

Abstract
The mammalian genome encodes two A-type cyclins, which are considered potentially redundant yet essential regulators of the cell cycle. Here, we tested requirements for cyclin A1 and cyclin A2 function in cerebellar development. Compound conditional loss of cyclin A1/A2 in neural progenitors resulted in severe cerebellar hypoplasia, decreased proliferation of cerebellar granule neuron progenitors (CGNP), and Purkinje (PC) neuron dyslamination. Deletion of cyclin A2 alone showed an identical phenotype, demonstrating that cyclin A1 does not compensate for cyclin A2 loss in neural progenitors. Cyclin A2 loss lead to increased apoptosis at early embryonic time points but not at post-natal time points. In contrast, neural progenitors of the VZ/SVZ did not undergo increased apoptosis, indicating that VZ/SVZ-derived and rhombic lip-derived progenitor cells show differential requirements to cyclin A2. Conditional knockout of cyclin A2 or the SHH proliferative target Nmyc in CGNP also resulted in PC neuron dyslamination. Although cyclin E1 has been reported to compensate for cyclin A2 function in fibroblasts and is upregulated in cyclin A2 null cerebella, cyclin E1 expression was unable to compensate for loss-of cyclin A2 function.
Keywords: Cyclin A2, external granule layer, Cyclin A1, Nestin, Cyclin E1, CNS development, DNA repair, cell cycle, brain malformation
Introduction
Normal central nervous system (CNS) development requires precise regulation of neural progenitor proliferation by extrinsic organizing signals, mitogens, and cell-intrinsic programs. Cell proliferation is driven by cyclins and their catalytic partners, the cyclin-dependent kinases (CDKs) (Bloom and Cross, 2007). Cyclin/CDK complexes, in concert with other proteins, control cell cycle through regulation of multiple cell cycle phases. In the classical model, particular cyclin/CDK complexes regulate transitions through G1, S, G2, and M cell cycle phases. This notion has been challenged by experiments demonstrating significant redundancy in cyclin-CDK binding and cell cycle progression (Aleem et al., 2005). Furthermore, cyclins have been shown to regulate other processes not traditionally associated with classical function, including CNS synapse development (Odajima et al., 2011) and DNA repair (Jirawatnotai et al., 2011).
The mammalian genome encodes two A-type cyclins, testis-specific cyclin A1 and ubiquitously expressed cyclin A2 (Sweeney et al., 1996; Yang et al., 1997a). Whereas male meiosis is dependent on cyclin A1 (Liu et al., 1998), cyclin A2 was originally shown to be required for the onset of DNA replication (Girard et al., 1991). Analyses of conventional cyclin A2 null mice revealed that these animals fail to develop after 5.5 days postcoitum, underscoring a critical function for this cyclin in cell proliferation (Murphy, 1999). In addition, conditional ablation of the A-type cyclins revealed that Cyclin A2 function is essential for cell-cycle progression of hematopoietic and embryonic stem cells, yet is redundant with cyclin E1 in mouse embryonic fibroblasts (Kalaszczynska et al., 2009). While cyclin A2 expression can be induced by Sonic hedgehog (SHH) signaling in cerebellar granule neuron precursors (CGNP) (Zhao et al., 2002), cyclin A2 function has not been investigated in cerebellar development.
The cerebellar external granule layer (EGL) is populated by cerebellar granule neuron precursors (CGNP). SHH signaling is required for both cerebellar morphogenesis and CGNP proliferation (Dahmane and Ruiz i Altaba, 1999; Wechsler-Reya and Scott, 1999). CGNP cells emerge from the rhombic lip and then migrate to the EGL, while Purkinje (PC) neurons originate in the hindbrain ventricular/subventricular zone (Hatten and Heintz, 1995; ten Donkelaar et al., 2003) and develop to become the source of SHH proteins (Wallace, 1999). We tested the hypothesis that proper cerebellar development generally requires function of A-type cyclins through a conditional gene targeting approach. Deleting cyclins A1 and A2 in neural progenitor cells results in dysmorphic cerebella characterized by reduced growth of the CGNP population, abnormal foliation, and PC dyslamination. Similar findings were obtained in nestin-cre, cyclin A2fl/fl animals showing that cyclin A1 is unable to compensate for loss of cyclin A2. Increased programmed cell death was noted during early embryonic development of cyclin A2-null neural progenitors. PC dyslamination and abnormal proliferation was also noted in Math1-cre, cyclin A2fl/fl mice, suggesting that these defects are intrinsic to the CGNP population.
Materials and Methods
Transgenic mice and animal husbandry
The generation of cyclin A1 and conditional cyclin A2fl/fl knockout mice is described in Kalaszczynska et al., 2009 and references therein (Kalaszczynska et al., 2009). These animals were then bred to cyclin A1 null mice (Liu et al., 1998), generating cyclin A1-/-A2fl/fl animals. A1-/-A2fl/fl and A2fl/fl mice were bred to nestin-cre mice (Tronche et al., 1999). Nestin-cre mice show recombination in the CNS, including all cells of the cerebellum by E13.5 (Huang et al., 2010) and Purkinje cells of adults (Jennemann et al., 2005). Verification of A2 loss was performed by western blot (Supplemental Fig. 2), in situ hybridization (Sup. Fig. 1), and immunofluorescence (Fig. 4). Math1-cre (Schuller et al., 2008) was crossed to A2fl/fl mice and Nmycfl/fl (Knoepfler et al., 2006) to generate Math1-cre, A2fl/fl mutants and Math1-cre, Nmycfl/fl mutants. Verification of Nmyc loss was performed by in situ hybridization. Developmental analyses in wild-type mice were performed in the CD-1 strain (Charles River). In BrdU pulsing experiments, animals were injected with 1 mg BrdU intraperitoneally 2 hours prior to sacrifice. All animal experimentation was performed in full compliance with the institutional review board requirements of Dana Farber Cancer Institute, University of California, San Francisco, and The Ohio State University.
Figure 4. Cytoarchitectural characterization of cerebellar cortex in nestin-cre, A1-/-A2fl/fl mice.
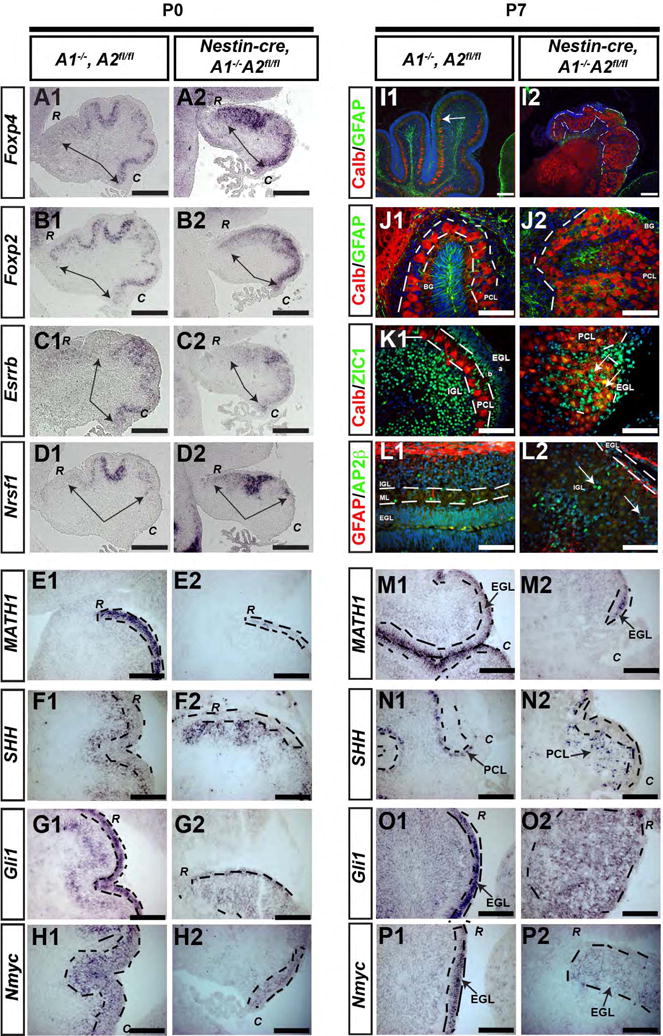
Genotypes and postnatal ages are denoted on the top, molecular markers are denoted on the left. (A-H) P0 mice were sacrificed from control and nestin-cre, A1-/- A2fl/fl mice and evaluated for expression of FoxP4 (A1,A2), FoxP2 (B1, B2), ESRRB (C1,C2), NRSF-1 (D1, D2), SHH (E1, E2), Gli (F1, F2), Math1 (G1, G2), and Nmyc (H1, H2) by in situ hybridization. In panels A-D, the black arrows delineate the expression domain of these patterning genes. In panels E1-H2 and M1-P2, EGL is outlined by the black dashed line. “C” denotes caudal cerebellum, “R” denotes rostral cerebellum. P7 mice were sacrificed from control (I1-P1) and conditional nestin-cre, A1-/-A2fl/fl mice (I2-P2). In panels I-L, blue is DAPI, and the red and green immunofluorescent signals are color coded within each panel. In I1, the linear Purkinje cell layer is denoted by the white arrows, whereas in I2, the patch-like Purkinje cell layer is outlined by white dashed lines. In J1-K2, the white dashed line delineates the PC cell layer. In K2 and L2, interneurons admixed within the patchlike PC layer are illustrated with white arrows. In M2 and O2, “C” denotes caudal cerebellum; in N2 and P2, “R” denotes rostral cerebellum. (In I-K, CALB = calbindin). Scale bars are as follows: A-D = 100 μm, E-H = 50 μm, I1-I2 = 100 μm, M-P = 50 μm. These images represent sections in the sagittal plane and are oriented such that dorsal is superior and caudal is on the bottom right. The abbreviations “R” and “C” in panels A-H and M-P represent high magnification images of rostral or caudal folia, respectively.
Histology, immunofluorescent staining, in situ hybridization, and photomicroscopy
To evaluate brain morphology, experimental and control mice were sacrificed at P0, P7, and adult ages, perfused, and heads or brains were dissected and incubated in 4% paraformaldehyde. Evaluation of embryonic time points was performed by sacrificing the pregnant mouse and drop-fixing whole embryos into 4% paraformaldehyde. Post-fixation processing, immunoblotting, and ISH were performed as described previously by our group (Fancy et al., 2011). Briefly, animals were perfused with 4% PFA, and brains were then drop fixed in 4%PFA overnight. Tissue was incubated in 30% sucrose-1XPBS at 4° C for 24-36 hours, embedded in OCT, and cryosectioned at 12-14 μm. Cre-negative littermates represent negative controls.
The following primary antibodies were used for immunohistochemistry and western blotting: GFAP (Abcam, ab7777-500), Calbindin (Sigma, C8666), AP2β (Santa Cruz Biotechnology, SC-8976), Zic-1 (kind gift from R. Sega, Dana Farber Cancer Center), cleaved Caspase 3 (Cell Signaling, 9664 and 9661), Ki67 (Vector Labs, VP-RM04), phosphohistone H3 (Cell Signaling, 2650S), BrdU (GeneTex, gtx26326), Tuj1 (Covance, MMS-435P-250), Pax6 (Santa Cruz Biotechnology, SC-7750), cyclin A (Santa Cruz, SC-53230 and SC-596), cyclin E1 (for IHC, antibody was provided by Dr. B. Clurman; for western blotting we used Santa Cruz, SC-481), cdk2 (Santa Cruz, sc-163 AC M2), cre (Millipore, MAB3120), GFP (Aves Lab, 1020), cdk1 (Abcam ab71939), synapsin-I (Millipore, AB1543). Appropriate secondary antibodies were purchased from Invitrogen/Molecular Probes. For brightfield and epifluorescence, a Zeiss Axioscope with an Axiocam HRC camera and a Nikon 80i microscope with Hamatasu Orca-R2 camera was used. Confocal microscopy instrumentation included Axioscope 2 mot plos with high-resolution color and monochrome camera and Axiovision 4.5 Image analysis software
In situ hybridization probe constructs for Gli1, Math1 and Shh were generously provided by A.L. Joyner (Memorial Sloan Cancer Center, New York, New York, USA), J Johnson (UT Southwestern, Dallas, USA) and A.P. McMahon (UCSF, USA), respectively. Probes for cyclinD1 (Sicinski et al., 1995) and Nmyc (Charron et al., 2002) and patterning genes Foxp4, Foxp2, Esrrb, Nrsf1 (Schuller et al., 2006) have been described and the hybridization protocols are specified therein. The cyclin A2 in situ hybridization probe was generated from cyclin A2 cDNA construct purchased from Open Biosystems (Clone BC052730 in pYX-Asc vector). This represents a 2770 bp fragment of cyclin A2 cDNA. To generate the antisense probe, the plasmid DNA was linearized with NheI (NEB) and transcribed with T3 polymease. Digoxigenin-labeled antisense RNA probes were made using plasmid DNA as the template (Roche). In situ hybridization with frozen sections was performed as described previously (Zhao et al., 2002). Western blotting was performed as described previously (Odajima et al., 2011). To evaluate protein levels across wells, band intensity was normalized to GAPDH band intensity determined by the densitometry function of ImageJ.
Unbiased stereology
Unbiased stereology was performed to determine estimates of volume and total number of cleaved-caspase 3 positive cells in the embryonic cerebellum (Stereoinvestigator™, MBF Biosciences, Fig. 6). Brains were cryosectioned at 50 μm (section cut thickness) and every 5th section (section evaluation interval) was immunostained with cleaved-caspase 3; Dako Envision™ kits developed the DAB reactions, nuclei were counterstained with hematoxylin (Electron Microscopy Solutions), and cover slips were mounted with Permount™ (Fisher). Note that peroxidase-DAB reactions using thick sections require increased incubation times in the final peroxidase reaction, and therefore it is common to see increased background when using a thick section. Cavalieiri estimation generated volume estimates with the following parameters: grid size = 30 m, shape factor = 4. Cleaved caspase-3 total cell number estimates were obtained using optical fractionator probes with the following parameters: counting was performed under oil immersion with 100X objective, dissector height = 20 μm, dissector volume = 50,000 μm3, counting frame height and width = 50 μm, sampling grid was 153.9 μm × 162.5 μm. We use the term ventricular zone/sub-ventricular zone (VZ/SVZ) to designate the 4th ventricular germinal neuroepithelium of the cerebellum, which for quantification purposes included the dense band of neural progenitor cells lining the 4th ventrical.
Figure 6. Distinct cerebellar neural progenitor compartments show differential requirements to cyclin A2.
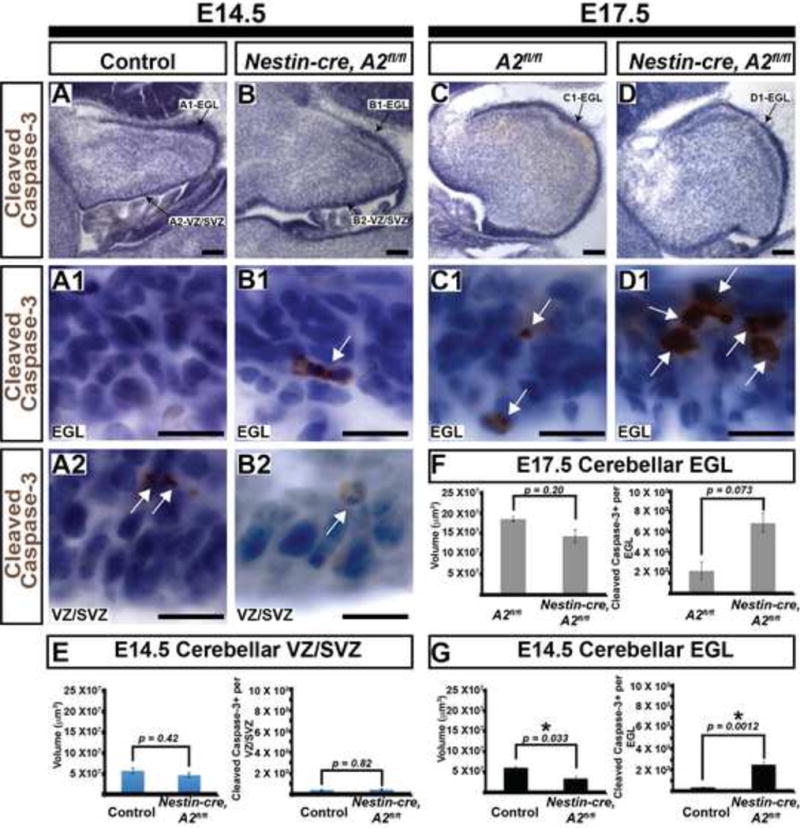
(A-D) Embryonic ages and genotypes are on the top and the molecular marker is on the left. Panels A, B, C, and D illustrate orientation of the pertinent structures quantified in E, F, and G. At E14.5, the EGL shows marked elevation in cleaved caspase-3 and a modest reduction in volume. No statistical difference between total volume and cleaved caspase-3+ cells is noted in the 4th ventricular VZ/SVZ at E14.5 or the EGL at E17.5. (Scale bars in A, B, C, D = 100 μm. Scale bars in A1, A2, B1, B2, C1, D1 = 12.5 μm, EGL = external granule layer, VZ/SVZ = ventricular zone/subventricular zone, * in G denotes statistical significance, scale bars in E, F, G= SEM, p values determined by 2-tailed homoscedastic T-test). White arrows in panels A1, A2, B1, B2, C1 and C2 illustrate cells that are positive.
Cerebellar GNP cultures
CGNP in vitro cultures were performed as described previously (Heine et al., 2011). Briefly, all cells were cultured in 37° C humidified incubators set at 5% CO2 and room air. Cerebellar Granule Progenitor Cell Cultures (CGNP's) were isolated from wild-type (WT) A1+/+A2fl/fl mice at postnatal day 5 (P5) and cultured overnight in the presence or absence of SHH (3 g/mL) in 24 well plates (Falcon) with cover slips treated with poly-ornithine (Kenney and Rowitch, 2000; Knoepfler et al., 2002). For infection, a replication incompetent adenovirus (Vector Labs, Catalogue # 1700) expressing cre and gfp on separate promoters was used; control was gfp-only adenovirus (Vector Labs, Catalogue # 1060). Protocols were adapted from others described previously (Kenney et al., 2003; Mills et al., 2003; Troussard et al., 2003). On day 1 post plating, half of the media was removed, and cells were incubated with 1 μl of adenovirus (titer = 1010 PFU/ml) per well of the 24 well plate for 1 hour at 37° C. Next, 250 l of media with SHH (or vehicle) was added directly to the cells, and the cells were re-incubated for three hours. After three hours, the cells were re-fed with fresh media (containing SHH or vehicle) and returned to the incubator. DMSO was added on day 2 post-plating, and BrdU was administered directly to the culture medium at 25 μg/ml for 2 hours on day 3 post-plating (day 2 post-infection). GFP+ and pH3 stains were used to quantify the mean proportion of GFP+pH3+ cells.
Statistical Analysis
Post-natal quantifications of cell number, cell density, and tissue area were obtained using NIH ImageJ. Statistical analysis was carried out using Microsoft Excel 2011 or R v2.12. The EGL was defined morphologically as a sub-pial dense band of neural cells separated from the deeper cerebellar cortex by a hypocellular molecular layer. Effort was made to quantify similar areas of EGL based on unaffected anatomical landmarks, despite the variations in cerebellar size amongst the experimental conditions. The EGL proliferation quantification was performed as described previously (Heine et al., 2011; Heine and Rowitch, 2009). For post-natal EGL area, at least three serial cross sectional areas at the center of the lobules from three mice per condition were obtained using ImageJ. Due to the difference in sizes between the controls and mutants, we used the olfactory bulb and lateral ventricles to confirm the similarity in anatomical area sampled. Similarly, proliferation was recorded by counting the number of proliferative cells in the EGL from at least three serial cross sctions from three mice per condition. Such “counts per section” are standard metrics used to evaluate post-natal cerebellar development (Chen et al., 2005; Falluel-Morel et al., 2008; Klein et al., 2002). The results were qualitatively verified by analysis of over three litters; the data illustrated was pooled from mice of two litters. Statistical hypothesis testing was performed using Student's T-test and ANOVA/Tukey HSD tests as indicated. The threshold for significance was set to α = 0.05. For these analyses, we defined the EGL as a cytoarchitectural structure characterized by a dense collection of cells between the cerebellar pial membrane and the hypocellular molecular layer.
Results
Proliferating neural progenitor cells and post-mitotic neurons express cyclin A2
To investigate the role of cyclin A2 during CNS development, we performed western blot analysis of cyclins and CDKs on total cerebral or cerebellar protein lysates at different postnatal ages. The ages represent proliferative ages (P0 to P7) and non-proliferative ages (8 months post-natal age) during cerebellar development. The cerebellar protein extracts were compared to cerebral cortex, which has significantly less proliferation at the early post-natal ages. Cyclin A2 protein can be observed in two isoforms, a full-length (“A60”) form and an N-terminal truncated form (“A38”) lacking the N-terminal “destruction” D-box (Welm et al., 2002) (Fig. 1A). We noted primarily the A60 isoform in cerebellum from P0 to P7, after which the smaller A38 isoform predominates (Fig. 1B). This is in direct contrast to cerebral cortex, where the A38 isoform was the predominant band throughout postnatal development. A38 protein levels showed positive correlation with synapsin levels (a marker of neuronal maturation in both cerebellar and cerebral cortex (Valtorta et al., 2011)). In contrast, E11.5 whole brain extracts (a time characterized by robust forebrain neural progenitor proliferation) showed high levels of A60. High expression of CDK1, CDK2, and CDK4 during early postnatal cerebellar development is consistent with high cerebellar proliferation at this stage. In contrast, cerebellar maturity (post-natal age over 1 month) was correlated with high protein levels of cdk5 and p27. We conclude that (1) A60 predominates during the time windows of cerebellar growth and (2) A38 predominates during times of cerebellar maturation.
Figure 1. Cyclin A2 expression during CNS development.
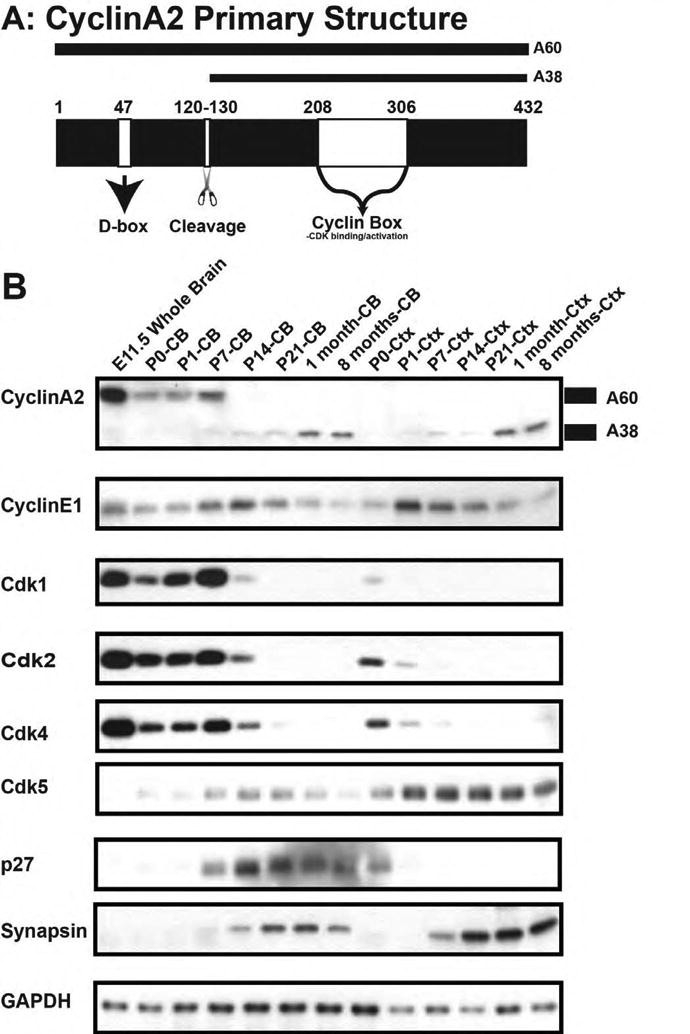
(A) Schematic of cyclin A2 protein structure illustrated D-box, cleavage point, and CDK interacting domain. (B) Western blotting of forebrain and cerebellum during post-natal development. Cyclin A2 becomes processed into two forms (denoted on right of blot as A60 and A38). Top shows labels of post-natal dates, and on the left side of panel is the antibody used in the blot. Note that markers of proliferation cyclin A2, cdk1, cdk2, and cdk4 are robustly expressed in post-natal cerebellum whereas synapsin (a marker of differentiation) is seen at later post-natal ages (CB = cerebellum, CTX = cerebral cortex).
The above findings suggested to us that A60 would be expressed in cycling progenitor cells whereas A38 would be found in differentiated cells. We tested this hypothesis by performing confocal microscopy of cyclin A2 expression in the developing murine CNS in CD1 mice at E11, P7, and adult (Fig. 2). We found at Ell.5 cyclin A2 protein expression in ki67-positive and pH3-positive cells of the VZ/SVZ, with some cyclin A2-positive cell processes oriented apically towards the pial surface. Of note, Tuj1-positive cells were cyclin A2-negative at E11 (Fig. 2C). We found no significant GFAP expressed in the VZ/SVZ before P0, which is concordant with other studies (Tramontin et al., 2003). The P7 cerebellum showed cyclin A2-positive cerebellar granule neuron progenitors in the proliferative EGL-a that were negative for Tuj1 and GFAP (Fig. 2G, H). Similar to other reports (Ikeda et al., 2011), we noted that mitotic cells in the EGL-a expressed cyclin A2 (Fig. 2D-F). Cyclin A2 did not colocalize with astrocytes at any time point investigated. Adult cerebral cortical neurons showed cyclin A2 localized to their nuclei and soma. Similarly, the adult cerebellum showed cyclin A2 localization in the neuronal somata and nuclei of PC cells and was expressed in the neuropil of the internal granule layer (IGL) (Fig.2I-L). We conclude that during neurogenesis, a period characterized by A60 predominance, proliferating progenitor cells express cyclin A2. In contrast, adult neurons, a period characterized by A38 predominance, express nuclear and cytoplasmic cyclin A2.
Figure 2. Cyclin A2 protein localization during DNS development.
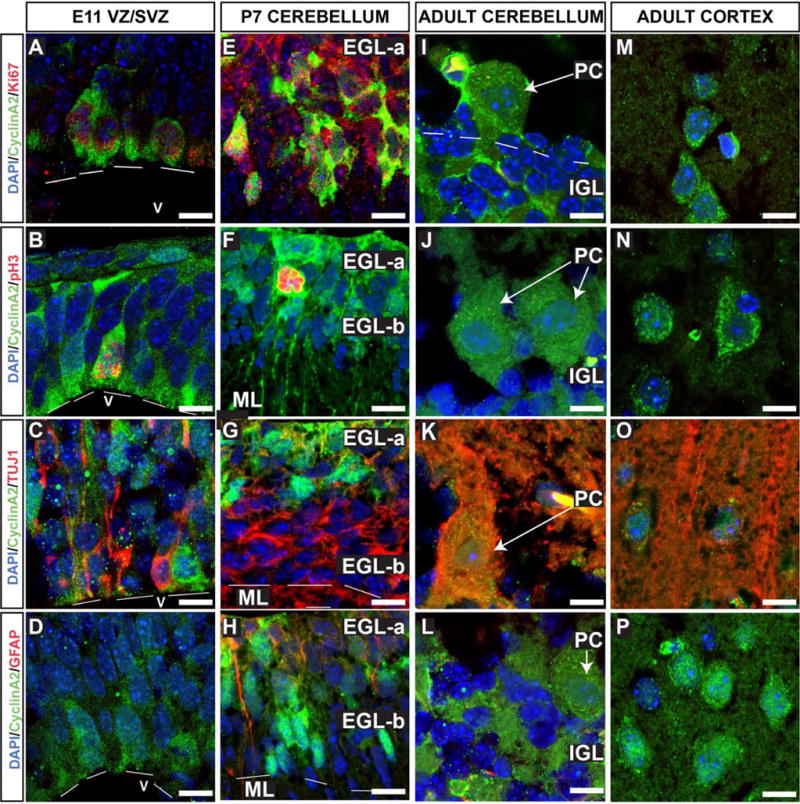
Cryosections of fixed CD1 mouse embryos were stained for cyclin A2 in cells undergoing cell cycle (Ki67), mitotic cells (pH3), neurons (TUJ1), and astrocytes (GFAP). Sections were counterstained with DAPI to visualize nuclei (Blue). Animal ages and anatomical locations are depicted in the top row, and molecular markers are color coded on the left hand side. Cyclin A2 expression is noted in proliferating, undifferentiated neural stem cells at E11.5 (A-D). In the P7 cerebellum (E-H), cyclin A2 localizes to proliferating and mitotic cerebellar granule neuron progenitor cells and is excluded from differentiated neurons and astrocytes (E-H). In adult CNS, cyclin A2 is expressed in neurons. In panel B2, cerebellar folia numbers are placed in quotations (eg., “X”). (Scale bars = 10 μm, EGL = external granule layer, PC = Purkinje neuron, IGL = internal granule layer, ML = molecular layer, V = ventricle).
Cerebellar cortical lamination and foliation require cyclin A2
We tested whether cerebellar development requires cyclin A function through a conditional gene targeting approach. Our analyses focused on post-natal cerebellar development when cerebellar growth is most robust. Cyclin A1-/-A2fl/fl (referred to hereafter as A1-/-A2fl/fl) or Cyclin A2fl/fl (referred to hereafter as A2fl/fl) compound mutant mice were bred to nestin-cre transgenic animals to induce recombination generally in neural progenitors. Although CGNP's and PC's are derived from distinct stem cell compartments, the progenitors of both pass through a nestin-positive stage, and therefore both precursor pools are expected to undergo recombination. Such nestin-cre, A1-/-A2fl/fl mice survived the perinatal period but showed delayed growth and gross motor deficits when compared to cre-negative littermates (Supplemental Fig. 1A-D). Postnatally, nestin-cre, A1-/-A2fl/fl and nestin-cre, A2fl/fl mice died by 1 month of age. We conclude that loss of cyclin A2 results in post-natal lethality.
We verified the loss of cyclin A2 in mice by in situ hybridization of nestin-cre, A1-/-A2fl/fl animals (Supplemental Fig. 1E-F). Control cerebella show high expression of cyclin A2 in the EGL; this expression is lost in the A2 null animals. Evaluation of the cyclin D1 ISH demonstrated robust expression of cyclin D1 in the EGL of control and nestin-cre, A1-/-A2fl/fl mice, but the size of the EGL was diminished relative to the control (Supplemental Fig. 1G-H). To investigate the effect of cyclin A2 loss in cerebellar development, we evaluated cerebellar morphology in nestin-cre, A1-/-A2fl/fl mice at P0 and P7. At P0, before normal foliation is apparent in the control, the EGL is significantly diminished in the nestin-cre, A1-/-A2fl/fl mice (Fig. 3 C1″-C2″). At P7, control cerebella show well-formed folia, whereas the nestin-cre, A1-/-A2fl/fl mice only folia IX and X are clearly apparent (Fig. 3 D1-D2). Of note, in the caudal cerebellum, some preservation of the EGL was noted, whereas the rostral EGL was more affected in the nestin-cre, A1-/-A2fl/fl mice (Fig. 3 B1′-B2″). In addition, the nestin-cre, A1-/-A2fl/fl mice showed marked dyslamination of the PC layer and no apparent molecular layer. We conclude that cerebellar cortical lamination and foliation require A-type cyclins.
Figure 3. Abnormal cerebellar morphogenesis in nestin-cre, A1-/-A2fl/fl animals.
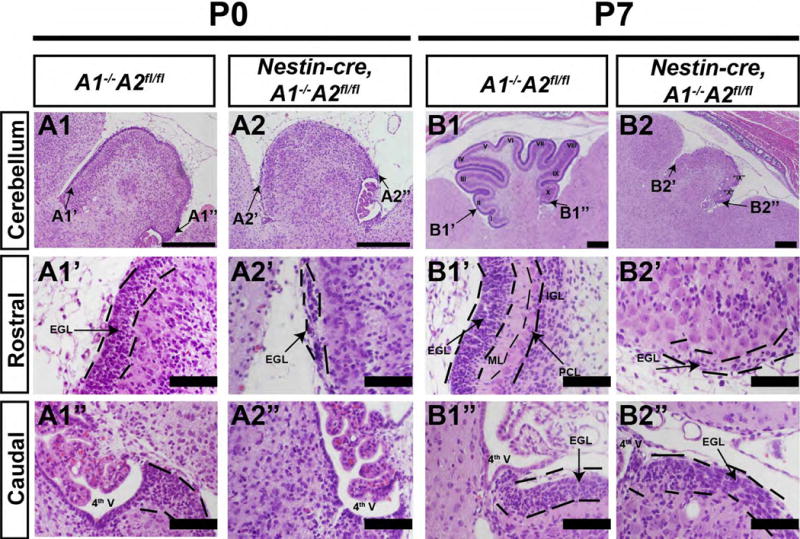
Sections represent sagittal sections stained with H&E, and the orientation is indicated to the left of the panels. Genotypes are denoted on the top. The most pronounced difference was noted in the P7 cerebellum, which was hypoplastic, lacked foliation, and showed a small EGL layer at the most caudal folia that dissipated on the rostral cerebellum. (Scale bars are as follows: A1, A2, B1, B2 scale bar = 100 μm. A1′, A1″, A2′, A2″, B1′, B1″, B2′, B2″ = 500 mm. 4th V = fourth ventrical, EGL = external granule layer, IGL = internal granule layer, ML = molecular layer, PCL = Purkinje cell layer)
Extensive crosstalk occurs between the diverse cellular constituents of the cerebellum during development. For example, PC's synthesize SHH for CGNP proliferation, and therefore intrinsic defects in PC patterning may cause the cytoarchitectural defects noted in the cyclin A2 mutants. We therefore tested for abnormal patterning of PC's in cyclin A-deficient mice. To investigate this, we performed in situ hybridization for the PC patterning genes Foxp4, Foxp2, Esrrb and Nrsf1 at P0 (Fig. 4 A-D) and P7 (Supplementary Fig. 2 C-F) (Schuller et al., 2006). Despite the their dysmorphic cerebella, nestin-cre, A1-/-A2fl/fl mice retained similar rostral-caudal expression domains for these genes at P0 with Foxp2 and Nrsf1 extending slightly more rostrally in the mutant (arrows in Fig. 4 A-D outline rostral-caudal expression domains). At P7, PC's showed developmentally appropriate expression of Foxp4, Foxp2, Esrrb and Nrsf1 in both control and Nestin-cre, A2fl/fl mice (Supplementary Fig. 2 C-F). We conclude that the PC patterning genes Foxp4, Foxp2, Esrrb and Nrsf1 show appropriate rostro-caudal expression domains in nestin-cre, A1-/-A2fl/fl mice at P0 and their developmentally appropriate expression at P7 is not disrupted despite PC dyslamination. These data suggest that defects in PC patterning do not mediate the dysmorphic cerebella in nestin-cre, A1-/-A2fl/fl mice.
To further investigate PC-CGNP crosstalk, we evaluated markers of EGL, PC, and interneurons using immunofluorescence and in situ hybridization. SHH, the major mitogen for CGNP's of the EGL, is produced by PC neurons (Lewis et al., 2004; Wechsler-Reya and Scott, 1999). SHH activates Smoothened signaling, leading to upregulation of the proliferative target genes Gli1 and Nmyc in CGNP cells (Kenney et al., 2003). As shown (Fig. 4 F,N), PC's in nestin-cre, A1-/-A2fl/fl mice continued to express SHH despite being dyslaminated. While drastically reduced in number, the remaining CGNPs in the conditional cyclin A2 knockout mice were located mainly in the caudal cerebellum of P0 and P7 animals, and were detected by their expression of Math1 (Fig. 4 E, M). Moreover, such CGNPs expressed of Gli1 and Nmyc, albeit at lower levels (Fig. 4 G,H,O,P, and Supplementary Fig. 3). In addition, CGNPs from cyclin A2fl/fl mice infected with cre-expressing adenovirus were able to show a proliferative response when treated with SHH in vitro (Supplemental Fig. 5). We conclude that the SHH signaling axis does not require cyclin A2 in CGNPs.
To characterize other cellular constituents of cerebellar cortex in the nestin-cre, A1-/-A2fl/fl mice, we performed immunofluorescent analysis of PC neurons (Calbindin-positive (Pogoriler et al., 2006)), cerebellar granule neurons (Zic1 (Nakata et al., 1998), interneurons (AP2β,(Schuller et al., 2006) and astrocytes (GFAP) at P7 in mutants and controls (Fig. 4 I-L). This revealed striking abnormalities in cell layering. We observed patches of densely packed, and dyslaminated PC cells (Fig. 4 I2, J2, K2, dashed white lines), oriented haphazardly throughout cerebellar cortex and lacking normal, complex “pia oriented” dendritic arborization (Fig. 4 J2). While packed at higher density, total PC numbers were not depleted (p > 0.3). While control cerebella show Bergmann glia with processes extending from the PC neuron layer to the pia (Fig. 4 J1), such GFAP+ fibers in nestin-cre, A1-/-A2fl/fl mice were diminished (Fig. 4 J2). In control mice, Zic1+ cells formed a band in the inner EGL (“EGLb”) and in the IGL, whereas no discrete EGLb or IGL was present in nestin-cre, A1-/-A2fl/fl mice (Fig. 4 K2). Cytoarchitectural disorganization of interneurons was illustrated by the pattern of AP2β immunofluorescence Fig. 4 L2). In summary, the nestin-cre, A1-/-A2f/fl mice show a striking loss of EGL cells, disorganized lamination of interneurons and PC cells, and poor arborization of PC dendrites.
Cyclin A2 loss in embryonic neural progenitor cells results in postnatal EGL proliferation defects
Defects in Pax6+ CGNP cell proliferation and/or apoptosis may cause the dysmorphic cerebella in the mutant mice (Engelkamp et al., 1999; Yamasaki et al., 2001). We therefore evaluated proliferation and apoptosis in CGNPs of nestin-cre, A1-/-A2fl/fl mice. Mice were injected with BrdU 2 hours prior to sacrifice, and evaluated by immunofluorescent staining using anti-BrdU and anti-pH3 antibodies (Fig. 5). Although greatly reduced in numbers, some Pax6-positive CGNPs in nestin-cre, A1-/-A2fl/fl cells showed immunoreactivity to BrdU or pH3 (Fig. 5 A-B, EGL is white dashed line), suggesting they could progress at least to S/M-phase despite cyclin A2 loss (Van Hooser et al., 1998). Such proliferating Pax6+ cells also showed aberrant location outside the EGL (white arrow, Fig. 5 A2). The proportion of cleaved-caspase 3+ cells at P7 (Fig. 5 C) did not demonstrate a significant increase in the proportion of apoptotic cells in the nestin-cre, A1-/-A2fl/fl mice relative to the control in the EGL (p > 0.3). These data suggest A-type cyclins are necessary for normal proliferation of CGNP cells, and suggests that the cerebellar dysmorphia is caused by abnormal proliferation of CGNPs.
Figure 5. External granule cell layer proliferation in nestin-cre, A1-/-A2fl/fl Mice.
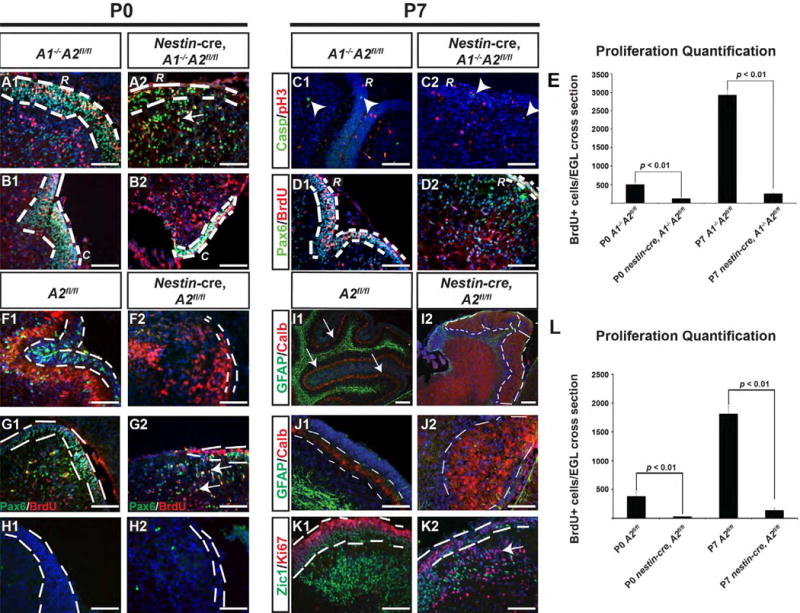
Genotypes and postnatal ages are denoted on the top, molecular markers are color coded and denoted on the left. Control and nestin-cre, A1-/-A2fl/fl mice were sacrificed at P0 and P7 and analyzed by immunofluorescent staining for Phosphohistone H3 (pH3), cleaved-caspase-3, Pax6, and BrdU. In all sections, nuclei are counterstained with DAPI in blue. “R” denotes rostral cerebella, and “C” denotes caudal cerebellum. The EGL is highlighted by white dashed lines in all panels except J1-J2, where the white dashed lines denote the PC neuron layer. In Panel A2, white arrow points to an IGL cell positive for Pax6 and BrdU. Arrowheads in C1 and C2 point to cleaved caspase-3+ cells. In panel E and L, the quantification of BrdU+ cells per cerebellar cross section at P0 and P7 is plotted for the nestin-cre A1-/-A2fl/fl and nestin-cre, A2fl/fl mice, respectively (Paired T-test) Panels F1-K2 illustrate data from nestin-cre, A2fl/fl mice. As expected, cyclin A2 expression, present in the EGL and IGL of controls, is lost in nestin-cre, A2fl/fl mice (F1-F2). No statistically significant difference in levels of apoptosis was noted by cleaved-caspase 3 immunofluorescence at P0 (H1-H2). The cytoarchitecture of the nestin-cre, A2fl/fl was characterized by disorganized PC neurons with poorly formed dendritic arborizations (I1-I2, J1-J2, white dashed line) and abnormal displacement of interneurons into the superficial cortex with displacement of proliferative cells (Pax6+Ki67+) into the IGL (K1-K2). Scale bars are as follows: I1-I2 = 100 μm, all remaining panels scale bar is 50 μm. These images represent sections in the sagittal plane and are oriented such that dorsal is superior and caudal is on the right. The abbreviations “R” and “C” in panels A-H and M-P represent high magnification images of rostral or caudal folia, respectively. (A2 = cyclin A2, casp = cleaved caspase-3, Calb = calbindin, error bars in E and L are SEM. Arrowheads in panel C1 and C2 point to cleaved caspase-3 positive cells).
Loss of one D-type cyclin will result in compensation by other D-type cyclins (Ciemerych et al., 2002). We therefore tested if cyclin A1 function was capable of compensating for cyclin A2 loss. Cyclin A1+/+ A2fl/fl animals (hereafter referred to as A2 fl/f1) were crossed with nestin-cre animals and evaluated by immunofluorescent staining for the cerebellar markers described above. As shown in Fig. 5 F-K, the cerebella of nestin-cre, A2fl/fl animals showed a phenotype that was indistinguishable to nestin-cre, A1-/-A2fl/fl (compare Fig. 5 F-K to Fig. 4). Thus, cyclin A1 cannot compensate for essential cyclin A2 functions during CNS development.
Total CGNP proliferation in the EGL was reduced in the nestin-cre, A2fl/fl mice similar to the nestin- cre, A1-/-A2fl/fl (Fig. 5 L). However, neither in the nestin-cre, A1-/- A2 fl/fl nor the nestin-cre, A2fl/fl mice was a complete block in proliferation noted (see BrdU and pH3 immunohistochemistry in Fig. 5 B2, D2, G2). Hence, cyclin A2 null cells are capable of progressing through S-phase and M-Phase, perhaps through redundancy of other cyclins. By western blot, the P1 cerebellum shows undetectable levels of cyclin A2 in the nestin-cre, A2fl/fl mice (A60 isoform is illustrated), whereas robust levels of A60 are seen in the control cerebellum (Supplemental Fig. 2A). The loss of cyclin A2 correlated with an elevation of cyclin E1 protein levels. Microscopic examination of the EGL demonstrated that CGNP s of control and nestin-cre, A2fl/fl mice show cyclin E1 in pH3+ cells (Supplemental Fig. 2B). These data indicate that cyclin A2 null cells enter S-phase and M-phase, perhaps through compensation by elevation in cyclin E1 protein.
EGL area is proportional to CGNP proliferation (Heine and Rowitch, 2009) and was dramatically reduced in nestin-cre, A1-/-A2fl/fl mice. To test grossly for postnatal expansion of the EGL, we measured total EGL cross-sectional area at P0 and P7 in control and nestin-cre, A2fl/fl mice. The EGL cross-sectional area was smaller in the mutants at P0 (nestin-cre, A1-/-A2fl/fl mice = 54208. 8 μm2 (SD = 6759); control = 302528.5 μm2 (SD = 17427), p = 0.01 by paired T-test), and at P7 (nestin-cre, A1-/-A2fl/fl mice = 266289 μm2 (SD = 27113); control = 3794192 μm2 (SD = 437382), p < 0.05 by paired T-test). This represents a 12.5-fold increase in EGL area increase from P0->P7 in control cerebella whereas the mutant cerebella showed only a 4.9 fold increase in EGL area. Hence, cyclinA2 null CGNP's cause growth of the EGL, albeit more slowly. We conclude that slower growth of the CGNPs in cyclinA2 null cells contributes to the dysmorphic cerebella.
Rhombic lip-derived and VZ/SVZ-derived neural progenitor cells show differentiation requirements to cyclin A2
To test if cerebellar development showed requirements of cyclin A2 at earlier embryonic ages, we evaluated EGL volume and programmed cell death in E14.5 and E17.5 embryos and 4th ventricular VZ/SVZ volume and programmed cell death at E14.5. Purkinje neurons are generated from the fourth ventricular VZ/SVZ during E13-E17 (Yuasa et al., 1991). Volumes were calculated by Cavalieri estimation, and total cleaved caspase-3 positive cells were quantified by optical fractionator (see Methods). These data are illustrated in Fig. 6 A-D. At E14.5, the EGL total volume was ∼40% reduced in nestin-cre, A2fl/fl mice. In contrast, the nestin-cre, A2 fl/fl EGL showed a significant increase in the number of cleaved caspase-3+ cells (Fig. 6 G). In contrast, the 4th ventricular VZ/SVZ showed no change in volume and no statistically significant difference in programmed cell death between control and Nestin-cre, A2fl/fl mice (Fig. 6 E). Although we noted a trend of increased cleaved caspase-3+ cells at E17.5, this finding did not meet our threshold for statistical significance (Fig. 6 F). We conclude that cyclin A2 loss in embryonic cerebellar VZ/SVZ neural progenitor cells does not induce programmed cell death, whereas cyclin A2 loss in embryonic CGNPs induces increased programmed cell death.
Intrinsic Proliferative Defects in CGNP's results in architectural cerebellar defects
Nestin-cre causes recombination in multipotent CNS progenitors in both forebrain and hindbrain progenitor compartments; thus, cre expression begins during early embryonic development amongst a broad pool of CNS progenitors. Thus, to investigate CGNP cell-type specific requirements for cyclin A2 function, we crossed conditional A2fl/fl mice to Math1-cre mice (Schuller et al., 2008). Overall, this yielded a relatively mild cerebellar hypoplasia phenotype relative to the Nestin-cre, A2 fl/fl mice with pathology confined to particular rostral cerebellar folia of Math1-cre, A2fl/fl mice. We found that this mild phenotype was due to preservation of cyclin A2 expression in the caudal folia, whereas the rostral folia showed cyclin A2 loss (Fig. 7, compare A2 and A3). PC dendrites in the rostral folia had poor arborizations and their somata failed to resolve into a monolayer (Fig. 7 A2-A2′). The underlying IGL Zic1+ cells were decreased in numbers (Fig. 7 B2), and we observed abnormal/ectopic sites of CGNP proliferation in the EGLb and IGL (Fig. 7 C2). Nevertheless, bergman glial processes were not disrupted in A2 null CGNPs (Fig. 7 B2). We conclude that cell-intrinsic cyclin A2 loss in CGNPs results in cerebellar cortical disorganization, including PC cell dyslamination. Furthermore, in areas were cyclin A2 was lost in CGNP's of Math1-cre, A2fl/fl mice, the cytoarchitectural abnormalities were similar to those found in the cerebella of Nestin-cre, A2fl/fl mice.
Figure 7. Intrinsic Proliferative Defects in CGNP's results in dyslamination of Purkinje neurons.
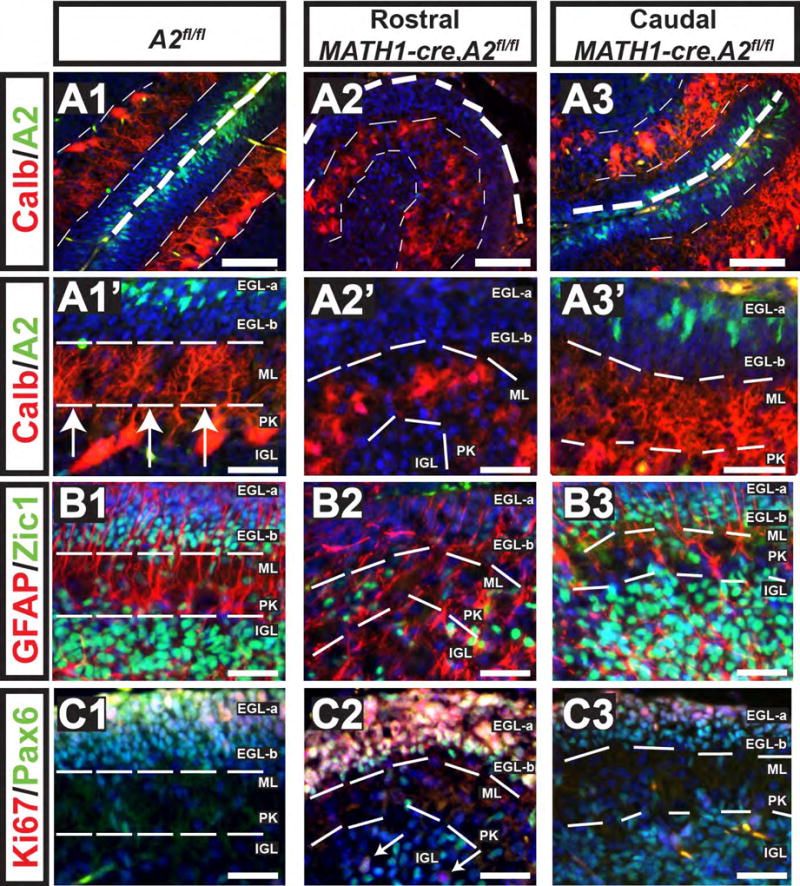
Genotypes and postnatal ages are denoted on the top, molecular markers are color coded and denoted on the left. Cyclin A2 was deleted in cerebellar granule precursor cells by intercrossing with Math1-cre mice and evaluated at P7 (genotypes are indicated atop each column). These mice showed incomplete cre-expression in caudal folia, which showed preserved cyclin A2 expression (A3). However, rostrally cyclin A2 was not detected in the EGL (A2). Calbindin staining in control shows laminar PC neurons, which in the cyclin A2 deleted areas showed PC neurons in dyslaminated aggregates (compare A1-A1′ to A2-A2′, and the internal control of A2, A3-A3′ ). Additional cytoarchitectural abnormalities included loss of Zic1+ cells in the EGL-b and a decrease in Zic1+ cells in the IGL (B1-B2). Pax6+Ki67+ cells were present in the full thickness of the EGL in the Math1-cre, cyclin A2fl/fl mice, which should normally show a band of Pax6+Ki67-negative cells in the EGL-b (C1-C2). White arrows in panel C3 point to Pax6+Ki67+ IGL cells. In the caudal folia of Math1-cre, A2fl/fl mice showed preserved cyclin A2 expression and showed cytoarchitecture similar to the control. Scale bars are as follows: A1-A3, B1-C3 = 50 μm, A1′-A3′ = 20 μm, The images show cerebella sectioned in the sagittal plane with the upper right being dorsal and the lower left being rostral (A2 = cyclin A2, Calb = calbindin).
The above findings suggest that normal PC lamination requires cyclin A2 expression in CGNP cells. This raised the possibility that PC lamination might require normal cell-intrinsic control of proliferation of CGNPs. As shown above (Figure 4), cyclin A2-deficient CGNP's show some Gli1 expression, indicating that a proliferative defect probably lies downstream of Shh-Smo signaling. With this in mind, we analyzed function of Nmyc, a factor known to be critical for the CGNP cell-intrinsic SHH proliferative response (Kenney et al., 2004). Math1-cre, Nmycfl/fl mice also showed relatively strong cre expression in the rostral, but not caudal folia (Supplemental Fig. 4 A). Consequently and similar to the Math1-cre, A2fl/fl mice, Nmyc expression was lost rostrally yet preserved caudally, supplying an internal control. Gli1 expression was preserved in the area of Nmyc loss (Supplemental Fig. 4 D), indicating that Shh-Smo signaling axis is intact in Nmyc null CGNP cells. Of note, cyclin A2 protein was detected in the areas of Nmyc loss, indicating that cyclin A2 expression does not require Nmyc expression (Supplemental Fig. 4 F). However, similar to our findings in the Math1-cre, A2fl/fl mice, we observed PC neuron dyslamination, poorly formed dendritic arborization and decreased IGL cells (Supplemental Fig. 4 G and H). In summary, loss of cyclinA2 in Math1 positive cells phenocopies Nmyc loss in Math1 positive cells. Together, these findings suggest that proper cerebellar cortical lamination requires normal proliferation of EGL, which requires intact CGNP cell-intrinsic proliferative response machinery. Indeed, we propose that any abnormality resulting in severely reduced CGNP proliferation, either extrinsic (e.g., loss of Smo) or intrinsic (e.g., loss of cyclin A2 or Nmyc) would cause a phenotype of PC dyslamination and other layering abnormalities noted above.
Discussion
Normal EGL Proliferation is Required for Cerebellar Cortical Lamination
The cyclin A2 cerebellar phenotype is different from that reported upon ablation of other cyclins. Loss of cyclin D1 leads to diminished post-natal cerebellar growth, diminished PC neuron dendritic arborization, and has a much milder CGNP proliferative phenotype than the cyclin A2 null mice presented here (Pogoriler et al., 2006). In contrast, although cyclin D2 null mice also have decreased EGL proliferation, they do not show such drastic abnormalities in PC dendritic arborization or cortical lamination, and Bergmann glial fibers appropriately extend to the sub-pial surface (Huard et al., 1999). Although concurrent loss of two D-type cyclins leads to small cerebella, these mice show better-formed folia and minimal PC displacement into the molecular layer (Ciemerych et al., 2002).
In contrast, the cyclin A2-null phenotype is more severe indicating its critical function downstream of SHH signaling. SHH treatment upregulates cyclin A2 in CGNP's (Zhao et al., 2002) and cyclin A2 is highly expressed in medulloblastoma (Thompson et al., 2006). Several studies show that cerebellar SHH loss decreases cerebellar foliation and induces PC dyslamination (Dahmane and Ruiz i Altaba, 1999; Wechsler-Reya and Scott, 1999), similar in severity to cyclin A2-null animals. In addition, seminal experiments by Atlman and Anderson have shown that prevention of EGL proliferation by brain irradiation leads to cerebellar pathology, including small cerebellar size and PC dyslamination (Altman and Anderson, 1971). However, it has been unclear as to whether these abnormalities in cerebellar lamination are due to absent SHH “morphogen” signaling or to just a consequence of decreased CGNP proliferation independent of SHH's morphogen activities. Indeed, it has been proposed that Shh has a direct role in cerebellar morphogenesis and foliation (Corrales et al., 2006; Corrales et al., 2004).
To address these questions, we targeted the A2 and Nmyc alleles using cre-loxp targeting technology, and our data show that reducing EGL proliferation intrinsically by targeted disruption of cyclin A2 and Nmyc also alters cerebellar cortical lamination. Furthermore, cyclin A2 null CGNP cells express Nmyc, and Nmyc CGNP cells express cyclin A2. Hence, the phenocopy between these Math1-cre, A2fl/fl and Math1-cre, Nmycfl/fl is not due to a direct mechanistic link between cyclin A2 and Nmyc. Although our in situ hybridization data demonstrating preserved Gli expression in cerebella of Nestin-cre, A2fl/fl mice my be due to expression in Bergman glia (Dahmane and Ruiz i Altaba, 1999), our in vitro CGNP cultures confirm the Math1-cre, A2fl/fl data in that they demonstrate that SHH treatment of A2-null CGNPs show a functional response to SHH. Taken together, our data suggest that intrinsic proliferative defects of the EGL leads to secondary abnormalities of cerebellar cortical lamination, and suggests that inhibition of CGNP proliferation—due to any cause—would lead to cortical dyslamination and reduced foliation (see Fig. 8 for schematic). One notable distinction however between the cyclin A2 null and SHH null phenotype in cerebellum is the presence of ectopic proliferative Pax6+ cells in the IGL, which suggests that cyclin A2 loss in CGNP's might effect placement of EGL cells or affect the response to SHH signaling in deeper layers the EGLb and IGL, which are normally non-proliferative.
Figure 8. CGNP Intrinsic Pathways are Required for Cerebellar Foliation and Purkinje Neuron Lamination.
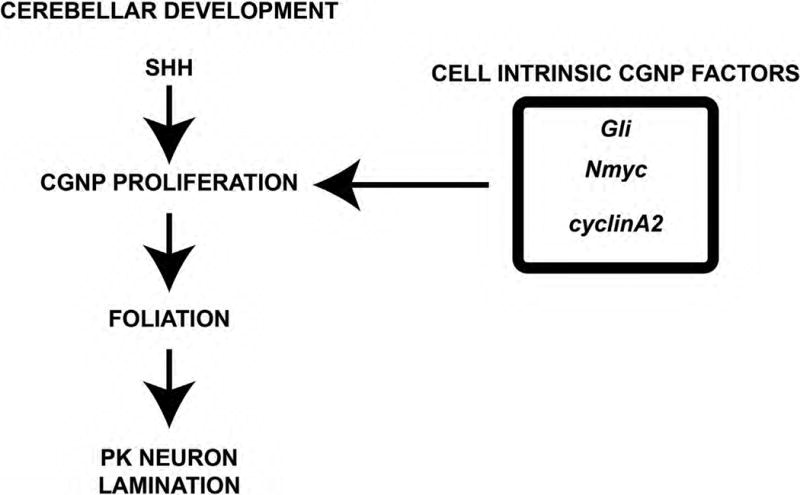
Proper cerebellar development requires SHH signaling in CGNPs. CGNP intrinsic factors (Gli, Nmyc, and cyclin A2) induce CGNP proliferation, resulting in appropriate foliation and PC neuron lamination.
Cyclin A2 regulatory functions in cerebellum
Under normal conditions, murine cyclin A1 is highly expressed in testicular germ cells (Sweeney et al., 1996) with only low expression levels reported in brain (Yang et al., 1997b). Furthermore, cyclin A1 is capable of interacting with critical cell cycle regulators including CDK2, the RB family of proteins, the E2F1 transcription factor, and p21 (Yang et al., 1999). These findings raised the possibility that cyclin A1 might show redundancy with cyclin A2. Our data demonstrate that cyclin A2 shares no redundancy with cyclin A1 in the developing cerebellum. This is in direct contrast to the D-type cyclins (cyclin D1, cyclin D2, and cyclin D3). D-type cyclins show a stereotypical tissue-specific expression pattern, yet under experimental conditions in which a D-cyclin is lost, the remaining D-type cyclins increase their expression and partially compensate for the deleted D-cyclin (Ciemerych et al., 2002).
Cyclin A2 has been shown to play a crucial role in cell cycle progression, where it interacts with CDK2 and CDK1 during S-phase and at the G2->M transition, respectively (Pagano et al., 1992); the cyclin A2/CDK1 complex in turn regulates cyclin B activity during the G2->M transition (Devault et al., 1992). Redundancy between E- and A-type cyclins was suggested by reports that both cyclins were capable of S-phase promoting activity (Strausfeld et al., 1996). In fibroblasts, loss of cyclin A2 does not prevent cell proliferation due to compensation by cyclin E1, which becomes expressed throughout the cell cycle. The finding that cyclin E1 protein levels are increased in cyclin A2 null cerebella suggested the presence of a similar redundancy. Nevertheless, the cerebella of cyclin A2 null mice shows drastic cytoarchitectural abnormalities. These data not only confirm that cyclin A2 shows incomplete redundancy with cyclin E1 in the EGL, but also that the nature of the cyclin A2 dependency in CGNP's is distinct from hematopoietic stem cells (Kalaszczynska et al., 2009). In fibroblasts, the A60 form of cyclin A2 is found and loss is compensated by Cyclin E1 (Kalaszczynska et al., 2009). Our data demonstrating increased levels of cyclin E1 in the cerebellum suggest that some compartments of the cerebellum show redundancy between cyclins A2 and E1. In particular, elevated apoptosis in E14.5 EGL without elevated apoptosis in the 4th ventricular neuroepithelium supports this notion. These data indicate that distinct stem cell compartments of the cerebellum show differential requirements to cyclin A2. Furthermore, our findings that post-natal CGNP's of the nestin-cre, cyclin A2fl/fl mice can incorporate BrdU and reach mitosis demonstrate that these cells are capable of proceeding through S-phase into M-phase. Our data suggest that the postnatal dysmorphic cerebellar phenotype may be due to a combination of early embryonic cerebellar apoptosis in the EGL anlage and due to stunted proliferation of the postnatal EGL. In contrast, other cell types in the cerebellum may not be affected because their ancestral progenitor cell compartments show a redundancy for cyclin A2 function by other cyclins such as cyclin E1.
Conclusions
We tested the hypothesis that cerebellar development requires A-type cyclin function through a conditional deletion approach. During this postnatal period, cytoarchitectural abnormalities of cerebellar cortex in conditional cyclin A2 null mice were noted and were dependent on cell intrinsic proliferative characteristics of CGNP's downstream of SHH signaling. Cyclin A2 loss cannot be compensated for by cyclin E1 expression in the EGL. Furthermore, distinct progenitor compartments of the cerebellum show differential requirements to cyclin A2. These data underscore novel roles of cyclin A2 in cerebellar morphogenesis and cortical organization.
Supplementary Material
Highlights.
The developing postnatal cerebellum shows robust expression of cyclin A2.
Cyclin A2 expression is present in cycling neural progenitor cells and in terminally differentiated neurons.
Cyclin A2 loss results in dysmorphic cerebellum and PC dyslamination.
Cyclin A1 cannot compensate for cyclin A2 loss.
We propose that any defect in CGNP proliferation may lead to PC dyslamination.
Acknowledgments
J.J.O acknowledges support from the California Institute for Regenerative Medicine (Grant Number TG2-01153) and NIH/NCRR (Grant Number UL1RR024131). W.M., A.G., and US were supported by the Foundation for Polish Science, American Brain Tumor Association, and the German Cancer Aid, respectively. This work was funded by NIH grants R01NS40511 (to D.H.R.) and R01 CA132740 (to P.S.). D.H.R. is a Howard Hughes Medical Institute Investigator.
The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH, CIRM or any other agency of the State of California.
Footnotes
Conflicts of Interest: The authors have no conflicts of interests with the work presented in this study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aleem E, Kiyokawa H, Kaldis P. Cdc2-cyclin E complexes regulate the G1/S phase transition. Nat Cell Biol. 2005;7:831–836. doi: 10.1038/ncb1284. [DOI] [PubMed] [Google Scholar]
- Altman J, Anderson WJ. Irradiation of the cerebellum in infant rats with low-level x-ray: histological and cytological effects during infancy and adulthood. Exp Neurol. 1971;30:492–509. doi: 10.1016/0014-4886(71)90150-6. [DOI] [PubMed] [Google Scholar]
- Bloom J, Cross FR. Multiple levels of cyclin specificity in cell-cycle control. Nat Rev Mol Cell Biol. 2007;8:149–160. doi: 10.1038/nrm2105. [DOI] [PubMed] [Google Scholar]
- Charron J, Gagnon JF, Cadrin-Girard JF. Identification of N-myc regulatory regions involved in embryonic expression. Pediatric research. 2002;51:48–56. doi: 10.1203/00006450-200201000-00010. [DOI] [PubMed] [Google Scholar]
- Chen YT, Collins LL, Uno H, Chang C. Deficits in motor coordination with aberrant cerebellar development in mice lacking testicular orphan nuclear receptor 4. Mol Cell Biol. 2005;25:2722–2732. doi: 10.1128/MCB.25.7.2722-2732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciemerych MA, Kenney AM, Sicinska E, Kalaszczynska I, Bronson RT, Rowitch DH, Gardner H, Sicinski P. Development of mice expressing a single D-type cyclin. Genes Dev. 2002;16:3277–3289. doi: 10.1101/gad.1023602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales JD, Blaess S, Mahoney EM, Joyner AL. The level of sonic hedgehog signaling regulates the complexity of cerebellar foliation. Development. 2006;133:1811–1821. doi: 10.1242/dev.02351. [DOI] [PubMed] [Google Scholar]
- Corrales JD, Rocco GL, Blaess S, Guo Q, Joyner AL. Spatial pattern of sonic hedgehog signaling through Gli genes during cerebellum development. Development. 2004;131:5581–5590. doi: 10.1242/dev.01438. [DOI] [PubMed] [Google Scholar]
- Dahmane N, Ruiz i Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126:3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- Devault A, Fesquet D, Cavadore JC, Garrigues AM, Labbe JC, Lorca T, Picard A, Philippe M, Doree M. Cyclin A potentiates maturation-promoting factor activation in the early Xenopus embryo via inhibition of the tyrosine kinase that phosphorylates cdc2. The Journal of cell biology. 1992;118:1109–1120. doi: 10.1083/jcb.118.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelkamp D, Rashbass P, Seawright A, van Heyningen V. Role of Pax6 in development of the cerebellar system. Development. 1999;126:3585–3596. doi: 10.1242/dev.126.16.3585. [DOI] [PubMed] [Google Scholar]
- Falluel-Morel A, Tascau LI, Sokolowski K, Brabet P, DiCicco-BIoom E. Granule cell survival is deficient in PAC1-/- mutant cerebellum. J Mol Neurosci. 2008;36:38–44. doi: 10.1007/s12031-008-9066-6. [DOI] [PubMed] [Google Scholar]
- Fancy SP, Harrington EP, Yuen TJ, Silbereis JC, Zhao C, Baranzini SE, Bruce CC, Otero JJ, Huang EJ, Nusse R, Franklin RJ, Rowitch DH. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat Neurosci. 2011;14:1009–1016. doi: 10.1038/nn.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard F, Strausfeld U, Fernandez A, Lamb NJ. Cyclin A is required for the onset of DNA replication in mammalian fibroblasts. Cell. 1991;67:1169–1179. doi: 10.1016/0092-8674(91)90293-8. [DOI] [PubMed] [Google Scholar]
- Hatten ME, Heintz N. Mechanisms of neural patterning and specification in the developing cerebellum. Annu Rev Neurosci. 1995;18:385–408. doi: 10.1146/annurev.ne.18.030195.002125. [DOI] [PubMed] [Google Scholar]
- Heine VM, Griveau A, Chapin C, Ballard PL, Chen JK, Rowitch DH. A small-molecule smoothened agonist prevents glucocorticoid-induced neonatal cerebellar injury. Sci Transl Med. 2011;3:105ra104. doi: 10.1126/scitranslmed.3002731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine VM, Rowitch DH. Hedgehog signaling has a protective effect in glucocorticoid-induced mouse neonatal brain injury through an 11betaHSD2-dependent mechanism. J Clin Invest. 2009;119:267–277. doi: 10.1172/JCI36376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Liu J, Ketova T, Fleming JT, Grover VK, Cooper MK, Litingtung Y, Chiang C. Transventricular delivery of Sonic hedgehog is essential to cerebellar ventricular zone development. Proc Natl Acad Sci U S A. 2010;107:8422–8427. doi: 10.1073/pnas.0911838107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard JM, Forster CC, Carter ML, Sicinski P, Ross ME. Cerebellar histogenesis is disturbed in mice lacking cyclin D2. Development. 1999;126:1927–1935. doi: 10.1242/dev.126.9.1927. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Matsunaga Y, Takiguchi M, Ikeda MA. Expression of cyclin E in postmitotic neurons during development and in the adult mouse brain. Gene expression patterns : GEP. 2011;11:64–71. doi: 10.1016/j.gep.2010.09.004. [DOI] [PubMed] [Google Scholar]
- Jennemann R, Sandhoff R, Wang S, Kiss E, Gretz N, Zuliani C, Martin-Villalba A, Jager R, Schorle H, Kenzelmann M, Bonrouhi M, Wiegandt H, Grone HJ. Cell-specific deletion of glucosylceramide synthase in brain leads to severe neural defects after birth. Proc Natl Acad Sci U S A. 2005;102:12459–12464. doi: 10.1073/pnas.0500893102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirawatnotai S, Hu Y, Michowski W, Elias JE, Becks L, Bienvenu F, Zagozdzon A, Goswami T, Wang YE, Clark AB, Kunkel TA, van Harn T, Xia B, Correll M, Quackenbush J, Livingston DM, Gygi SP, Sicinski P. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature. 2011;474:230–234. doi: 10.1038/nature10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaszczynska I, Geng Y, lino T, Mizuno S, Choi Y, Kondratiuk I, Silver DP, Wolgemuth DJ, Akashi K, Sicinski P. Cyclin A is redundant in fibroblasts but essential in hematopoietic and embryonic stem cells. Cell. 2009;138:352–365. doi: 10.1016/j.cell.2009.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003;130:15–28. doi: 10.1242/dev.00182. [DOI] [PubMed] [Google Scholar]
- Kenney AM, Rowitch DH. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol. 2000;20:9055–9067. doi: 10.1128/mcb.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney AM, Widlund HR, Rowitch DH. Hedgehog and PI-3 kinase signaling converge on Nmycl to promote cell cycle progression in cerebellar neuronal precursors. Development. 2004;131:217–228. doi: 10.1242/dev.00891. [DOI] [PubMed] [Google Scholar]
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002;16:2699–2712. doi: 10.1101/gad.1021202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, McMahon SB, Eisenman RN. Myc influences global chromatin structure. Embo J. 2006;25:2723–2734. doi: 10.1038/sj.emboj.7601152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PM, Gritli-Linde A, Smeyne R, Kottmann A, McMahon AP. Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Developmental biology. 2004;270:393–410. doi: 10.1016/j.ydbio.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Liu D, Matzuk MM, Sung WK, Guo Q, Wang P, Wolgemuth DJ. Cyclin Al is required for meiosis in the male mouse. Nat Genet. 1998;20:377–380. doi: 10.1038/3855. [DOI] [PubMed] [Google Scholar]
- Mills J, Digicaylioglu M, Legg AT, Young CE, Young SS, Barr AM, Fletcher L, O'Connor TP, Dedhar S. Role of integrin-linked kinase in nerve growth factor-stimulated neurite outgrowth. J Neurosci. 2003;23:1638–1648. doi: 10.1523/JNEUROSCI.23-05-01638.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M. Delayed early embryonic lethality following disruption of the murine cyclin A2 gene. Nat Genet. 1999;23:481. doi: 10.1038/70612. [DOI] [PubMed] [Google Scholar]
- Nakata K, Nagai T, Aruga J, Mikoshiba K. Xenopus Zic family and its role in neural and neural crest development. Mech Dev. 1998;75:43–51. doi: 10.1016/s0925-4773(98)00073-2. [DOI] [PubMed] [Google Scholar]
- Odajima J, Wills ZP, Ndassa YM, Terunuma M, Kretschmannova K, Deeb TZ, Geng Y, Gawrzak S, Quadros IM, Newman J, Das M, Jecrois ME, Yu Q, Li N, Bienvenu F, Moss SJ, Greenberg ME, Marto JA, Sicinski P. Cyclin E constrains Cdk5 activity to regulate synaptic plasticity and memory formation. Developmental cell. 2011;21:655–668. doi: 10.1016/j.devcel.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is required at two points in the human cell cycle. The EMBO journal. 1992;11:961–971. doi: 10.1002/j.1460-2075.1992.tb05135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogoriler J, Millen K, Utset M, Du W. Loss of cyclin Dl impairs cerebellar development and suppresses medulloblastoma formation. Development. 2006;133:3929–3937. doi: 10.1242/dev.02556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, Ma Q, Alvarez-Buylla A, McMahon AP, Rowitch DH, Ligon KL. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller U, Kho AT, Zhao Q, Ma Q, Rowitch DH. Cerebellar ‘transcriptome’ reveals cell-type and stage-specific expression during postnatal development and tumorigenesis. Mol Cell Neurosci. 2006;33:247–259. doi: 10.1016/j.mcn.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, Haslam SZ, Bronson RT, Elledge SJ, Weinberg RA. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell. 1995;82:621–630. doi: 10.1016/0092-8674(95)90034-9. [DOI] [PubMed] [Google Scholar]
- Strausfeld UP, Howell M, Descombes P, Chevalier S, Rempel RE, Adamczewski J, Mailer JL, Hunt T, Blow JJ. Both cyclin A and cyclin E have S-phase promoting (SPF) activity in Xenopus egg extracts. J Cell Sci. 1996;109(Pt 6):1555–1563. doi: 10.1242/jcs.109.6.1555. [DOI] [PubMed] [Google Scholar]
- Sweeney C, Murphy M, Kubelka M, Ravnik SE, Hawkins CF, Wolgemuth DJ, Carrington M. A distinct cyclin A is expressed in germ cells in the mouse. Development. 1996;122:53–64. doi: 10.1242/dev.122.1.53. [DOI] [PubMed] [Google Scholar]
- ten Donkelaar HJ, Lammens M, Wesseling P, Thijssen HO, Renier WO. Development and developmental disorders of the human cerebellum. J Neurol. 2003;250:1025–1036. doi: 10.1007/s00415-003-0199-9. [DOI] [PubMed] [Google Scholar]
- Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, Chintagumpala M, Adesina A, Ashley DM, Kellie SJ, Taylor MD, Curran T, Gajjar A, Gilbertson RJ. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24:1924–1931. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- Tramontin AD, Garcia-Verdugo JM, Lim DA, Alvarez-Buylla A. Postnatal development of radial glia and the ventricular zone (VZ): a continuum of the neural stem cell compartment. Cereb Cortex. 2003;13:580–587. doi: 10.1093/cercor/13.6.580. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Troussard AA, Mawji NN, Ong C, Mui A, St Arnaud R, Dedhar S. Conditional knock-out of integrin-linked kinase (ILK) demonstrates an essential role in PKB/Akt activation. J Biol Chem. 2003 doi: 10.1074/jbc.M303083200. [DOI] [PubMed] [Google Scholar]
- Valtorta F, Pozzi D, Benfenati F, Fornasiero EF. The synapsins: multitask modulators of neuronal development. Seminars in cell & developmental biology. 2011;22:378–386. doi: 10.1016/j.semcdb.2011.07.008. [DOI] [PubMed] [Google Scholar]
- Van Hooser A, Goodrich DW, Allis CD, Brinkley BR, Mancini MA. Histone H3 phosphorylation is required for the initiation, but not maintenance, of mammalian chromosome condensation. J Cell Sci. 1998;111(Pt 23):3497–3506. doi: 10.1242/jcs.111.23.3497. [DOI] [PubMed] [Google Scholar]
- Wallace VA. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol. 1999;9:445–448. doi: 10.1016/s0960-9822(99)80195-x. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- Welm AL, Timchenko NA, Ono Y, Sorimachi H, Radomska HS, Tenen DG, Lekstrom-Himes J, Darlington GJ. C/EBPalpha is required for proteolytic cleavage of cyclin A by calpain 3 in myeloid precursor cells. The Journal of biological chemistry. 2002;277:33848–33856. doi: 10.1074/jbc.M204096200. [DOI] [PubMed] [Google Scholar]
- Yamasaki T, Kawaji K, Ono K, Bito H, Hirano T, Osumi N, Kengaku M. Pax6 regulates granule cell polarization during parallel fiber formation in the developing cerebellum. Development. 2001;128:3133–3144. doi: 10.1242/dev.128.16.3133. [DOI] [PubMed] [Google Scholar]
- Yang R, Morosetti R, Koeffler HP. Characterization of a second human cyclin A that is highly expressed in testis and in several leukemic cell lines. Cancer Res. 1997a;57:913–920. [PubMed] [Google Scholar]
- Yang R, Morosetti R, Koeffler HP. Characterization of a second human cyclin A that is highly expressed in testis and in several leukemic cell lines. Cancer research. 1997b;57:913–920. [PubMed] [Google Scholar]
- Yang R, Muller C, Huynh V, Fung YK, Yee AS, Koeffler HP. Functions of cyclin Al in the cell cycle and its interactions with transcription factor E2F-1 and the Rb family of proteins. Molecular and cellular biology. 1999;19:2400–2407. doi: 10.1128/mcb.19.3.2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa S, Kawamura K, Ono K, Yamakuni T, Takahashi Y. Development and migration of Purkinje cells in the mouse cerebellar primordium. Anat Embryol (Bed) 1991;184:195–212. doi: 10.1007/BF01673256. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Kho A, Kenney AM, Yuk Di DI, Kohane I, Rowitch DH. Identification of genes expressed with temporal-spatial restriction to developing cerebellar neuron precursors by a functional genomic approach. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:5704–5709. doi: 10.1073/pnas.082092399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.