun. 1989:616–7. [Google Scholar]; (c) Amon C, Banwell MG, Gravatt GL. J Org Chem. 1987;52:4851–5. [Google Scholar]
- 15.Zinser J, Henkel S, Föhlisch B. Eur J Org Chem. 2004:1344–56. [Google Scholar]
- 16.(a) Baldwin JE, Mayweg AVW, Neuman K, Prichard GJ. Org Lett. 1999;1:1933. doi: 10.1021/ol991067y. [DOI] [PubMed] [Google Scholar]; (b) Adlington RM, Baldwin JE, Mayweg AVW, Pritchard GJ. Org Lett. 2002;4:3009. doi: 10.1021/ol026467r. [DOI] [PubMed] [Google Scholar]; (c) Baldwin JE, Mayweg AVW, Pritchard GJ, Adlington RM. Tetrahedron Lett. 2003;44:4543. [Google Scholar]; (d) Graening T, Bette V, Neudorfl J, Lex J, Schmalz HG. Org Lett. 2005;7:4317. doi: 10.1021/ol051316k. [DOI] [PubMed] [Google Scholar]
- 17.Volkmann RA, Weeks PD, Kuhla DE, Whipple EB, Chmurny GN. J Org Chem. 1977;42:3976.Garst M, El McBride BJ, Douglass J. Tetrahedron Lett. 1983;24:1675.Wender PA, McDonald FE. J Am Chem Soc. 1990;112:4956.Rumbo A, Mouriño A, Castedo L, Mascareñas JL. J Org Chem. 1996;61:6114. doi: 10.1021/jo960854v.Mascareñas JL, Perez I, Rumbo A, Castedo L. Synlett. 1997:81.Rodriguez JR, Rumbo A, Castedo L, Mascareñas JL. J Org Chem. 1999;64:4560. doi: 10.1021/jo982050g.Wender PA, D’Angelo N, Elitzin VI, Ernst M, Jackson-Ugueto EE, Kowalski JA, McKendry S, Rehfeuter M, Sun R, Voigtlaender D. Org Lett. 2007;9:1829. doi: 10.1021/ol0705649.For some recent reviews covering oxidopyrylium cycloadditions, see: Harmata M, editor. Advances in Cycloaddition. Vol. 6 Jai Press; Stamford, CT: 1999. Singh V, Krishna UM, Vikrant, Trivedi GK. Tetrahedron. 2008;64:3405.Pellissier H. Adv Synth Catal. 2011;353:189.McBride BJ, Garst ME. Tetrahedron. 1993;49:2839.Lopez F, Castedo L, Mascareñas JL. Chem Eur J. 2002;8:884. doi: 10.1002/1521-3765(20020215)8:4<884::aid-chem884>3.0.co;2-q.
- 18.Wender PA, Mascareñas JL. Tetrahedron Lett. 1992;33:2115.For related studies in an intramolecular context, see: Wender PA, Mascareñas JL. J Org Chem. 1991;56:6267.
- 19.Meck C, Mohd N, Murelli RP. Org Lett. 2012;14:5988. doi: 10.1021/ol302892g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fakih S, Podinovskaia M, Kong X, Collins HL, Schaible UE, Hider RC. J Med Chem. 2008;51:4539. doi: 10.1021/jm8001247. [DOI] [PubMed] [Google Scholar]
- 21.3- and 7-Methoxytropolones interconvert through rapid tautomerization. The usual designation in the literature is as 3-methoxytropolone. Our assignment as the 7-methoxytropolone in the current manuscript reflects our HMBC/HSQC NMR data of 6f. See the supporting information for detail.
- 22.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 23.Padwa A, Zhi L, Fryxell GE. J Org Chem. 1991;56:1077. [Google Scholar]
- 24.Song ZZ, Ho MS, Wong HNC. J Org Chem. 1994;59:3917. [Google Scholar]
- 25.Mann J, Wilde PD, Finch MW. Tetrahedron. 1987;43:5431. [Google Scholar]
- 26.(a) Limpricht H. Ber Dtsch Chem Ges. 1870;3:90. [Google Scholar]; (b) Paal C. Ber Dtsch Chem Ges. 1884;17:2756–67. [Google Scholar]; (c) Knorr L. Ber Dtsch Chem Ges. 1884;17:2863–70. [Google Scholar]
- 27.For some recent examples, see: Kramer S, Skrydstrup T. Angew Chem, Int Ed. 2012;51:4681–4684. doi: 10.1002/anie.201200307.Fournier J, Arseniyadis S, Cossy J. Angew Chem, Int Ed. 2012;51:7562–66. doi: 10.1002/anie.201202486.Raimondi W, Dauzonne D, Constantieux T, Bonne D, Rodriguez J. Eur J Org Chem. 2012:6119–23.For a recent review, see: Sabounchei SJ, Bagherjeri FA, Panahimehr M. Adv Chem Res. 2012;15:31–55.
- 28.For examples, see: Gotthardt H, Huisgen R, Bayer HO. J Am Chem Soc. 1970;92:4340–4.Koenig H, Graf F, Weberndoerfer V. Liebigs Ann. 1981:669–82.Wong MK, Leung Y, Wong HNC. Tetrahedron. 1997;53:3497–512.Wolkenberg SE, Boger DL. J Org Chem. 2002;67:7361–4. doi: 10.1021/jo020437k.Dolbier WR, Jr, Mitani A, Xu W, Ghuviriga I. Org Lett. 2006;24:5573–75. doi: 10.1021/ol0622662.
- 29.Triflate salt 2c was prepared by mixing pyromeconic acid (500 mg, 4.46 mmol) and methyl triflate (759 μL, 6.69 mmol) in CH2Cl2 (2.25 mL, 1.98 M) and heating under reflux for 3 hours. After 3 hours, solvent was removed under reduced pressure, and recrystallized from CHCl3. Unlike other salts however, 2c decomposes over time (a few hours) as a solid, and thus it must be stored as a suspension in chloroform to prevent decomposition.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.