

Abstract
In this study, we have developed a multiscale systems model of interleukin (IL)-6–mediated immune regulation in Crohn's disease, by integrating intracellular signaling with organ-level dynamics of pharmacological markers underlying the disease. This model was linked to a general pharmacokinetic model for therapeutic monoclonal antibodies and used to comparatively study various biotherapeutic strategies targeting IL-6–mediated signaling in Crohn's disease. Our work illustrates techniques to develop mechanistic models of disease biology to study drug–system interaction. Despite a sparse training data set, predictions of the model were qualitatively validated by clinical biomarker data from a pilot trial with tocilizumab. Model-based analysis suggests that strategies targeting IL-6, IL-6Rα, or the IL-6/sIL-6Rα complex are less effective at suppressing pharmacological markers of Crohn's than dual targeting the IL-6/sIL-6Rα complex in addition to IL-6 or IL-6Rα. The potential value of multiscale system pharmacology modeling in drug discovery and development is also discussed.
Introduction
Among the factors impeding discovery of new drugs is a lack of understanding of the complex biological systems underlying diseases and systemic repercussions of drug candidate–target interactions.1 Model-based drug discovery and development has been increasingly utilized to address this challenge2; in recent years, the multidisciplinary approach of systems pharmacology that combines systems biology with pharmacokinetic/pharmacodynamic (PK/PD) modeling principles has progressively drawn attention in the pharmaceutical industry.3 Systems pharmacology holds promise as a discipline that can help develop holistic understanding of interactions between drugs and complex biological systems underlying disease and has been termed the “next iteration” of model-based drug discovery and development.4,5,6 In this study, we have developed a multiscale systems model of Crohn's disease to investigate candidate therapeutic strategies and to demonstrate the potential of quantitative systems pharmacology in drug discovery and development.
Crohn's disease is an inflammatory bowel disease that is thought to be caused by a combination of genetic and environmental factors,7 resulting in a proinflammatory environment in the mucosal layer of the gastrointestinal (GI) tract.8 Along with several other cytokines that are perturbed in Crohn's disease, interleukin (IL)-6 has been shown to be increased in intestinal mucosa and in peripheral blood.9,10,11,12 IL-6 signaling is thought to be an important player in Crohn's disease contributing to enhanced T-cell survival and apoptosis resistance in the lamina propria along with elevated chemokine secretion.13
IL-6 signaling can occur via membrane-bound IL-6 receptor (IL-6Rα)–mediated classical pathway as well as soluble IL-6 receptor (sIL-6Rα)–mediated trans-signaling pathway (Figure 1a). The membrane-bound IL-6/IL-6Rα dimer or the circulating preformed complex of IL-6/sIL-6Rα can recruit a membrane-bound gp130 coreceptor, forming a heterotrimer which subsequently dimerizes, resulting in an active hexameric IL-6R receptor complex.14,15,16 This complex initiates phosphorylation of gp130-bound Janus kinase (Jak) family proteins and subsequent signal transducer and activator of transcription 3 (STAT3) phosphorylation.17 Trans-signaling may be important in disease because transmembrane IL-6Rα is expressed only in a small number of cell types, e.g., hepatocytes and a subset of leukocytes, whereas gp130 is almost ubiquitously expressed.18 Alternative gene splicing and shedding of transmembrane IL-6Rα are thought to be responsible for production of sIL-6Rα.19 IL-6Rα shedding was suggested to be accelerated by proteins including C-reactive protein (CRP).20 The coreceptor gp130 can also exist in a soluble form (sgp130), acting as a natural inhibitor of trans-signaling by sequestering the IL-6/sIL-6Rα complex16 (Figure 1a). The downstream effects of IL-6 signaling are multifarious, ranging from differentiation of lymphocytes to expression of acute-phase proteins such as CRP.18 The reported increase in serum concentrations of CRP,21 sIL-6Rα,22 and pSTAT3 in colon biopsies23 of Crohn's disease patients may be driven by increased IL-6, acting through the complex network of IL-6 signaling (Figure 1c).
Figure 1.

Schematic of multiscale model construction. (a) IL-6 signals via classical and trans-signaling pathways. Both routes activate the same downstream signaling. IL-6/sIL-6Rα can be sequestered by sgp130. (b) The model of IL-6 signaling was reduced using a rationale-based approach (see Supplementary Text S1). (c) The signaling model was embedded in compartments representing liver and GI tract. The organs were connected by a third compartment representing circulation through which they exchange serum-soluble molecules. All molecules had basal production and decay terms; sIL-6Rα was also produced by CRP-mediated shedding. CRP, C-reactive protein; GI, gastrointestinal; IL, interleukin.
A pilot clinical trial with anti–IL-6R antibody, tocilizumab, has shown positive clinical activity in Crohn's disease,24 suggesting inhibition of IL-6 signaling as a therapeutic strategy. Several other strategies that modulate IL-6 signaling have been proposed, which include targeting IL-625 or IL-6R,24 using sgp130 as a natural antagonist of IL-6 trans-signaling,15,16 or simultaneously inhibiting classical (target IL-6 or IL-6R) and trans (target IL-6/sIL-6R complex) IL-6 signaling.15 Antibodies against these targets in Crohn's disease are in early clinical development; however, clinical biomarker or efficacy data are currently unavailable.
Systems biology models of IL-6–mediated immune signaling have been published previously,26,27,28,29 but these are single-scale models at the cellular level, and their translation to clinical end points has not been established. At a very different level of detail, PK/PD relationships of Ab–receptor interaction and biomarker or efficacy modulations are characterized in vitro and in vivo in preclinical and clinical studies. However, the PK/PD understanding is often limited by models based on limited data and lack of incorporation of biology. With various new IL-6–related therapeutic agents in the pipeline, we have developed a multiscale system model that integrates current knowledge about the biology of IL-6–mediated immune response in Crohn's and used it for mechanistic assessment and comparison of several proposed therapeutic strategies. We have further used the model to explore the value of quantitative system pharmacology modeling in discovery and development, particularly in the areas of target selection, candidate optimization, and dose selection.
Results
Construction of the multiscale model of IL-6 signaling in Crohn's disease
The multiscale model comprises three overlapping structural modules spanning spatial scales from cellular to organ levels. The first module describes events at the cellular level and consists of a reduced model of IL-6–mediated signal transduction. The second module is made up of target organs relevant in Crohn's disease, and the first module (the cell signaling model) is embedded within these organs. The third and final module is a general PK model for monoclonal antibodies, including specific target binding that is linked to the first two modules to study the action of drug on the system. Each of these modules is described below.
Module 1: IL-6–mediated cellular signal transduction. Detailed models of IL–mediated Janus kinase-Signal Transducer and Activator of Transcription (Jak-STAT) signaling, including IL-6/STAT3 signaling, have been described previously.26,30,31,32 Starting from the canonical structure of these models (schematic diagram in Figure 1a), we reduced the signaling network using a rationale-based approach to obtain a more parsimonious description while retaining the essential features of the IL-6 signaling pathway (Figure 1b). Simplifying assumptions used to reduce the model are listed in Supplementary Text S1.
Module 2: integrating cell signaling with organ-level kinetics. Instead of constructing a large-scale, physiologically based model accounting for all major organs in the body, we chose a parsimonious description by identifying the two most important target organs in Crohn's disease and modeling them as separate, communicating compartments (Figure 1c). The first of these compartments represents the GI tract, which is the organ affected directly by Crohn's disease.8,13 The second compartment models the liver, which was included for its prominent role in the upregulation of acute-phase proteins associated with Crohn's disease, such as CRP.27 We further assumed that each of these organs was separately made up of homogeneous cell populations with identical signaling dynamics with minor differences (see Supplementary Text S1 and S2). Assuming that the IL-6 signaling network topology and reaction rate parameters remain conserved across different organs, we embedded the IL-6 signaling module into the cells of these organs. The organs were allowed to exchange soluble components through blood serum, which was modeled as a separate compartment representing the circulation. Briefly, starting from a single cell model, we scaled up to organs, modeling them as compartmentalized collections of cells and connected the organs through blood circulation (Figure 1c). Assumptions accompanying this module are listed in Supplementary Text S1.
Module 3: PK of monoclonal antibodies. To study the effects of different therapeutic agents, we developed a module to incorporate drug PK. The antibody PK model comprises target-binding, dynamic exchange of free and bound drug between organ compartments, and clearance of free as well as bound antibody (Supplementary Figure S1). This model of drug PK describes the equilibration between multiple compartments incorporating both linear and target-mediated disposition of the antibody.
Parameter estimation
We exploited the modular structure of our model to break down the parameter estimation problem into a two-step process. In the first step, the model was divided into structural modules, and parameters were either assigned values available from the literature or estimated for each module in isolation using any published data we could find (example in Figure 2a). The model was reassembled by putting the modules together, and the parameters were assigned values obtained from the first step as initial estimates. It was expected that the estimates obtained from isolated modules would no longer be accurate with complex interactions between the modules. Accordingly, in the second step, the initial parameter estimates were further fine-tuned to fit the system-level behavior of the whole model to available data.
Figure 2.

Parameter estimation. (a) As part of fitting individual modules to available data, the dynamics of the cell-surface reactions of IL-6 signaling were optimized by fitting to in vitro data from HepG2 cells (see Supplementary Figure S7). (b) In the second step of parameter optimization, the complete model, except drug PK, was assembled. Parameter estimates from the first step were refined to fit steady-state outputs to values reported in the literature (Table 1). The y-axis shows ratios of fitted values to representative values from the literature. Healthy subject model (Healthy) and Crohn's disease model (CD) were fitted to get the ratio on y-axis close to 1 (shaded area shows a 10-fold range on either side of measured value). (c) PK profile of anti–IL-6 antibody at three doses (solid lines) matched well with the median behavior expected of monoclonal antibodies28 (dashed lines). (d) PK parameters were optimized so that antibody concentration in liver at steady state was nearly 100% of serum concentration and that in the gut was 50%. PK profiles at 100 mg drug dose are shown. IL, interleukin; PK, pharmacokinetics.
When optimizing the parameters in step 2, we hypothesized that observed differences in concentrations of molecular markers between healthy subjects and Crohn's disease patients are a systemic outcome of the differences in IL-6 concentrations. Therefore, although the model does not account for the mechanism of increased IL-6 in Crohn's patients, it explains differences in other molecular markers as a consequence of IL-6 signaling. Given the large variability in measurements of biomarkers in human subjects and the sparsity of data, we chose parameter values that produced reasonable fits across all data points rather than parameter sets that resulted in over fitting of some data points at the cost of others (Supplementary Text S3). Using baseline concentrations of a set of biomarkers available from healthy subjects and Crohn's disease patients, the model was optimized to fit its steady-state values to measured typical values, and the ratio of fitted value to measured value was close to 1 for all molecular markers (Table 1 and Figure 2b).
Table 1. Concentrations of biomarkers used to optimize model parameters.
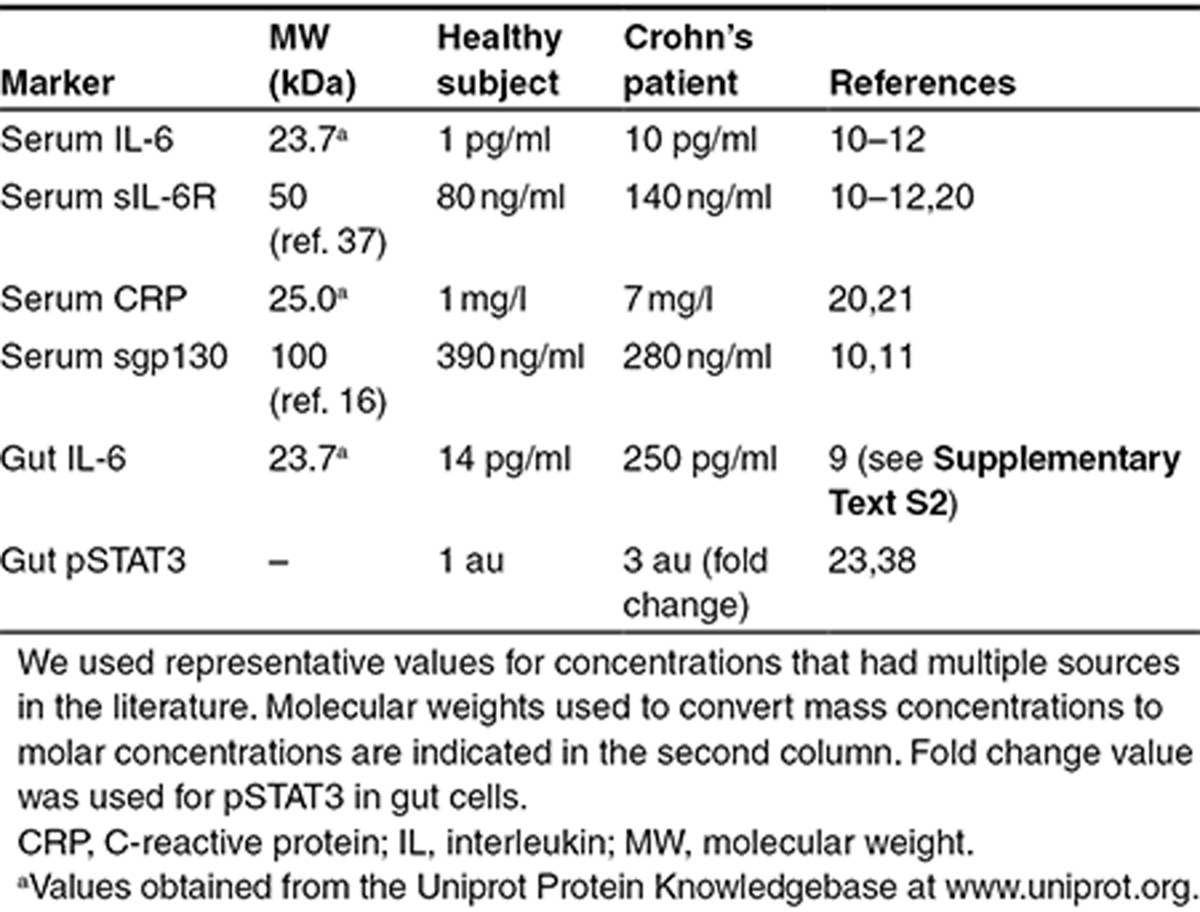
Since our study was largely focused on assessing the effects of treatment with monoclonal antibodies, we calibrated the drug PK parameters to fit the serum concentration of drug to median PK of monoclonal antibodies as reported by Dirks and Meibohm28 (Figure 2c,d).
Model validation
Data from a previously published pilot trial of a humanized anti–IL-6Rα monoclonal antibody, tocilizumab, in Crohn's subjects24 was used to validate the model. Using the optimized model, we simulated the pharmacological effects of treatment with placebo or 8 mg/kg anti–IL-6Rα antibody at intervals of 4 or 2 weeks (Figure 3a). The simulated anti–IL-6Rα antibody was assigned a Kd of 1 nmol/l as an order of magnitude estimate based on target-binding studies on tocilizumab.29 The PK profile of anti–IL-6Rα antibody in our simulation was in agreement with the reported PK profile of tocilizumab24 (Supplementary Figure S2). Comparing the simulation results with the mean CRP levels measured by Ito et al. under identical dose, we found that the model was successful at capturing the trend shown by the clinical study even though it could not reproduce the data quantitatively (Figure 3b). Similar to the clinical data, the model showed that an every 4-week intravenous (i.v.) administration of tocilizumab transiently reduced serum CRP, only to be restored to baseline level by the end of each 4-week cycle. For an every 2-week i.v. dosing schedule, both the model and clinical data showed a sustained reduction in serum CRP levels. However, under both dose schedules, the model underestimated the reduction in serum CRP when compared with the clinical data (compare Figure 3a and 3b).
Figure 3.

Model validation. Effect of treatment by anti–IL-6Rα antibody on serum CRP concentration was used to validate the trained model by comparing it against data from a pilot clinical trial with tocilizumab.24 Data in both plots are normalized so that serum CRP at time 0 is 100 across treatment conditions. (a,b) Simulation with placebo or 8 mg/kg intravenous dose of anti–IL-6Rα antibody at every 4-week (Q4W) and 2-week (Q2W) intervals matches qualitatively with clinical data. y-axis scale is not identical in a and b. CRP, C-reactive protein; IL, interleukin.
Dose–response characteristics of anti–IL-6 and anti–IL-6R antibodies are distinct
Using the optimized model, we simulated the effects of anti–IL-6 and anti–IL-6Rα on PD biomarkers including serum CRP, pSTAT3 activity in the GI tract, and IL-6 levels in the GI tract and serum after every 4-week i.v. dosing for a 12-week period. To examine the role of drug dose and antibody target–binding affinity in determining drug efficacy, we ran multiple simulations while systematically varying the dose and binding affinities of the antibodies. When administered at sufficiently high doses, both anti–IL-6 and anti–IL-6Rα (assumed to target both transmembrane and soluble IL-6Rα) treatments lead to the suppression of serum CRP and gut pSTAT3 in a time-dependent manner (Figure 4a,d, respectively). Measured in terms of CRP suppression at the end of the 12-week treatment, anti–IL-6 antibody had a graded dose response tending toward saturation at high dose (Figure 4b). Anti–IL-6Rα antibody, on the other hand, remained ineffective over a large dose range but suddenly transitioned to high efficacy at a certain dose threshold (Figure 4e). We found that this qualitative difference of graded vs. switch-like response was not CRP specific and was maintained across multiple biomarkers (Supplementary Figure S3). Furthermore, both antibodies showed increased effectiveness with increasing target-binding affinity.
Figure 4.

Response to treatment with anti–IL-6 or anti–IL-6Rα antibody. All panels show results from simulation of intravenous dosing at 4-week intervals for a 12-week period. Anti–IL-6Rα targeted both transmembrane and soluble IL-6Rα. Time courses of a set of biomarkers after treatment with (a) 300 mg anti–IL-6 antibody or (d) anti–IL-6Rα antibody. Dose–response of (b) anti–IL-6 and (c) anti–IL-6Rα antibodies over varying target-binding affinities. The plotted values represent percent decrease in serum CRP from the baseline level after a 12-week treatment. Concentration of free drug in blood serum is maintained at higher levels for (c) anti–IL-6 at 200 and 500 mg doses but (f) anti–IL-6Rα is depleted in about 2 weeks at 200 mg dose. Higher dose of 500 mg helps maintain free anti–IL-6Rα at notable concentration. CRP, C-reactive protein; IL, interleukin.
To explain this difference in response to the two antibodies, we examined their PK and found that despite identical parameters of distribution and linear clearance, the two types of antibodies could have very different PK time profiles depending on dose. For example, at 200 mg dose administered every 4 weeks, the anti–IL-6 antibody concentration was sustained at high enough levels to suppress IL-6 signaling (Figure 4c), whereas the anti–IL-6Rα antibody was severely depleted in 2 weeks (Figure 4f). Increasing the dose to 500 mg sustained anti–IL-6Rα over the dose cycle in a manner similar to anti–IL-6, resulting in higher efficacy (Figure 4c,f). Increasing the dosing frequency also resulted in greater efficacy at lower doses of anti–IL-6Rα (Supplementary Figure S4).
Targeting the IL-6/sIL-6R complex in addition to free IL-6 or IL-6R as a therapeutic approach
In all the simulations presented so far, it was assumed that the anti–IL-6 and anti–IL-6Rα antibodies bind only to free IL-6 and IL-6Rα, respectively. Given the hypothesized importance of the role of IL-6/sIL-6Rα complex in Crohn's disease,15 we ran simulations to assess the effects of targeting this complex in addition to free IL-6 or IL-6Rα.
When anti–IL-6 was allowed to bind to IL-6 and IL-6/sIL-6Rα complex with the same affinity, the dose–response characteristics of the antibody showed improved pharmacological efficacy over a range of doses and binding affinities in a dose-dependent manner (Figure 5a). However, similar improvement was not observed in the case of anti–IL-6Rα treatment, and the dose response remained largely unchanged (Figure 5b).
Figure 5.

Changes in dose response on targeting IL-6/sIL-6Rα complex along with IL-6 or IL-6Rα are shown. The data points show simulated decrease in serum CRP as percent of the baseline value after a 12-week treatment with intravenous drug administration at 4-week intervals. (a) Compared with the case when only IL-6 was targeted (solid lines), the efficacy of the drug was predicted to be higher when both IL-6 and IL-6/sIL-6Rα were targeted (dashed lines). (b) Targeting the complex (dashed lines) did not cause any significant improvement in CRP suppression compared with the case when only IL-6Rα was targeted (solid lines). CRP, C-reactive protein; IL, interleukin.
Evaluating alternative therapeutic approaches using the model
We performed global sensitivity analysis on the optimized model to assess the sensitivity of steady-state values of a panel of biomarkers to parameters of the model (see Methods section). The parameters to which the output was found to be least and most sensitive are shown in Figure 6a (see Supplementary Figure S5 for a complete list). The parameters that most strongly influenced the output biomarkers were rate of synthesis of IL-6 in the GI tract, rate of degradation of soluble receptor, parameters associated with IL-6–mediated production of CRP and CRP degradation. The production and degradation rates of sgp130 were among the least influential parameters. The sensitivity analysis suggests that while IL-6 and sIL-6Rα are among the best targets, sgp130-based therapies may not be as effective.
Figure 6.

Evaluating other targeting strategies. (a) Sensitivity analysis was performed on the model to assess which parameters had the greatest influence on the steady-state values of a set of output biomarkers. Parameters to which the output was least and most sensitive are shown. (b) The response to sgp130Fc in terms of serum CRP suppression from baseline increased linearly with dose except for the Q1W schedule where it showed a tendency to saturate at higher doses. CRP, C-reactive protein; Q1W, every 1 week.
A fusion protein combining the extracellular portion of gp130 with the Fc region of human IgG1 (sgp130Fc) has been shown to inhibit IL-6 trans-signaling in cultured cells.16 To study the potential of sgp130 as a therapeutic approach in Crohn's disease, we simulated i.v. dosing of sgp130Fc at 4-week intervals. We assumed that sgp130Fc binds the IL-6/sIL-6Rα complex with the same affinity as natural sgp130 and that the PK of sgp130Fc is identical to that of a typical monoclonal antibody with linear clearance. Dose–response curves in terms of CRP suppression for several dose schedules are shown in Figure 6b. Similar dose–response curves were obtained for pSTAT3 suppression in gut (Supplementary Figure S6). In agreement with the sensitivity analysis which suggested relative insensitivity of the output to sgp130, simulations showed sgp130Fc to be effective only at very high and frequent doses.
Discussion
We have constructed a system-level, multiscale, deterministic model of IL-6 signaling in Crohn's disease that elucidates how ideas from traditional pharmacometrics can be combined with strategies from systems biology to design disease models in systems pharmacology. Our work advances existing systems pharmacology modeling methodologies and illustrates model development strategies with particular focus on integrating the role of cell signaling networks in disease. Although highly detailed quantitative models of system dynamics are desirable for several reasons,33 the construction of such models is often hindered by the sparsity of pertinent molecular-level kinetic measurements. Therefore, a balance needs to be struck between the level of detail in the model against the quality and quantity of data available to reliably incorporate such detail. To attain this balance, whenever possible, we have chosen to favor parsimony in describing the biological processes in our model. Notably, despite the simplifying assumptions and limited amount of data available to train the model, we were able to qualitatively validate the model against an independent clinical data set (Figure 3). Particularly, even though the CRP time course was not exactly replicated quantitatively by the model in the validation step, the model was successful at qualitatively predicting the long-term behavior of CRP following drug treatment in the typical Crohn's disease patient showing the model's potential as a tool to study the effects of perturbation through various treatments.
We compared four different strategies that have been proposed to treat Crohn's disease by inhibition of IL-6 signaling through targeting IL-6, IL-6Rα, IL-6/sIL-6Rα in addition to IL-6 or IL-6Rα, or using sgp130Fc as an antagonist of IL-6 trans-signaling. The model predicts that two of these strategies, targeting IL-6 or IL-6Rα, can effectively suppress markers of inflammation (CRP) and IL-6 signaling in target organs. Using sgp130Fc to exploit the natural ability of sgp130 to inhibit IL-6 signaling was predicted to be effective only at very high and frequent doses. Simulated dose–response curves as well as sensitivity analysis done on the model suggest that targeting IL-6 requires lower and less frequent dosing as compared with anti–IL-6Rα to achieve similar reduction of the systemic effects of IL-6 signaling. This statement must be qualified, however, by our observation that the model underestimated the effect of anti–IL-6Rα treatment when compared with clinical data (Figure 3). Nonetheless, the dose–response characteristics of anti–IL-6Rα are clearly distinct from those of anti–IL-6 (Figure 4b,e). This difference arises due to the immense disparity between the levels of free IL-6 (order of 10 pg/ml ≈ 10−4 nmol/l)12,13 and free sIL-6Rα (order of 100 ng/ml ≈ 1 nmol/l) in circulation.22 At doses below a certain threshold, the antibody becomes the limiting factor in the anti–IL-6Rα/sIL-6Rα interaction as most of the antibody is sequestered by sIL-6Rα (Figure 4f, 200 mg curve). Once this threshold is crossed, the antibody overwhelms sIL-6Rα production and accumulates in the system in sufficient amount (Figure 4f, 500 mg curve) to suppress IL-6 signaling, resulting in the switch-like dose–response curve seen in Figure 4e.
The PK profiles of anti–IL-6 and anti–IL-6Rα antibodies are different in our model (compare Figure 4c and 4f) due to differences in the kinetics of their interactions with their targets. Nonlinear clearance very similar to that predicted by the model for the anti–IL-6Rα antibody has been observed for tocilizumab at therapeutic doses in clinical studies in rheumatoid arthritis.34,35 This provides further validation to our modeling approach and shows that a well-constructed model of the underlying biological system can automatically accommodate target-mediated disposition of the antibody, reducing the need for case-by-case optimization of drug PK.
The IL-6/sIL-6Rα complex is an important intermediate in IL-6 signaling and has been proposed as a therapeutic target.15 We tested the relative gain in the efficacy of biomarker modulation from combined targeting of the complex in addition to IL-6 or IL-6Rα. Greater dose-dependent modulation of the immunological biomarkers was suggested in the case of anti–IL-6 but not for anti–IL-6Rα. As argued above, at small drug doses, substantial concentration of sIL-6Rα remains in circulation and any IL-6/sIL-6Rα complex blocked by the antibody is quickly replenished, negating the effect of targeting the complex. At larger drug doses when sIL-6Rα is effectively suppressed by the antibody, IL-6/sIL-6Rα complex is naturally depleted because reduced sIL-6Rα concentration drives the IL-6/sIL-6Rα binding equilibrium toward the unbound state. Taken together, at both high and low concentrations, the effect of targeting the complex does not add much to the efficacy of IL-6Rα targeting.
Targeting IL-6/sIL-6Rα alone to inhibit trans-signaling by sgp130Fc was found to be effective only at very high and frequent doses (Figure 6b). This result was supported by sensitivity analysis using the model, which indicated that altering the levels of sgp130 has very little effect on steady-state values of a set of output parameters. This is explained by the high baseline level of sgp130 (≈ 300 ng/ml or 3 nmol/l) as compared with free IL-6 (≈ 10 pg/ml or 4 × 10−4 nmol/l).10,11,12 This means that sgp130 is basally present in large excess when compared with the IL-6/sIL-6Rα complex, which is limited by the low IL-6 concentration. The implication of this concentration difference is that small perturbations in sgp130 level have minimal effect on system dynamics, and any observable effects require large changes in sgp130.
One of the limitations of the current model is that it underestimates the CRP response under anti–IL-6R treatment. The reasons for this are twofold. First, there was limited data available to both train and validate the model, meaning that estimates of some parameters could be inaccurate. Although it may ultimately be possible to simulate first use of a drug in humans in silico, achieving this routinely and with confidence requires more two-way flow of information from clinical trial back to the model for model training, and from model-generated hypothesis to the clinic for testing. Currently, we do not have sufficient dose–response clinical data available in the literature to warrant model refinement based on the data. The model will be refined to incorporate emerging data from ongoing anti-IL-6–related clinical studies to improve its predictive power. Second, the structure of our model is limited by our biological knowledge about the system, which leaves room for improvement. For example, while we have singled out IL-6 signaling to study the Crohn's disease system, other cytokines such as IL-10 and tumor necrosis factor-α are known to be important in this disease. Future models could also include distinct subpopulations of cells, such as T-lymphocyte subsets in the GI tract which differentially influence the disease state.
While admitting scope for improvement, the present work demonstrates the potential of the systems pharmacology approach in drug discovery and development. As demonstrated by model-based comparison of the competing IL-6 signaling–related therapeutic strategies, the system model can be tremendously valuable at the drug discovery stage. Whereas intuition, simplified experimental systems, and single-scale computational models are limited in their scope, a multiscale systems pharmacology model is better able to predict outcomes arising from system-level interactions and combination treatment, and has enormous potential in facilitating target selection and candidate optimization in drug discovery. The applications of systems pharmacology are not limited to the discovery stage. Phase I studies in patients are particularly relevant to the development of a quantitative systems pharmacology model, because it is at this stage that the human PK and PD information of the new therapeutic agent becomes available. Once a system model structure is in place, integration of phase I biomarker data can facilitate model refinement, resulting in more quantitative prediction of clinical efficacy and/or safety responses based on correlation with biomarker response. The refined model can aid optimal proof-of-concept phase II design and support an optimal dosing regimen. It is at this stage that one can truly test the correct selection of targets, drugs, dosing, and therapeutic strategy. Model development during phase II may be used to address different questions related to the understanding of drug effect for both safety and efficacy. Overall continuous integration of system-level models with data from multiple sources and various development stages would permit more complete exploration and interpretation of clinical response data for new and existing therapeutic agents and could enhance decision making throughout the development process.
Methods
Model implementation and parameter optimization. All modeling, simulation and analysis was done in Matlab (R2012b). The SimBiology toolbox of Matlab was used to implement the model (see Supplementary file 1 CD model and example Matlab script to run the model in Supplementary file 2 Sample Matlab code). To facilitate platform-independent exchange, Systems Biology Markup Language (SBML) versions of the models are also provided (Supplementary Files 3–6). Enzymatic reactions in the signaling pathway and pSTAT3-induced protein expression were modeled using Michaelis–Menten kinetics. All other events were modeled using mass action kinetics. All first-order rates have units of 1/hr, second-order rates are in l/(nmol·hr), and all concentrations are defined in nmol/l. All simulations were performed by first running the model to steady state and then simulating the effects of exogenous perturbations. Parameter estimation was performed using a combination of fitting by hand and functions from the optimization toolbox of Matlab. Further details of the fitting process are described in Supplementary Text S3.
Global sensitivity analysis. Partial rank correlation coefficients between the steady-state output and input parameters were calculated (Supplementary Text S4).36 To get a single sensitivity measure per parameter, the partial rank correlation coefficientss for each parameter were summed over the output species and divided by a constant to scale the highest sensitivity to 1 (see Supplementary Figure S5).
Author Contributions
C.L. and G.D. wrote the manuscript. C.L. and G.D. designed the research. C.L., G.D., L.F., M.H., J.H., and A.H. performed the research. G.D. analyzed the data.
Conflict of Interest
The authors declared no conflict of interest.
Study Highlights

Supplementary Material
References
- Butcher E.C., Berg E.L., Kunkel E.J. Systems biology in drug discovery. Nat. Biotechnol. 2004;22:1253–1259. doi: 10.1038/nbt1017. [DOI] [PubMed] [Google Scholar]
- Derendorf H., et al. Pharmacokinetic/pharmacodynamic modeling in drug research and development. J. Clin. Pharmacol. 2000;40:1399–1418. [PubMed] [Google Scholar]
- van der Graaf P.H., Benson N. Systems pharmacology: bridging systems biology and pharmacokinetics-pharmacodynamics (PKPD) in drug discovery and development. Pharm. Res. 2011;28:1460–1464. doi: 10.1007/s11095-011-0467-9. [DOI] [PubMed] [Google Scholar]
- Iyengar R., Zhao S., Chung S.W., Mager D.E., Gallo J.M. Merging systems biology with pharmacodynamics. Sci. Transl. Med. 2012;4:126ps7. doi: 10.1126/scitranslmed.3003563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano H. Computational systems biology. Nature. 2002;420:206–210. doi: 10.1038/nature01254. [DOI] [PubMed] [Google Scholar]
- Allerheiligen S.R. Next-generation model-based drug discovery and development: quantitative and systems pharmacology. Clin. Pharmacol. Ther. 2010;88:135–137. doi: 10.1038/clpt.2010.81. [DOI] [PubMed] [Google Scholar]
- Baumgart D.C., Carding S.R. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–1640. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- Baumgart D.C., Sandborn W.J. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet. 2007;369:1641–1657. doi: 10.1016/S0140-6736(07)60751-X. [DOI] [PubMed] [Google Scholar]
- Mitsuyama K., et al. Colonic mucosal interleukin-6 in inflammatory bowel disease. Digestion. 1991;50:104–111. doi: 10.1159/000200747. [DOI] [PubMed] [Google Scholar]
- Narazaki M., et al. Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood. 1993;82:1120–1126. [PubMed] [Google Scholar]
- Mahida Y.R., Kurlac L., Gallagher A., Hawkey C.J. High circulating concentrations of interleukin-6 in active Crohn's disease but not ulcerative colitis. Gut. 1991;32:1531–1534. doi: 10.1136/gut.32.12.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustot T., et al. Profile of soluble cytokine receptors in Crohn's disease. Gut. 2005;54:488–495. doi: 10.1136/gut.2004.043554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudter J., Neurath M.F. Il-6 signaling in inflammatory bowel disease: pathophysiological role and clinical relevance. Inflamm. Bowel Dis. 2007;13:1016–1023. doi: 10.1002/ibd.20148. [DOI] [PubMed] [Google Scholar]
- Boulanger M.J., Chow D.C., Brevnova E.E., Garcia K.C. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science. 2003;300:2101–2104. doi: 10.1126/science.1083901. [DOI] [PubMed] [Google Scholar]
- Rose-John S., Waetzig G.H., Scheller J., Grötzinger J., Seegert D. The IL-6/sIL-6R complex as a novel target for therapeutic approaches. Expert Opin. Ther. Targets. 2007;11:613–624. doi: 10.1517/14728222.11.5.613. [DOI] [PubMed] [Google Scholar]
- Jostock T., et al. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 2001;268:160–167. doi: 10.1046/j.1432-1327.2001.01867.x. [DOI] [PubMed] [Google Scholar]
- Heinrich P.C., Behrmann I., Haan S., Hermanns H.M., Müller-Newen G., Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggio M., Guralnik J.M., Longo D.L., Ferrucci L. Interleukin-6 in aging and chronic disease: a magnificent pathway. J. Gerontol. A Biol. Sci. Med. Sci. 2006;61:575–584. doi: 10.1093/gerona/61.6.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S.A., Horiuchi S., Topley N., Yamamoto N., Fuller G.M. The soluble interleukin 6 receptor: mechanisms of production and implications in disease. FASEB J. 2001;15:43–58. doi: 10.1096/fj.99-1003rev. [DOI] [PubMed] [Google Scholar]
- Jones S.A., Novick D., Horiuchi S., Yamamoto N., Szalai A.J., Fuller G.M. C-reactive protein: a physiological activator of interleukin 6 receptor shedding. J. Exp. Med. 1999;189:599–604. doi: 10.1084/jem.189.3.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber S., et al. CDP870 Crohn's Disease Study Group A randomized, placebo-controlled trial of certolizumab pegol (CDP870) for treatment of Crohn's disease. Gastroenterology. 2005;129:807–818. doi: 10.1053/j.gastro.2005.06.064. [DOI] [PubMed] [Google Scholar]
- Mitsuyama K., et al. Soluble interleukin-6 receptors in inflammatory bowel disease: relation to circulating interleukin-6. Gut. 1995;36:45–49. doi: 10.1136/gut.36.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey R., et al. Activation of an IL-6:STAT3-dependent transcriptome in pediatric-onset inflammatory bowel disease. Inflamm. Bowel Dis. 2008;14:446–457. doi: 10.1002/ibd.20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H., et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn's disease. Gastroenterology. 2004;126:989–996; discussion 947. doi: 10.1053/j.gastro.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Nishimoto N. Interleukin-6 as a therapeutic target in candidate inflammatory diseases. Clin. Pharmacol. Ther. 2010;87:483–487. doi: 10.1038/clpt.2009.313. [DOI] [PubMed] [Google Scholar]
- Moya C., Huang Z., Cheng P., Jayaraman A., Hahn J. Investigation of IL-6 and IL-10 signalling via mathematical modelling. IET Syst. Biol. 2011;5:15. doi: 10.1049/iet-syb.2009.0060. [DOI] [PubMed] [Google Scholar]
- Vermeire S., Van Assche G., Rutgeerts P. C-reactive protein as a marker for inflammatory bowel disease. Inflamm. Bowel Dis. 2004;10:661–665. doi: 10.1097/00054725-200409000-00026. [DOI] [PubMed] [Google Scholar]
- Dirks N.L., Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin. Pharmacokinet. 2010;49:633–659. doi: 10.2165/11535960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Mihara M., et al. Tocilizumab inhibits signal transduction mediated by both mIL-6R and sIL-6R, but not by the receptors of other members of IL-6 cytokine family. Int. Immunopharmacol. 2005;5:1731–1740. doi: 10.1016/j.intimp.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Yamada S., Shiono S., Joo A., Yoshimura A. Control mechanism of JAK/STAT signal transduction pathway. FEBS Lett. 2003;534:190–196. doi: 10.1016/s0014-5793(02)03842-5. [DOI] [PubMed] [Google Scholar]
- Zi Z., Cho K.H., Sung M.H., Xia X., Zheng J., Sun Z. In silico identification of the key components and steps in IFN-gamma induced JAK-STAT signaling pathway. FEBS Lett. 2005;579:1101–1108. doi: 10.1016/j.febslet.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Singh A., Jayaraman A., Hahn J. Modeling regulatory mechanisms in IL-6 signal transduction in hepatocytes. Biotechnol. Bioeng. 2006;95:850–862. doi: 10.1002/bit.21026. [DOI] [PubMed] [Google Scholar]
- Hlavacek W.S. How to deal with large models. Mol. Syst. Biol. 2009;5:240. doi: 10.1038/msb.2008.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto N., et al. Toxicity, pharmacokinetics, and dose-finding study of repetitive treatment with the humanized anti-interleukin 6 receptor antibody MRA in rheumatoid arthritis. Phase I/II clinical study. J. Rheumatol. 2003;30:1426–1435. [PubMed] [Google Scholar]
- Frey N., Grange S., Woodworth T. Population pharmacokinetic analysis of tocilizumab in patients with rheumatoid arthritis. J. Clin. Pharmacol. 2010;50:754–766. doi: 10.1177/0091270009350623. [DOI] [PubMed] [Google Scholar]
- Marino S., Hogue I.B., Ray C.J., Kirschner D.E. A methodology for performing global uncertainty and sensitivity analysis in systems biology. J. Theor. Biol. 2008;254:178–196. doi: 10.1016/j.jtbi.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa K., et al. Purification and characterization of soluble human IL-6 receptor expressed in CHO cells. J. Biochem. 1990;108:673–676. doi: 10.1093/oxfordjournals.jbchem.a123261. [DOI] [PubMed] [Google Scholar]
- Mudter J., et al. Activation pattern of signal transducers and activators of transcription (STAT) factors in inflammatory bowel diseases. Am. J. Gastroenterol. 2005;100:64–72. doi: 10.1111/j.1572-0241.2005.40615.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.