

Abstract
The level of the endocannabinoid anandamide is controlled by fatty acid amide hydrolase (FAAH). In 2011, PF-04457845, an irreversible inhibitor of FAAH, was progressed to phase II clinical trials for osteoarthritic pain. This article discusses a prospective, integrated systems pharmacology model evaluation of FAAH as a target for pain in humans, using physiologically based pharmacokinetic and systems biology approaches. The model integrated physiological compartments; endocannabinoid production, degradation, and disposition data; PF-04457845 pharmacokinetics and pharmacodynamics, and cannabinoid receptor CB1-binding kinetics. The modeling identified clear gaps in our understanding and highlighted key risks going forward, in particular relating to whether methods are in place to demonstrate target engagement and pharmacological effect. The value of this modeling exercise will be discussed in detail and in the context of the clinical phase II data, together with recommendations to enable optimal future evaluation of FAAH inhibitors.
The identification of cannabinoid receptors CB1 and CB21,2,3 and their endogenous lipid ligands has triggered an exponential growth of studies exploring the endocannabinoid (EC) system and its regulatory functions in health and disease.4 The EC system has two endogenous ligands, 2-arachidonoyl glycerol and N-arachidonoyl ethanolamide (alternatively known as arachidonoylethanolamide, anandamide, or AEA).5,6,7 AEA was the first endogenous ligand found to bind to CB's5 and is biosynthesized by the activities of multiple enzymes. N-acyl transferases (NATs) catalyze the synthesis of the precursor N-arachidonoyl phosphatidylethanolamine (NAPE) from lecithin and cephalin. NAPE-phospholipase D then catalyzes the conversion of NAPE into AEA. AEA is in turn degraded by fatty acid amide hydrolase (FAAH), which also catalyzes the degradation of other fatty amides, including N-palmitoyl ethanolamide (PEA), N-oleoyl ethanolamide (OEA), N-stearoyl ethanolamide (SEA), and N-linoleoyl ethanolamide (LEA). It has been reported that AEA is not stored in “resting” cells but is, instead, synthesized and released “on demand” following physiological and pathological stimuli, such as neuronal depolarization,8 and thus rapid coordinated synthesis and removal of AEA is required. The first step in the inactivation of ECs requires fast (half-life t1/2 = 5 min) clearance from the milieu around their molecular target. Due to its high lipophilicity, AEA can, in principle, diffuse through the plasma membrane. Indeed, there is some evidence that AEA binds its cognate receptor via the plasma membrane (Figure 1).9 However, to explain the fast decay of effect, the transport of AEA needs to be mediated to some extent by controllable and selective mechanisms, such as either a membrane transporter protein or an intracellular enzymatic process capable of rapidly reducing the intracellular concentrations. The majority of the available evidence supports the hypothesis that AEA is taken up by cells via a selective, saturable, temperature-dependent, and Na-independent “facilitated transport” mechanism, known as the anandamide membrane transporter.10,11,12,13,14,15,16,17 However, to date, the putative transporter or transporters remains to be isolated or cloned.
Figure 1.

Cannabinoid receptor (CB) anandamide (AEA) binding model. AEA partitions into the cell membrane due to its high lipophilicity (log D7.4 = 5.7). It then diffuses laterally to the binding site of the cannabinoid receptor CB1 (after Tian et al.). Observed binding can then be given by the product of the partition coefficient and the binding affinity, as shown in the figure (after Vauquelin and Packeu). Hence, observed affinity (dissociation constant KD) is approximately the receptor binding affinity KD′(200 nmol/l) × Kp−1 (2.1 × 10−6) = 0.4 pmol/l.
The receptor CB1 is considered to be the most abundant G-protein-coupled receptor in mammalian brain. Its activation can typically initiate responses such as the closure of Ca2+ channels, opening of K+ channels, inhibition of adenylyl cyclase activity, and stimulation of kinase. Furthermore, CB1 agonists inhibit N- and P/Q-type voltage-activated Ca2+ channels.18,19,20 This effect is proposed to underlie the CB1-mediated depression of transmitter release at the γ-aminobutyric acid synapses in the CA1 field of the hippocampus21 and at the glutamatergic synapses in the dorsal striatum22,23; this has led to the idea that CB1 agonism may have benefits in some pain states.24 Importantly, however, ECs such as AEA may also activate ion channels, including those of vanilloid transient receptor potential cation channel subfamily V member 1 (TRPV1) receptors25,26 and thus, potentially at least, AEA may also enhance nociception.
CB1 is a key mediator of the effects of cannabinoid drugs such as tetrahydrocannabinol,27 which, in addition to the psychotropic effects, has historically been speculated to have analgesic and other medicinal effects.28 In addition, there are some preclinical data suggesting that EC-modulating drugs such as FAAH inhibitors have efficacy in disease models of pain,24 and hence many pharmaceutical companies are exploring FAAH inhibitors and CB1 agonists as drug targets.29
To this end, PF-04457845, an irreversible potent FAAH inhibitor, was developed and subsequently found to have good in vivo efficacy in an animal model of pain.30 PF-04457845 was progressed to phase I clinical trials and it was found that FAAH activity was inhibited >97% in plasma and significant increases in plasma AEA and other biomarkers of pharmacology were achieved.31 This article discusses the use of a systems pharmacology approach to understanding the wealth of complex and sometimes apparently contradictory data relating to the use of FAAH inhibitors for the treatment of pain. This work was carried out before the clinical trials of PF-04457845 and was previously reported briefly elsewhere.32 By using physiologically based pharmacokinetic and pharmacological models as a tool, in tandem with the available preclinical and clinical data, this approach was used (i) to develop a model of the human EC system to understand the outcome of intervening pharmacologically and (ii) to identify key missing data and knowledge. These quantitative analyses will be discussed in detail in the context of clinical efficacy data, together with recommendations for optimal development of EC-modulating drugs.
Results
To integrate all available data regarding the fatty acid ethanolamide metabolism and distribution in the human body, we created a mechanistic ordinary differential equation-based model as shown in Figure 2. To account for the known mutually exclusive binding of ethanolamides to FAAH and to enable the use of all clinical biomarker data for parameter estimation, the model included not only AEA, but also OEA, PEA, SEA, and LEA. The resulting model was fit to the individual clinical data for a single oral dose of 10 mg of PF-04457845. Supplementary Table S1 and Model S1 show the ordinary differential equations, parameters, and data sources. Values were estimated from the clinical trial data31 with the remainder fixed, based on literature or in-house laboratory data. It was found that a good fit to the data could be achieved, provided FAAH-independent clearance was included in the model (Figure 3). Figure 3 also illustrates that removal of the FAAH-independent clearance from the model (for example, assuming maximal rate of the process equal to zero) resulted in both substantial changes in the shape of the time–response curve and a significant increase of more than 10-fold the maximal biomarker concentration. The model was also able to accurately describe the dose–response kinetics of FAAH activity in human plasma at 1 and 10 mg of PF-04457845. Furthermore, this model also accurately simulated the observed human dynamics of AEA, LEA, PEA, and OEA at both 1 and 10 mg of PF-04457845 (Figure 4). We used this model to simulate the brain CB1 occupancy in hypothetical patients at doses of 0.1–40 mg. These simulations showed that the projected occupancy saturated at ~25%, independent of the drug dose. However, increasing dose did prolong the time at peak receptor occupancy (Figure 5). To obtain insight into the most influential parameters in the model, we carried out a sensitivity analysis.33 Integrating the resulting values yielded a representation of the influence of each parameter on the CB occupancy in brain. The results are summarized in Figure 6 and show that the five most influential parameters in their order of influence, respectively, were kdeg_FAAH (the degradation rate constant for FAAH), p_A (the precursor substrate concentration for NAPE synthesis), b_FAAH_brain (brain FAAH concentration), a-NAT_A (the brain N-acyltransferase catalyzing the synthesis of the AEA precursor N-arachidonoyl phosphatidylethanolamine from p_A), and Kp_b (the brain partition coefficient).
Figure 2.

Schematic representation of the systems pharmacology model. Arrows represent binding, synthesis, degradation, and transport processes. XEA (X = A, O, P, and S) relates to the various ethanolamides (A – anandamide, O – oleoylethanolamide, P – palmitoylethanolamide, and S – stearoylethanolamide). The terms CB1 and CB1–AEA designate cannabinoid receptor CB1 and its complex with anandamide, respectively. Empty circles indicate fatty acid amide hydrolase (FAAH)-catalyzed XEA degradation and PF-04457845 inhibition of FAAH. The question mark shows the FAAH-independent clearance process. BBB, blood–brain barrier.
Figure 3.

Initial simulations related to example clinical data. The observed plateau in anandamide (AEA) response cannot be adequately described without evoking an additional fatty acid amide hydrolase (FAAH)-independent clearance process. The open circles are mean clinical plasma AEA data, following single oral administration of 10 mg of PF-04457845. The dotted line shows simulations without a FAAH-independent clearance and the solid line shows the results after including it.
Figure 4.

Simulations of the model compared with clinical data assuming fatty acid amide hydrolase (FAAH)-independent clearance. (a,b) Time dependence of FAAH activity and (c,d) concentrations of ethanolamides after single oral administration of (a,c) 1 mg and (b,d) 10 mg of PF-04457845. AEA, OEA, PEA, and LEA: anandamide, oleoylethanolamide, palmitoylethanolamide, and stearoylethanolamide, respectively. Dots indicate clinically measured data, and solid curves are simulation results.
Figure 5.

Simulations of percentage cannabinoid receptor (CB) occupancy in the brain according to the model at doses of 0.1–40 mg.
Figure 6.

Sensitivity analysis plot showing the integral of the sensitivity for each parameter in the model, calculated as described in the Methods section without normalization. kdeg_FAAH (the degradation rate constant for FAAH), p_A (the precursor substrate concentration for N-arachidonoyl phosphatidylethanolamine (NAPE) synthesis), b_FAAH_brain (brain FAAH concentration), a-NAT_A (the brain N-acyltransferase catalyzing the synthesis of the anandamide (AEA) precursor N-arachidonoyl phosphatidylethanolamine from p_A), and Kp_b (the brain partition coefficient) are shown. FAAH, fatty acid amide hydrolase.
Discussion
Typically, preclinical species data are used as indicators of probable drug efficacy in patients, with arguably some successes in pain.34 However, the utility of animal models of pain to predict outcome in humans in general is controversial and in the case of FAAH inhibitors, there are reports of both clear efficacy in inflammatory pain30 and, conversely, no activity.35 In this context, we decided to use a quantitative systems pharmacology approach to exploring the likely risks for PF-04457845 before progression to patient trials. This approach has the benefit that the model produced can enable complex interactions to be accounted for, allow key assumptions to be systematically identified, and furthermore provide a tool using which these assumptions can be made explicit and easily communicated.
Supplementary Table S1 and Model S1 summarize the model, together with the equations and parameters used. Due to a lack of human data, some of the parameters necessary to construct the model were drawn from a range of sources and species. Hence, a key caveat is that these may not accurately represent the patient population values. Nevertheless, we have shown that the current model adequately describes the observed clinical end points in our data set. From a sensitivity analysis of the data, we concluded that parameters controlling the level of FAAH enzyme in the brain were ranked most highly. To date, as far as we are aware, there are limited data on this in humans, and estimates of the relevant parameters in human brain would be useful to validate our model estimates. From a pharmacology perspective, the observation of the importance of brain levels of FAAH to CB occupancy in brain implies that optimal efficacy may be dependent on the extent of the delivery of drug to FAAH in the brain. Hence, a hypothesis on the basis of our model results is that drugs with the maximum blood–brain barrier penetration will arguably have the highest probability of delivering optimal efficacy. The available data suggest that PF-04457845 exhibits good blood–brain barrier permeability.36 The substrate concentrations and enzyme involved in the synthesis of NAPE were also influential, consistent with the AEA substrate in brain being a limiting factor. Thus, our model suggests that relevant contextual data on the precursor substrate levels could be valuable to enhance our understanding. The partition coefficient for AEA into lipid was also found to be very important to the prediction of CB occupancy. Given this, data were generated in our laboratories to define this as accurately as possible. These data were consistent with previous estimates and confirmed the high lipophilicity of AEA.
Although the highest ranked parameters related to AEA mainly, our sensitivity analysis showed that parameters influencing other ethanolamides did also influence the outcome for CB occupancy by AEA (not shown). This observation, together with our assumption that the PEA, OEA, LEA, and PEA ethanolamide substrates bind mutually exclusively to FAAH, would suggest that including all known substrates for FAAH in the model may be important for accurately describing the receptor occupancy dynamics. Nevertheless, it may also be possible to simplify this aspect. The model is a tool that can enable investigation of this important question.
We used the model to compare the observed clinical biomarker data with predictions. A key conclusion drawn from this modeling exercise was that the initial iteration of the model could not explain the plateau observed in the data and, therefore, a FAAH-independent clearance mechanism must exist. A literature survey revealed that there are numerous candidates for this process, including cyclooxygenases, cytochromes P450, and hydrolases, in addition to nonenzymatic routes such as renal clearance. By including an additional clearance process in the model, estimating the parameters for the clearance process from our data, and comparing these values with those in the available literature, we were able to quantitatively rank the possible mechanisms. This highlights a key benefit of the systems pharmacology approach, specifically that the additional dynamic information can be used to prioritize hypotheses. Table 1 summarizes enzymes that can degrade AEA and, consequently, can be considered possible candidates for role of “the unknown enzyme.” Selection of the best candidate is based on the reasoning that “the unknown enzyme” should start to contribute substantially to AEA degradation when FAAH is completely (or almost completely) inhibited with PF-04457845. This requirement yields a plateau in the clinically measured time dependences of AEA following PF-04457845 administration, as observed. The reciprocal contribution of FAAH and “the unknown enzyme” to AEA degradation can be interpreted in the following manner: (i) in the absence of PF-04457845, FAAH is the main enzyme responsible for AEA degradation and the contribution of “the unknown enzyme” is negligible and (ii) when PF-04457845 inhibits FAAH, “the unknown enzyme” becomes the main contributor to AEA degradation. One of the possible ways to meet with these requirements is to assume that the Km value of “the unknown enzyme” with respect to AEA is greater than that of FAAH. In this case, inhibition of FAAH with PF-04457845 results in an increase in AEA and, as a consequence, acceleration of its degradation by “the unknown enzyme.” We have found Km values of all enzymes with respect to AEA in the literature (Table 1) and have concluded that cyclooxygenase-2 and N-acylethanolamine-hydrolyzing acid amidase only have Km values greater than that of FAAH. However, similar to AEA, the time dependence of PEA resulting from PF-04457845 administration also has a plateau (Figure 4). This means that cyclooxygenase-2 cannot be a candidate for “the unknown enzyme” because it cannot metabolize XEA with saturated fatty acid (for example, PEA) and, as a result, must not contribute to PEA degradation to provide a “plateau” time response to PF-04457845 administration. Hence, we concluded that the most likely FAAH-independent route was via the enzyme N-acylethanolamine-hydrolyzing acid amidase. This enzyme has structural and functional similarity to acid ceramidase but no homology to FAAH. It belongs to the choloylglycine hydrolase family and has an acidic pH optimum in contrast with FAAH. The organ distribution of the messenger RNA in rats revealed that it is widely distributed.37 The development of specific inhibitors of N-acylethanolamine-hydrolyzing acid amidase, along with appropriate pharmacokinetics, will be useful to test the hypothesis that this enzyme is in part responsible for FAAH-independent clearance in vivo. This may be important if combinations of FAAH inhibitors with other drugs are required for efficacy in pain. For example, the use of R-profens in combination with FAAH inhibitors has recently been proposed for pain indications.38 Our hypothesis suggests that this may not be an optimal approach, unless R-profens can significantly inhibit N-acylethanolamine-hydrolyzing acid amidase in vivo.
Table 1. List of possible candidates for “the unknown enzyme”.
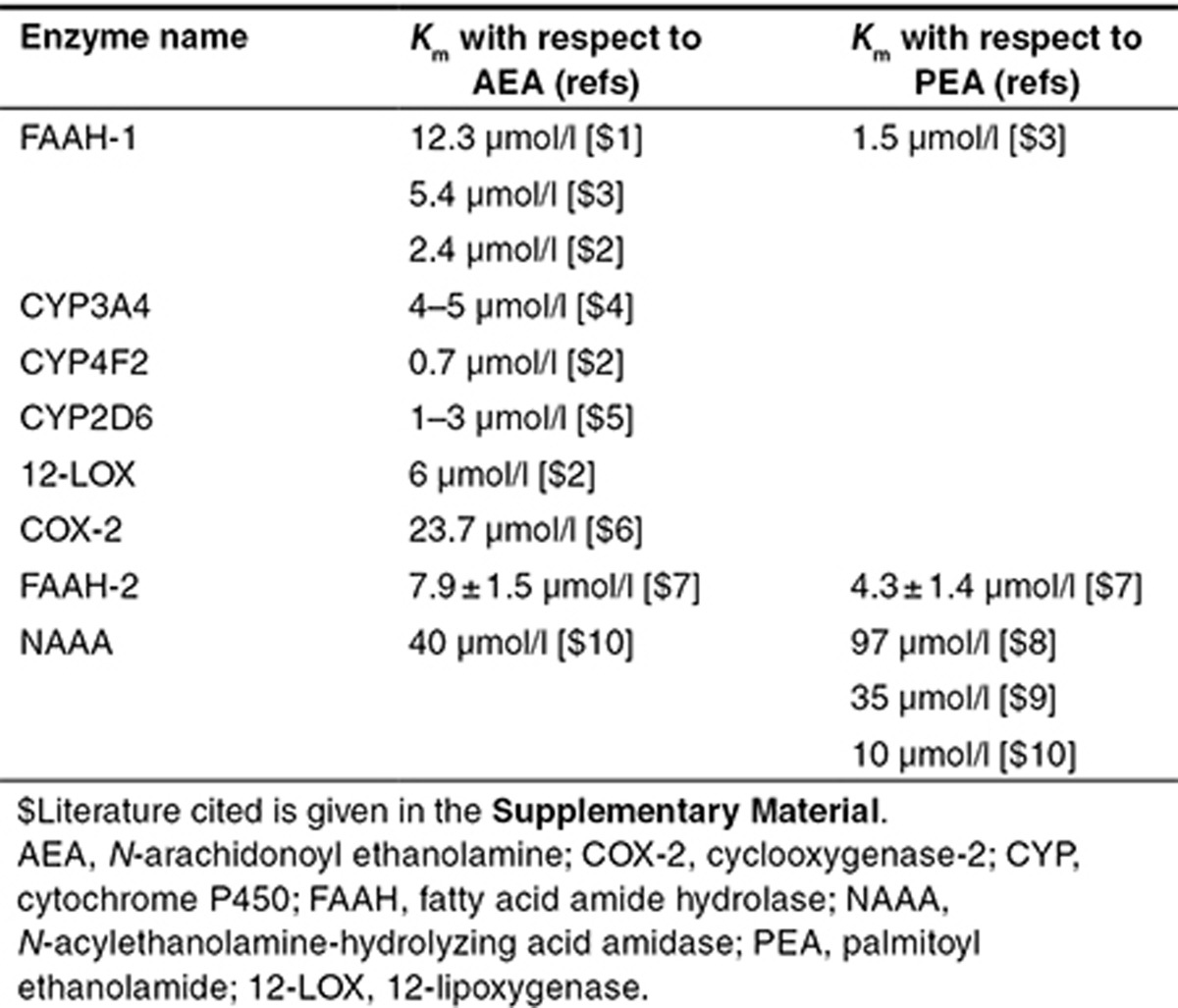
Figure 4 shows that the maximum plasma AEA achieved was of the order of 10 nmol/l. The primary in vitro binding affinity for AEA is reported as ~200 nmol/l.39 Given that the free fraction for AEA in plasma is ~0.0001, this gives a maximum free concentration of ca 1 pmol/l. According to standard free drug hypothetical assumptions around equilibrium of free drug between compartments, this yields an occupancy estimate of 0.0005%. Alternatively, it could be argued that AEA is subject to effects that invalidate typical free drug assumptions. In our modeling exercise, we used the following distinctions; first, AEA accesses its receptor only after partitioning to the plasma membrane and that the Kd,observed can be calculated according to ligand partitioning theory and Kd,obs = Kd · Kp−1 where Kd,obs = observed Kd, Kd = local receptor binding affinity, and Kp−1 = partition coefficient between the aqueous and internal membrane environment;40 and second, AEA clearance between the plasma and brain is transporter mediated. Taking into account these considerations, our simulations indicate a maximum occupancy in the region of 25%. This model does not, however, account for any localized concentrations occurring, for example, due to physical separation of AEA and its clearance mechanisms, and local occupancy in excess of this value may be possible. One clear conclusion however is that 25% or higher receptor occupancy implies that imaging technology41 may be an option to measure occupancy and resolve these questions.
A limitation of our model is that it does not include the available data on the CB agonist 2-arachidonoyl glycerol, which is thought to be mainly hydrolyzed in vivo by monoacylglycerol lipase.42 Monoacylglycerol lipase has attracted interest as a target for pharmacological intervention for various indications including pain, and inhibitors of the enzyme have been developed.43 Thus, an interesting next step would be to develop a model that includes 2-arachidonoyl glyceroland hence more fully reflects all of the relevant biology. This model could be used to explore questions relating to, for example, the benefits of combinations of FAAH and monoacylglycerol lipase inhibitors.
Of critical importance for drug discovery in the endocannabinoid pathway is the relationship between CB occupancy, the extent of nerve firing attenuation, and in turn the impact of this on pain. There are some data, for example, in rat brain slices, showing that AEA substantially depresses glutamatergic synaptic transmission in the striatum.23 Indeed, in part this observation has led to the hypothesis that CB1 agonists may potentially have an antinociceptive effect. However, to our knowledge, there are no quantitative data relating the reported substantial depression of synaptic transmission (i) with an analgesic effect or (ii) the level of receptor occupancy to give a specific level of synaptic transmission attenuation. Hence, a key conclusion from the systems pharmacology perspective was that it is not possible at this point to draw any conclusions on the extent of receptor occupancy required to deliver sufficient decrease in nerve firing to affect nociception.
In summary, this quantitative systems pharmacology–based evaluation of FAAH as a target for pain led to the following conclusions; firstly, although some significant and perhaps detectable CB1 occupancy could be expected by inhibiting FAAH in humans, the quantitative data to predict an effect in pain are lacking. Secondly, the impact of FAAH inhibitors is limited by a FAAH-independent mechanism. Finally, the incomplete understanding of redundancies in the biology (for example, with respect to effects of AEA not only on CB1 but also on TRPV1) imply the presence of risks in interpreting the steady-state outcome of perturbing this system with a drug. Notably, when PF-04457845 was evaluated recently in osteoarthritis patients, it exhibited no discernible analgesic effect.44
Taken together, these conclusions lead to the recommendations that any future progression of FAAH inhibitors for human disease should be accompanied by efforts to develop technologies that will enable demonstration of pharmacology at CB1, as advocated more generally by others.45 In addition, a better quantitative understanding of the relationship between receptor occupancy and attenuation of synaptic transmission should be developed, together with an improved knowledge of how the components of the EC system behave in humans. Together, these should enable more optimal development of modulators of the EC system in the future. More generally, this example shows the benefit of a systems pharmacology approach as an adjunct to traditional approaches to developing confidence in a target preclinically, rather than focusing mainly on animal models of disease. The key added value of the systems pharmacology discipline is to highlight questions that may be critical to a positive outcome but that are obscured from a traditional drug discovery perspective due to, for example, system complexity.46 The mathematical representation of a model enables explicit communication of assumptions and these can be challenged with data and alternative hypotheses, in contrast with other approaches. In addition, although such models undoubtedly have uncertainties, this does not mean they are not useful because the model was created to enable an enquiry into our understanding, rather than specifically for the purpose of describing data, as has been discussed recently.47 Furthermore, a model is not static and can be improved by integrating appropriate data. If this can be executed in a timely way, in tandem with model construction, as has been successfully used in pharmacokinetic/pharmacodynamic model development, this will provide a novel methodology to tackle attrition in drug discovery.
Methods
The FAAH systems model
All models were developed and analyzed using DBSolve Optimum (ISBSPb, Moscow, Russia) and Matlab/simbiology version 2010b, 2011b, or 2013b (Matlab, Natick, MA). The model comprised 39 variables and 75 reactions (for summary of model, reactions, parameters, and sources, see Supplementary Table S1 and Model S1). Four physiological compartments were introduced to model the behavior: the brain, the plasma, the remaining parts of the body, and an additional compartment representing the blood–brain barrier formed by the microvascular endothelial cells. The blood–brain barrier was introduced as the pathway for the clearance of XEA (AEA, OEA, PEA, and LEA) between the brain and the plasma. Clearance of ethanolamides between model compartments and tissue binding was modeled according to standard physiologically based pharmacokinetic techniques48 taking into account a logD value for AEA of ~7.2 (Critchell K, personal communication). The organ volumes and blood flow rates were those for an average 70-kg human.49 The lipophilic properties of AEA and other ethanolamides results in high-affinity binding to both human serum albumin and lipoproteins. Hence, the free fraction of XEA was very low and fixed at fu ~ 0.0001 (Pfizer in-house data). EC production was assumed to be due to the NAPE-phospholipase D biosynthetic pathway. FAAH hydrolyzes both AEA and other noncannabinoid ethanolamides and this was modeled assuming competitive inhibition at the FAAH active site. Rate equations for these enzymes were derived in accordance with standard enzyme kinetics techniques,50 as given in Supplementary Table S1. Parameters of the rate equations were identified on the basis of in vitro experimental data available in literature. Using in vitro binding data from literature, AEA was assumed to bind to CB1 in the brain following initial partitioning of the agonist into the plasma membrane before receptor engagement. The effective dissociation constant was proposed to be ~200 nmol/l as was reported in ref. 39. Parameters for the pharmacokinetics and irreversible inhibition of FAAH by PF-04457845, basal levels of anandamide synthesis, and FAAH activity were identified using clinical phase I trial data31 and assuming that steady-state FAAH and AEA are given by the respective ratios of synthesis and degradation rates. These data include (i) PF-04457845 pharmacokinetics, (ii) FAAH activity, and (iii) time response in concentrations of ethanolamides, measured in blood, resulting from single administration of compound. Sensitivity analysis was carried out using Matlab as follows. Assuming a model has a species of interest, x, and two parameters, y and z, the time-dependent sensitivities of x with respect to each parameter value are the time-dependent derivatives,
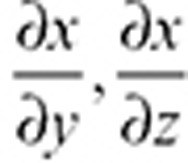 |
where the numerator is the sensitivity output and the denominators are the sensitivity inputs. This principle can be applied to n parameters. The absolute time integral was calculated as in the manufacturers' software.
Author Contributions
N.B., E.M., O.D., G.L.L., D.N., and P.H. van der G. wrote the manuscript, and E.M. and O.D. analyzed the data.
Conflict of Interest
Neil Benson, Piet van der graaf, and Gai Ling Li have been employees of Pfizer. Don Nichols is an employee of Pfizer. As Editor-in-Chief for CPT: Pharmacometrics and Systems Pharmacology, P.H. van der G. was not involved in the review or decision process for this article.
Study Highlights

Supplementary Material
References
- Matsuda L.A., Lolait S.J., Brownstein M.J., Young A.C., Bonner T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Gérard C.M., Mollereau C., Vassart G., Parmentier M. Molecular cloning of a human cannabinoid receptor which is also expressed in testis. Biochem. J. 1991;279 (Pt 1):129–134. doi: 10.1042/bj2790129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S., Thomas K.L., Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Pacher P., Bátkai S., Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devane W.A., et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Mechoulam R., et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- Sugiura T., et al. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- Di Marzo V., Melck D., Bisogno T., De Petrocellis L. Endocannabinoids: endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998;21:521–528. doi: 10.1016/s0166-2236(98)01283-1. [DOI] [PubMed] [Google Scholar]
- Tian X., Guo J., Yao F., Yang D.P., Makriyannis A. The conformation, location, and dynamic properties of the endocannabinoid ligand anandamide in a membrane bilayer. J. Biol. Chem. 2005;280:29788–29795. doi: 10.1074/jbc.M502925200. [DOI] [PubMed] [Google Scholar]
- Fowler C.J., Jacobsson S.O. Cellular transport of anandamide, 2-arachidonoylglycerol and palmitoylethanolamide–targets for drug development. Prostaglandins Leukot. Essent. Fatty Acids. 2002;66:193–200. doi: 10.1054/plef.2001.0357. [DOI] [PubMed] [Google Scholar]
- Hillard C.J., Jarrahian A. Cellular accumulation of anandamide: consensus and controversy. Br. J. Pharmacol. 2003;140:802–808. doi: 10.1038/sj.bjp.0705468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L., Bisogno T., Davis J.B., Pertwee R.G., Di Marzo V. Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett. 2000;483:52–56. doi: 10.1016/s0014-5793(00)02082-2. [DOI] [PubMed] [Google Scholar]
- Di Marzo V., et al. Highly selective CB(1) cannabinoid receptor ligands and novel CB(1)/VR(1) vanilloid receptor “hybrid” ligands. Biochem. Biophys. Res. Commun. 2001;281:444–451. doi: 10.1006/bbrc.2001.4354. [DOI] [PubMed] [Google Scholar]
- López-Rodríguez M.L., et al. Design, synthesis and biological evaluation of novel arachidonic acid derivatives as highly potent and selective endocannabinoid transporter inhibitors. J. Med. Chem. 2001;44:4505–4508. doi: 10.1021/jm015545y. [DOI] [PubMed] [Google Scholar]
- Ortar G., Ligresti A., De Petrocellis L., Morera E., Di Marzo V. Novel selective and metabolically stable inhibitors of anandamide cellular uptake. Biochem. Pharmacol. 2003;65:1473–1481. doi: 10.1016/s0006-2952(03)00109-6. [DOI] [PubMed] [Google Scholar]
- Kathuria S., et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Chicca A., Marazzi J., Nicolussi S., Gertsch J. Evidence for bidirectional endocannabinoid transport across cell membranes. J. Biol. Chem. 2012;287:34660–34682. doi: 10.1074/jbc.M112.373241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K., Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc. Natl. Acad. Sci. USA. 1992;89:3825–3829. doi: 10.1073/pnas.89.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield M.P., Brown D.A. Cannabinoid receptor agonists inhibit Ca current in NG108-15 neuroblastoma cells via a pertussis toxin-sensitive mechanism. Br. J. Pharmacol. 1992;106:231–232. doi: 10.1111/j.1476-5381.1992.tb14321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twitchell W., Brown S., Mackie K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J. Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- Hoffman A.F., Lupica C.R. Mechanisms of cannabinoid inhibition of GABA(A) synaptic transmission in the hippocampus. J. Neurosci. 2000;20:2470–2479. doi: 10.1523/JNEUROSCI.20-07-02470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman G., Lovinger D.M. CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J. Neurophysiol. 2001;85:468–471. doi: 10.1152/jn.2001.85.1.468. [DOI] [PubMed] [Google Scholar]
- Huang C.C., Lo S.W., Hsu K.S. Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. J. Physiol. (Lond.) 2001;532:731–748. doi: 10.1111/j.1469-7793.2001.0731e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg J.E., Kinsey S.G., Lichtman A.H. Targeting fatty acid amide hydrolase (FAAH) to treat pain and inflammation. AAPS J. 2009;11:39–44. doi: 10.1208/s12248-008-9075-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movahed P., et al. Endogenous unsaturated C18 N-acylethanolamines are vanilloid receptor (TRPV1) agonists. J. Biol. Chem. 2005;280:38496–38504. doi: 10.1074/jbc.M507429200. [DOI] [PubMed] [Google Scholar]
- Zygmunt P.M., et al. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]
- Herkenham M., et al. Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. USA. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christison R. Pharmacopoeias of Great Britain (and the United States) Lea and Blanchard; Philadelphia, PA; 1848. [Google Scholar]
- Blankman J.L., Cravatt B.F. Chemical probes of endocannabinoid metabolism. Pharmacol. Rev. 2013;65:849–871. doi: 10.1124/pr.112.006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K., et al. Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J. Pharmacol. Exp. Ther. 2011;338:114–124. doi: 10.1124/jpet.111.180257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.L., et al. Assessment of the pharmacology and tolerability of PF-04457845, an irreversible inhibitor of fatty acid amide hydrolase-1, in healthy subjects. Br. J. Clin. Pharmacol. 2012;73:706–716. doi: 10.1111/j.1365-2125.2011.04137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan P.A., et al. Model-based drug development: a rational approach to efficiently accelerate drug development. Clin. Pharmacol. Ther. 2013;93:502–514. doi: 10.1038/clpt.2013.54. [DOI] [PubMed] [Google Scholar]
- Hu D., Yuan J.M. Time-dependent sensitivity analysis of biological networks: coupled MAPK and PI3K signal transduction pathways. J. Phys. Chem. A. 2006;110:5361–5370. doi: 10.1021/jp0561975. [DOI] [PubMed] [Google Scholar]
- Whiteside G.T., Adedoyin A., Leventhal L. Predictive validity of animal pain models? A comparison of the pharmacokinetic-pharmacodynamic relationship for pain drugs in rats and humans. Neuropharmacology. 2008;54:767–775. doi: 10.1016/j.neuropharm.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Okine B.N., et al. Lack of effect of chronic pre-treatment with the FAAH inhibitor URB597 on inflammatory pain behaviour: evidence for plastic changes in the endocannabinoid system. Br. J. Pharmacol. 2012;167:627–640. doi: 10.1111/j.1476-5381.2012.02028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks J.W., et al. Synthesis and preclinical evaluation of [11C-carbonyl]PF-04457845 for neuroimaging of fatty acid amide hydrolase. Nucl. Med. Biol. 2013;40:740–746. doi: 10.1016/j.nucmedbio.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi K., Sun Y.X., Okamoto Y., Araki N., Tonai T., Ueda N. Molecular characterization of N-acylethanolamine-hydrolyzing acid amidase, a novel member of the choloylglycine hydrolase family with structural and functional similarity to acid ceramidase. J. Biol. Chem. 2005;280:11082–11092. doi: 10.1074/jbc.M413473200. [DOI] [PubMed] [Google Scholar]
- Piscitelli F., Di Marzo V. “Redundancy” of endocannabinoid inactivation: new challenges and opportunities for pain control. ACS Chem. Neurosci. 2012;3:356–363. doi: 10.1021/cn300015x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPartland J.M., Glass M., Pertwee R.G. Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: interspecies differences. Br. J. Pharmacol. 2007;152:583–593. doi: 10.1038/sj.bjp.0707399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vauquelin G., Packeu A. Ligands, their receptors and. plasma membranes. Mol. Cell. Endocrinol. 2009;311:1–10. doi: 10.1016/j.mce.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns H.D., et al. [18F]MK-9470, a positron emission tomography (PET) tracer for in vivo human PET brain imaging of the cannabinoid-1 receptor. Proc. Natl. Acad. Sci. USA. 2007;104:9800–9805. doi: 10.1073/pnas.0703472104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savinainen J.R., Saario S.M., Laitinen J.T. The serine hydrolases MAGL, ABHD6 and ABHD12 as guardians of 2-arachidonoylglycerol signalling through cannabinoid receptors. Acta Physiol. (Oxf) 2012;204:267–276. doi: 10.1111/j.1748-1716.2011.02280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowska-Jankowska B.M., et al. In vivo characterization of the highly selective monoacylglycerol lipase inhibitor KML29: Antinociceptive activity without cannabimimetic side effects. Br. J. Pharmacol. 2013. [DOI] [PMC free article] [PubMed]
- Huggins J.P., Smart T.S., Langman S., Taylor L., Young T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain. 2012;153:1837–1846. doi: 10.1016/j.pain.2012.04.020. [DOI] [PubMed] [Google Scholar]
- Morgan P., et al. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival. Drug Discov. Today. 2012;17:419–424. doi: 10.1016/j.drudis.2011.12.020. [DOI] [PubMed] [Google Scholar]
- Benson N., Cucurull-Sanchez L., Demin O., Smirnov S., van der Graaf P. Reducing systems biology to practice in pharmaceutical company research; selected case studies. Adv. Exp. Med. Biol. 2012;736:607–615. doi: 10.1007/978-1-4419-7210-1_36. [DOI] [PubMed] [Google Scholar]
- Hendriks B. Negative modeling results: a dime a dozen or a stepping stone to scientific discovery. CPT. Pharmacometrics Syst. Pharmacol. 2013;2:e48. doi: 10.1038/psp.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlowski L.E., Jain R.K. Physiologically based pharmacokinetic modeling: principles and applications. J. Pharm. Sci. 1983;72:1103–1127. doi: 10.1002/jps.2600721003. [DOI] [PubMed] [Google Scholar]
- Peters S.A., Hultin L. Early identification of drug-induced impairment of gastric emptying through physiologically based pharmacokinetic (PBPK) simulation of plasma concentration-time profiles in rat. J. Pharmacokinet. Pharmacodyn. 2008;35:1–30. doi: 10.1007/s10928-007-9073-1. [DOI] [PubMed] [Google Scholar]
- Demin O., Goryanin I. Kinetic Modelling in Systems Biology. Taylor & Francis; London, UK; 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.