

Abstract
BACKGROUND
Recurrent prostate cancer can be osseous, androgen independent and lethal. The purpose is to discern the efficacy of synthetic small molecule telomerase enzyme inhibitors (TEI) alone or in combination with other cytotoxic therapies in controlling metastatic osseous prostate cancer.
METHODS
C4-2B was pre-treated with a match or mismatch TEI for 6 weeks and then inoculated into nude mice subcutaneously or intraosseously. In a separate experiment, untreated C4-2B was injected into femur of nude mice. The mice were divided into seven systemic “combination” treatment groups of control, Ad-BSP-E1a virus, docetaxel, mismatch and match TEI. Serum PSA was followed longitudinally. Histology analyses and histomorphometry were performed. Repeated measure analysis was applied for statistical analysis and Bonferroni method was used in multiple comparisons.
RESULTS
In the pre-treated study, the PSA of match treated cells in subcutaneous or intraosseous model was significantly lower than mismatch TEI or PBS treated group (P <0.05). Histology revealed increased fibrosis, apoptosis and decreased PSA staining in the match TEI treated subcutaneous xenografts. In the combination treatment study, the PSA was significantly lower in single/double treatment and triple treatment than control (P <0.05). Histology revealed that triple therapy mice had normal femur architecture. Histomorphometrics revealed that the area of femur tumor and woven bone was significantly positively correlated (P =0.007).
CONCLUSIONS
Multiple lines of data point toward the efficacy of systemically administered telomerase inhibitors. Combining cytotoxic regimens with telomerase inhibitors could be beneficial in controlling prostate cancer. Clinical trials are warranted to explore the efficacy of TEI in prostate cancer.
Keywords: hormone refractory prostate cancer, bone metastasis, small molecule telomerase inhibitors, oligonucleotides, combination therapy, adeno-viral vectors
INTRODUCTION
Prostate cancer, the most commonly diagnosed non-dermatological malignancy worldwide, still ranks second among cancer-related deaths in the United States with an estimation of 193,000 new cases and an expectation of 27,000 deaths in 2009 [1]. With the advent of PSA screening and increased public awareness and education, early stage prostate cancer is being increasingly diagnosed and managed with the various options of local definitive therapy [2]. Recurrence after definitive therapy, both at local and distant sites, can be as high as 25% after 15 years [3–6]. Hormonal modulation is often the first choice for managing distant metastatic disease. Prostate cancer cells eventually acquire androgen independency, becoming refractory to hormonal ablation [7,8]. In the past, the development of “androgen refractory or independent prostate cancer” limited options to control both cancer and associated morbidity. Historically, the median survival of a patient after developing hormone refractory prostate cancer (HRPC) is approximately 12–24 months [8].
Telomeres are non-coding (TTAGGG)n tandem repeats at the end of mammalian chromosomes. Telomeres shorten with every cell cycle until they reach a critical length at which time the cell goes into senescence. The regeneration of telomeres by telomerase, a RNA-dependent DNA-polymerase, allows a cell to undergo extended cell proliferation resulting in a potentially immortal cell line [9–11]. Telomerase activity has been reported in germ cells, stem cells, and cancers but not in other somatic cell types. Its reactivation in differentiated cells is associated with immortalization and malignant transformation [10,11]. Structurally, telomerase consists of two subunits: a protein reverse transcriptase (hTERT) containing the enzymatic activity [12–14] and an RNA component (hTR) functioning as a template for binding to DNA and for the addition of new repeats [15]. The vast majority of all prostate carcinomas and nearly all of the high-grade cancers (Gleason score of 7 or more) have reactivated telomerase [9,16–18].
Blockade of telomerase can be accomplished by multiple means [19–27]. Genistein, a naturally occurring soy isoflavone, is thought to act by decreasing the transcriptional activity of hTERT. This may possibly explain the growth inhibitory effect genistein has on prostate cancer cells [22–24]. Another promising family of small molecule telomerase-template inhibitors includes two oligonucleotides: 2′-O-(2-methox-yethyl) (2′-MOE) RNA 13-oligomer (ISIS 24691, ISIS, Carlsbad, CA) [25,27] and a N3′ → P5′ thio-phosphoramidate (NPS) oligonucleotide (GRN163, Geron Corp. Menlo Park, CA) [26]. These are oligonucleotides targeting the RNA template (hTR) of the human telomerase. The oligonucleotides act, not by activating the RNase-H-based degradation of their target sequence, but rather as direct template enzyme inhibitors (TEI) of the enzyme’s active site [25–27,28]. They cause telomere shortening, followed by cell senescence or apoptosis in prostate cancer cell lines [25–27]. Unlike almost all other oligonucleotides or siRNAs, 13-mer base template antagonists, employed in the studies herein, do not require carrier molecules in vivo to be active inside cells [29]. Some of these small molecule telomerase-template inhibitors are in early-stage clinical trials, and offer the possibility of systemic administration, such as GRN163L (Geron Corp.).
In the past, chemotherapy has been considered to be relatively ineffective in HRPC. However, taxanes possess the ability to block microtubule degradation, and have both mitotic and non-mitotic effects, a feature that has proved to be of importance with tumors known to have a low mitotic rate, such as prostate cancer. Recently, multiple studies investigating taxanes (doce-taxel and paclitaxel) alone, or in combination with other drugs have demonstrated PSA reduction, and some tumor response in advanced prostate cancer patients [30–33], and in model systems [28].
Oncolytic viral vectors under the control of a tissue specific promoter have been tested in clinical trials [34–36]. Adenovirus-based vectors with a “suicide” gene under the control of a prostate cancer specific promoter, such as a bone sialoprotein (BSP), osteocalcin, or a mutated androgen receptor-type promoter, show tumor targeting, relative tissue specificity, and the ability to infect both dividing and non-dividing cells [34–36]. The E1a gene of adenovirus is responsible for adenovirus lytic cycle and is engineered in the replication competent adenovirus vector.
With limited means to control HRPC, the idea of a combination approach targeting different vital mechanisms of the aggressive tumor, diminishing its ability to develop resistance, and promoting eventual apoptosis, has become plausible. Initial research combining telomerase enzyme inhibitors (TEI) and anti-proliferative agents has shown encouraging results in vitro and in subcutaneous in vivo models [28]. Telomerase inhibition by RNA interference sensitizes cancer cells to ionizing radiation and chemotherapeutic agents, especially when using topoisomerase inhibitors or bleomycin agents known to cause DNA double-strand breaks [37]. In early studies, a telomerase-selective oncolytic adenoviral agent, OBP-401, revealed encouraging therapeutic synergism when used with docetaxel and other chemotherapeutic agents [38].
To better evaluate the role of small molecule telomerase-template inhibitors, in the present studies, HRPC intraosseous models are targeted with a combination of docetaxel (Taxotere®), ocolytic adenoviral vector (Ad-BSP-E1a), and a TEI (ISIS 24691) in single and combination treatment groups.
EXPERIMENTAL PROCEDURES
Cell Lines and Oligonucleotides
C4-2B lineage cells are an androgen-independent prostate cancer cell line of the LNCaP lineage, derived initially from a cervical lymph node metastasis of human prostate cancer patient and developed by Dr. Leland Chung (Dianon Systems, Stratfort, CT) [39–42]. C4-2B cells were plated on 10 cm plates (Cornig, Acton, MA) in T medium (Invitrogen Corp., Carlsbad, CA) enriched with 5% FBS (Invitrogen Corp.) and 1% penicillin/streptomycin. Incubation for cell culture utilized a humidified incubator at 37°C with 5% CO2. Two oligonucleotides were used, ISIS 24691 and ISIS 125628 (ISIS, Oakland, NJ). ISIS 24691 is a telomerase enzyme inhibitor, 13-base-long oligomer containing MOE (methoxyethyl) bases with phosphorothioate backbone linkages in which a sulfur atom replaces non-bridging oxygen [25,27,28]. ISIS 24691 is complementary to hTR, blocks binding of primer DNA, and inhibits telomerase with an IC50 value of ~3 nM in cell-free assays [25,27,28]. ISIS 125628 is also a MOE oligomer with two mismatched bases relative to ISIS 24691, inhibits telomerase activity 200-fold less potently than ISIS 24691, and is used as a negative control. C4-2B cells were treated with either 5 μM of ISIS 24691, ISIS 125628 or phosphate buffer saline (PBS) for 42 days with fresh drugs added every 3 days along with media changes. When near confluent, cells were harvested, counted, and reseeded in new dishes.
Subcutaneous and Intraosseous Pretreated Models
1 × 106 cells pretreated for 42 days as above were injected either subcutaneously, or intraosseously (into the right femur) into nude irradiated mice (Harland, Indianapolis, IN) after animal protocol approval was obtained from the University of Texas, Southwestern Animal Care and Usage Committee, in conformity with all regulations. The mice underwent bilateral orchiectomy 2–3 weeks after cell inoculation to simulate the androgen-independent state seen in human patients treated for advanced stage HRPC. Subcutaneous tumors were followed by tumor volume and PSA serum level measurements. Intraosseous xenografts were followed by PSA serum levels only. PSA measurements were done using Abbott IMx enzymatic assay (IMx PSA reagent kit, Abbott Laboratories, Irving, TX), utilizing a blood sample taken every week from retro-orbital puncture. The mice were sacrificed at ~12–15 weeks from inoculation, or when tumor burden was judged too large based upon Animal Care and Usage Committee guidelines.
Histological analysis of xenografts utilized gross inspection, H&E staining, trichrome staining, PSA immunohistochemistry, and terminal deoxynucleotidyl transferase (TdT) dUTP nick end-labeling (TUNEL) staining. Tissues were harvested and fixed with 10% buffered formalin (Fisher Scientific, Pittsburgh, PA) and embedded in paraffin. Five micrometer consecutive sections were dewaxed and rehydrated by sequential treatment with xylene, and 100%, 95%, 70%, and 50% EtOH. Slides were stained either with H&E (Sigma–Aldrich, St. Louise, MO) according to the universal standard protocol or with a Sigma trichrome staining kit (Sigma–Aldrich) according to the manufacturers’ protocols. For PSA immunostaining, tissue sections were dewaxed and rehydrated, endogenous peroxidase was quenched with 3% H2O2 in methanol at room temperature for 30 min. After washing 3 times with PBS, sections were blocked with 0.05% BSA and 0.2% (v/v) normal goat serum (Sigma–Aldrich) in PBS at room temperature for 30 min. A polycolonal rabbit anti-human PSA antibody (BioGenex, San Ramon, CA) was used as the primary antibody at a 1:200 dilution in phosphate buffered saline (PBS). PBS without antibody was used as negative control. Tissue sections with either primary PSA antibody or PBS alone were inoculated at 4°C overnight in a humidified chamber, then washed with 0.05% Tween 20 (Sigma–Aldrich) in PBS 3 times and incubated with biotin-conjugated goat anti-rabbit IgG (Sigma–Aldrich) at room temperature for 60 min. After washing 3 times with 0.05% PBS-Tween 20, VECTOR-STAIN Elite ABC reagent (Vector Laboratories, Burlin-game, CA) was applied on tissues for 30 min at room temperature and brown color positive staining was developed with the Sigma FAST 3-3′-diaminobenzi-dine tablet set at room temperature for 10 min. The reaction was stopped by gently flushing with tap water for 10 min. Harris hematoxylin (Sigma–Aldrich) was used for counter-staining and cover glass was mounted with Permount (Biomeda, Foster City, CA).
Generation of Tissue Specific, Replication-Competent Virus Ad-BSP-E1a and Control Virus Ad-BSP-TK
Virus was engineered by inserting the human BSP promoter (Bone Sialoprotein, ~1.4 kb) into the shuttle vector ΔpBPAE1 containing the adenovirus E1a region. This BSP-E1a shuttle vector and pJM17, the plasmid that harbors the backbone of adenovirus, were co-transfected into 293 cells to obtain the recombinant virus Ad-BSP-E1a (similar to 34, 35). PCR and sequencing confirmed the correct clone, and viruses were amplified by growth in 911 cells (generously provided by Chinghai Kao, Indiana University), which cause no, or a very low level of wild-type virus formation. The virus was purified by CsCl-gradient ultra-centrifugation. The titer of each batch of virus was determined using a plaque-forming assay.
Ad-BSP-TK is a replication-defective adenovirus in which the E1 gene was knocked out and replaced by thymidine kinase (TK) gene. BSP promoter was inserted into pE1sp1BTKPA shuttle vector and then co-transfected with pJM17 plasmid into 293 cells to yield the replication-deficient virus Ad-BSP-TK. PCR and sequencing confirmed the correct clone. This virus is inactive in the absence of gancilcovir and serves as a virus negative control (dummy virus).
In Vivo Xenograft Prostate HRPC Model
After obtaining animal usage approval in accordance with the rules and regulations of the University of Texas Southwestern, male nude mice (nu/nu) (Harland, Indianapolis, IN) underwent whole body irradiation (400 Rad/mouse) 48–72 hr prior to tumor cell injection. 1 × 106 native C4-2B cells were injected using a 26 or 27 gauge needle into the right femur through a small skin incision. All mice underwent bilateral orchiectomy 2–3 weeks after tumor injection. When PSA was detectable, the mice were divided into one of seven groups. Mice with PSA values of more than 10 ng/ml (n =4) were thought to have a more advanced disease compared to the rest of experimental subjects and were excluded from our analysis to ensure homogeneity among the treatment groups in accordance with our preset exclusion criteria. Two control groups had either IV PBS, or IV dummy virus (Ad-BSP-TK) (dose 1 × 109 pfu, IV through tail vein) plus docetaxel (Taxotere®, Sanofi-Aventis, Bridgewater, NJ) (2 mg/kg, IV through tail vein) 3 times over 14 days. The third group of mice was a single treatment arm treated with Ad-BSP-E1a (dose 1 × 109 pfu, IV through tail vein) 3 times over 14 days. The double treatment arms were three different groups and these had E1a virus plus docetaxel, E1a virus plus docetaxel plus mismatch TEI (ISIS 125628) or E1a virus plus match TEI (ISIS 24691). The dose of either match or mismatch TEI was 25 mg/kg administered via intraperitoneal injection every Monday, Wednesday, and Friday for 6 weeks (42 days). The final treatment group was a triple treatment group and these mice were treated with E1a virus, Taxotere and Match TEI. A summary of the treatment groups is found in Table I.
TABLE I.
Intraosseous Model and Systemic Combination Therapy
| Categories | Treatment schedule | IV (tail vein) 3 times over 14 days | IP (3 times per week for 6 weeks) | Number of each treatment for PSA analysis | Number of tissue for OM | |
|---|---|---|---|---|---|---|
| I | Control (n =17) | Group 1 | PBS | — | 9 | 9 |
| Group 2 | TK +Tax | — | 8 | 4 | ||
| II | Single treatment (n =10) | Group 3 | Ad-BSP-E1a | — | 10 | 4 |
| Double treatment (n =22) | Group 4 | Ad-BSP-E1a +Tax | — | 9 | 7 | |
| Group 5 | Ad-BSP-E1a +Tax | MM-TEI | 5 | 5 | ||
| Group 6 | Ad-BSP-E1a | M-TEI | 8 | 7 | ||
| III | Triple treatment (n =9) | Group 7 | Ad-BSP-E1a +Tax | M-TEI | 9 | 4 |
| IV | Normal femur (n =19) | 19 | ||||
The seven different treatment arms for the intraosseous prostate cancer cell model. Groups 1 and 2 are the control groups. Group 3 is the single cytotoxic treatment group (Ad-BSP-E1a virus). Groups 4, 5, and 6 are the “double treatment cytotoxic” groups. Group 7 is the triple treatment group. IV, intravenous; IP, intraperitoneal; PBS, phosphate buffered saline; Ad-BSP-TK, recombinant “dummy” adenoviral vector with thymidine kinase gene (negative control for treatment virus); Tax, Taxotere®; Ad-BSP-E1a, adenoviral vector, replication competent, with E1a gene under control of BSP promoter; MM-TEI, mismatch small molecule telomerase-template inhibitor; M-TEI, match small molecule telomerase-template inhibitor.
OM, osteomeasure.
Serum PSA measurements were performed every week of the experiment, using the Abbott IMX technique and utilizing a blood sample taken from retro-orbital puncture. Physical signs and weight were monitored weekly. The experiment was terminated after ~15 weeks from the start of the treatment (~24 weeks after tumor cell implantation). Mice were sacrificed and tumor tissues were harvested. Histological analysis utilized gross inspection, H&E, TUNEL, PSA and trichrome staining.
Histomorphometric Evaluation of Femurs
A total of 59 femur samples were used for histomorphometry. Nineteen normal femur samples were harvested from the mice. Forty treated femur samples were from the various combinations of treatment experiments and are outlined in Table I (some femurs were excluded because they were not acceptable after full tissue processing). Femurs were cut in the full coronal plane, and bone prostate cancer tumor area and newly formed woven bone area were measured using OsteoMeasure™, version 3.0 (OsteoMetrics, Inc., Atlanta, GA) on the femur H&E staining slide. The tissue was measured at 16× magnification under a Leica DM4000B light microscope (Leica Microsystems, Bannockburn, IL) in a 9.7656 mm3 square area directly below the growth plate at distal end of each femur. Figure 6 illustrates the area that was measured.
Fig. 6.
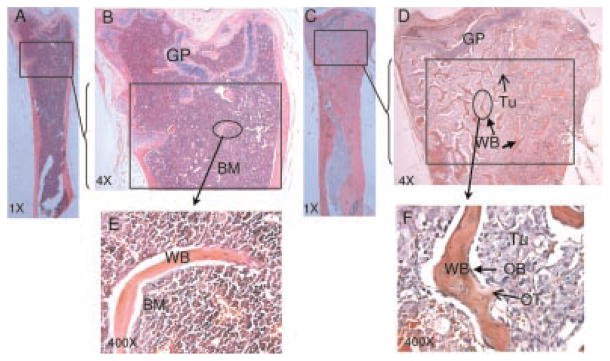
Example of mice femur H&E staining used for tumor and osteoid measurement in Osteomeasure™ system. A and C are at 1× magnification, B and D are at 4× magnification. A and B are femurs without tumor, C and D are femurs filled with C4-2B tumor. E and F are at 20 × magnification enlargements of normal and tumor bearing area that circled in B and D. The area used to perform the measurement is boxed. Tum: tumor (open arrow in D), OT: osteoid (open arrow in F), OB: osteoblast (arrow in F), BM: bone marrow, GP: growth plate, WB: newly formed woven bone (arrow in D).
Statistical Analysis
Repeated measure analysis was used to evaluate the tumor size and PSA data. PSA serum level measurements were logarithmically transformed to better resemble a normal distribution. Because the minimum value of PSA is zero, the natural logarithm of PSA level plus 1 [ln(PSA +1)] was used in the transformation. The analysis was based on transformed data. Time was treated as a discrete variable. Akaike Information Criterion (AIC) was used for the choice of covariance matrix. Potential covariates include groups of treatment, time, and their interaction. In intraosseous injection and combined treatment analysis, we also adjusted for baseline PSA levels. The Bonferroni method was used for adjusting P values in multiple comparisons. Alpha was set at 5%. All statistical analyses were implemented using SAS version 9.1.
For histomorphometry evaluation of tumor area, ANOVA was first used to examine the difference of tumor area among control, single/double and triple treatment groups. Then, t-test was applied to test the hypothesis that mean value of tumor area in each group is equal to zero (normal). Square root of mean square error (MSE) from ANOVA analysis is used for estimated standard deviation. Bonferroni method was used to maintain overall level of significance.
For osteoid area, ANOVA was used to compare osteoid area in control, single/double and triple treatment groups with that in normal group. P-value is adjusted by Dunnett method for multiple comparisons.
RESULTS
Subcutaneous Model
Cells were treated in vitro for a period of 6 weeks with a match small molecule telomerase template inhibitor (TEI) or a mismatch TEI. After this in vitro treatment period, cells were then injected into the nude mice, PSA and tumor volume measurements were taken on a weekly basis beginning at week 8 post-injection (Fig. 1a). At week 8 after tumor inoculation in vivo, the average PSA for the match TEI treatment group was 0.1 ng/ml whereas for the mismatch TEI group it was 30.3 ng/ml. The repeated measure model was used to study the association of ln(1 +PSA) with week and group up to week 12. There was no significant interaction between treatment group and time. Results from unstructured covariance matrix showed that PSA levels for the match TEI treated group (n=8) were significantly lower than the mismatch TEI (n=7) treated group with P-value <0.0001. There was also a significant time (week) effect, P-value =0.0001, indicating a change in PSA values across the different time points.
Fig. 1.

a: Logarithmic conversion of PSA values [ln(1 +PSA)] from subcutaneous tumor models originating from C4-2B cells pretreated in vitro with match TEI or mismatch TEI. Match TEI values were significantly lower than mismatch, P <0.0001. b: Logarithmic conversion of volumes of subcutaneous tumor model (vertical axis) originating from C4-2B cells pretreated in vitro with match TEI or mismatch TEI, versus time (horizontal axis). Tumor volumes were significantly lower for match TEI mice, than mismatch TEI mice on weeks 8, 10, 11, and 12 (P <0.05).
Due to a significant statistical interaction between treatment group and time (weeks) for the match TEI (n=8) and mismatch TEI (n =7) treatment groups were compared using results from unstructured covariance matrix at each week separately (Fig. 1b). At the 5% confidence level, tumor volume was significantly lower for the match TEI treatment group when compared to mismatch TI group on weeks 8, 10, 11, and 12 (P-value <0.05).
Histologic examination and TUNEL staining of the harvested subcutaneous tumors revealed increased apoptosis in match TEI treated inoculants when compared to PBS and mismatch TEI (Fig. 2A,B,G,H). PSA staining revealed decreased PSA production (Fig. 2C,D), and trichrome stain revealed increased fibrosis in the match TEI treated cells (Fig. 2E,F). The level of tissue PSA immunostaining corresponded to tumor volume and serum PSA. Thus, much of the “tumor volume” of match TEI treated mice was non-PSA producing, nonviable cellular debris.
Fig. 2.

These panels represent subcutaneous tumor of pre-treated C4-2B cells. Panels A,C,E,G are mismatch telomerase inhibitor (MM-TI) treated and panels B,D,F,H are match therapeutic drug telomerase inhibitor (M-TEI) treated. Panels A–F were taken at 200× magnification. A and B are hematoxylin and eosin stained. Panels C and D are PSA immunohistochemistry. Panel C reveals the speckled brown staining representing PSA expression throughout the tumor. Arrow indicated PSA positive cells. However, in panel D, the match treated only showed few PSA positive cells. Most of the tumor mass was negative for PSA cells. The corresponding mouse serum PSA in the match treated was also much lower, as seen in Figure 1. Panels E and F are trichrome staining, and Panel E, the mismatch treated, shows viable C4-2B cells. However, in panel F, there were more are as in blue (arrow) represent cellular debris and fibrosis than in panel E. It is apparent that much of this subcutaneous tumor mass was non-viable and filled with fibrosis. Panels G and H are TUNEL staining taken at 40× magnification. Small 200× power enlargements were embedded in the upper right corner of panels G and H, the arrows indicated the apoptotic cells. As seen in panel G, the mismatch treated, there are small brown areas of cell turnover and apoptosis, the majority of the tumor section is viable. However, in panel H, match TEI treated, most of the tumor mass has brown positive staining from TUNEL, indicative of likely non-viable cells. The subcutaneous tumors depicted herein of mismatch and match treated mice are quite representative of all the experimental specimens.
Pre-Treated Intraosseous Model
To establish “proof of principle,” obviating the variability of delivery issues, cells were “pre-treated” in vitro. According to our previous experience, these tumors do not produce consistently measurable serum PSA until approximately at week 8 in vivo, thus the graphs start at the most informative juncture, approximately at week 8. The PSA values from the three treatment groups, match TEI, mismatch TEI, and PBS (control) were compared pair-wisely; P values were adjusted by the Bonferroni method. There was no significant interaction between treatment group and time. Figure 3a shows the ln(1 +PSA) for each group as a function of time. The match TEI treatment group (n =15) had PSA values significantly lower than the PBS treatment group (n =7; P-value 0.033), and marginally lower than the mismatch TEI treated group (n =15; P-value 0.069). Since there was no statistically significant difference between mismatch TEI and PBS treated groups (P-value 1.000), the two groups were combined into one group (n =22), plotted as a new line in Figure 3a, and compared to the match TEI treated group. Match TEI treated mice had a significantly lower PSA values than the PBS and mismatch TEI combined groups (P-value =0.0055). Figure 3b reveals all the three groups, saline control (left), mismatch TEI, and match TEI (right), with SEM bars—the observed visual discrepancy of saline versus mismatch TEI is because of the slightly greater variance of saline control size (statistically these are equivalent groups).
Fig. 3.

a: Logarithmic conversion of PSA values [ln(1 +PSA)] from intraosseous tumor models originating from C4-2B cells treated invitro with match TEI, mismatch TEI, or PBS control. There is no statistical difference between mismatch TEI, and PBS treated groups (P-value =1.000). Match TEI treated mice had significantly lower PSA values than PBS, and mismatch TEI combined groups (P-value 0.0055). Panel b reveals all the three groups, saline control (left), mismatch TEI, and match TEI(right), with SEM bars.
Histologic examination of the femur implants revealed little bone destruction, and decreased PSA production by match TEI treated inoculants when compared to mismatch TEI treated group (Fig. 4). Mismatch TEI and PBS treated groups showed destruction of normal bone architecture caused by tumor cell proliferation (Fig. 4G,H), and a resultant partial and focal osteolytic, and net osteoblastic effect (Fig. 4A,B). Most specimens from match TEI treated subjects (mice) showed no evidence of tumor growth and the preservation of normal bone architecture. In the match TEI tumor mice, which had low, but detectable serum PSA, most times the C4-2B cells could not be found in the bone (Fig. 4E,F).
Fig. 4.
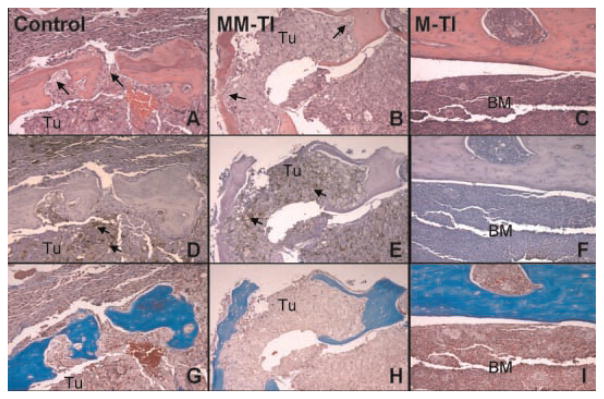
Femurs from PBS treated (control), mismatch telomerase inhibitor, which also is another control, and match telomerase inhibitor. Control PBS is the far left column. Mismatch telomerase inhibitor is the middle column, and match telomerase inhibitor is the far right column. All photos were taken at 200× magnification. Row 1 constituting A, B, and C is hematoxylin and eosin stain. Row 2 consisting of D,E, and F is PSA immunohistochemistry stain. Row 3 consisting of G,H, and I is trichrome stain. BM, bone marrow; Tum, tumor. In A and B control treated mice panels, there is obvious bone destruction and C4-2B cells are seen through out the marrow cavity and invading into the bone (arrow). However, panel C, match treated mouse reveals normal bone marrow and normal calcified osseous bone structures. In panels D and E, on the PSA immunohistochemistry stain and adjacent slices, the brown PSA staining is seen throughout the marrow space and within the bone(arrow), in both the control treated PBS and mismatch telomerase in panels D and E, respectively. However, once again, in panel F, no PSA immunostain is seen in the match treated mouse. Lastly, in panels G and H, adjacent sections, control treated animals reveal bone destruction, whereas match treated animals in panel I reveal a normal architecture. This pattern in these panels was consistent in all the respective mice treatment groups.
Intraosseous Model and Systemic Administration of Combination Treatment
Non-treated native C4-2B tumor cells were injected into the femur, mice were followed until they started having detectable serum PSA levels (usually 8–12 weeks). They were not pre-treated cells. As soon as the PSA serum levels were detected, mice were randomized into one of the seven systemic intravenous or intraperitoneal delivery treatment groups in three different categories, control (group I), single/double combination treatment (group II), or triple combination treatment (group III), and treatment was started according to the specifications mentioned in Table I, and as described previously in the methods. PSA values were followed for ~14 weeks after the start of the treatment (~24 weeks after tumor inoculation). The average starting PSA for all groups was 3.66 ng/ml (range 0.8–4.7 ng/ml, SD 2.7). Comparisons of ln(1 +PSA) between these groups were done using an unstructured covariance matrix model and P values were adjusted by the Bonferroni method.
There was no statistically significant difference in pair-wise comparisons between any of the groups at the start of treatment. When comparing single, double and triple treatments in pairs, there was no significant difference between single versus double, single versus triple, or double versus triple treatment categories over the period of follow-up. To better evaluate the effect of triple treatment, the seven treatment groups were combined into three categories, (I) control/PBS, (II) single/double, and (III) triple treatment. Due to significant interaction with the data sets between I, II, and III, a point-by-point pair-wise comparison was done. PSA values were statistically significantly higher in the control category (I) than in either single/double (II) treatment, or triple (III) treatment at almost all time points after week 2 of treatment, (P-value <0.05) (Fig. 5a). As shown in Figure 5b, which plots the average PSA values in a bar graph, the average PSA for the triple treatment group was much lower than that of either single/double treatment or control groups at weeks 14 and 16. Figure 5c is presented to show the seven individual therapy group PSA values at weeks 0, 8, and 16, with associated standard error (of the mean) bars.
Fig. 5.

a:Logarithmic conversion of PSA values [ln(1 +PSA)] for Groups I (top), Group II (middle), and Group III (lower) treatment categories, which correspond to control versus single/double versus triple treatment over 16 weeks (see Table I). b:PSA averages for treatment categories Group I, II, and III at weeks 0, 8, 14, and 16. Standard error bars a represent. The triple treatment group had the lowest values, and is seen on the far right. c: PSA averages for the seven treatment groups at weeks 0, 8, and 16, with standard error of the mean (SEM) bars present.
On histologic examination, there was obvious evidence of PSA IHC staining in mice with high serum PSA (Fig. 4). In these mice with high serum PSA, bone architecture resembled the net osteoblastic reaction interwoven with tumor cells (C4-2B), similar to that seen in Figure 4A,D,G.
Osteomeasure Evaluation of Tumor and New Woven Bone Area
Using the OsteoMeasure™ software, the tumor volume and the area of new woven bone formation were quantified under the light microscope, and associations and possible relationship between these parameters were evaluated. In the systemic “combination therapy” in vivo osseous model (Table I), the “normal” femur had very little new bone formation. The tumor area and woven bone area were significantly and positively correlated in all specimens.
The femurs with tumor growth had much more new bone formation. Tumor area and the area of new woven bone formation were compared between femurs from different treatment groups versus normal femurs. For all 59 specimens, the correlation coefficient (r) equals 0.35 (P-value =0.007), which indicated that the tumor area and woven bone area were significantly and positively correlated. Figure 6 illustrates a representative area of both normal, and tumor filled architecture. The new woven bone/osteoid is clearly visualized in the tumor filled bone (Fig. 6C,D) in contrast to the normal femur on the left side (Fig. 6A,B) with little new bone. Tumor and woven bone area were compared among all treatment groups and the normal femurs. The mean value of tumor area in control and single/double groups is significantly higher (different from) than that in the normal group, which had no tumor in the femur (P values are less than 0.0001), while its value in “triple treatment” is similar, in most of the aspects, to that in the normal group (P-value =0.17). These P values are adjusted by Bonferroni method. This implies that the tumor area in bone in the triple treatment group is much less, and overall this triple treatment cohort is, in most of the aspects, similar to the normal femur. This result provides another avenue of evidence that the triple treatment regime of therapy is more effective than single/double treatment in tumor killing.
The woven bone area evaluation revealed that the mean value of woven bone areas in control, single/double and triple treatment is not significantly higher than that in normal group (P values are 0.11, 0.15, and 1.00, respectively, as adjusted by the Dunnett method). This result indicated, even though the woven bone area is positively correlated to the tumor area, the treatment effect on new woven bone formation may not be as significant as tumor per se. The bone turnover in the prostate bone metastasis is complicated and the mechanism likely involves complex interactions between osteoblasts, osteoclasts, and tumor cells. In addition, the relationship between the tumor and new bone formation during the tumor killing by treatment agent is likely a complex and an active process.
DISCUSSION
C4-2B is an androgen-independent PSA producing prostate cancer cell line, closely mimicking advanced metastatic prostate cancer. The validity of this model has been used in several studies [39–43]. Telomeres are ~2.3–2.7 kb long in C4-2B cells, which are significantly shorter than other prostate cancer cell lines, making them a useful model to study the effect of TEI [28].
Telomerase inhibition in this study, using a small molecule telomerase template inhibitor, appears to retard the ability of C4-2B cells to grow and establish tumors after being treated in vitro as evident from the reduced PSA levels and tumor volumes in the subcutaneous model. Furthermore, there was a statistically significant difference at most time points with match TEI treated groups having a lower PSA levels and tumor volume, indicating a slower growth rate. A similar effect was observed in the “pretreatment intraosseous model” for cells treated in vitro with match TEI, versus the control groups: mismatch TEI, or PBS.
The trend in the management of advanced androgen-independent prostate cancer is shifting toward combination therapy. Trials combining multiple modalities of treatment with different mechanisms of actions are expected to be more effective in achieving tumor control, which may later translate to decreased morbidity, increased life expectancy and potential cure of metastatic disease. Initial pre-clinical attempts in combining telomerase inhibition with chemotherapeutic modalities have been encouraging. Several studies have demonstrated that the inhibition of telomerase sensitizes tumors, and has a synergistic effect in vitro with an antiproliferative agent [9], UV irradiation [44], or the tyrosine kinase inhibitor imatinib [45]. Ward and Autexier [46] have shown that pharmacological telomerase inhibition using BIBR1532, a small-molecule inhibitor of telomerase catalytic activity, in combination therapy succeeded in sensitizing otherwise chemotherapy resistant cells.
Docetaxel (Taxotere®), a semi-synthetic taxane that binds to β tubulin and causes cell cycle arrest by inhibiting microtubule disassembly, has been shown to improve survival in metastatic androgen-independent prostate cancer when combined with estramustine and corticosteroids [32,33]. Chemotherapy in conventional doses is associated with a wide spectrum of potential side effects. These side effects are usually dose related. The dose of docetaxel used in this experiment was much lower than what is usually given (2 mg/kg), even in animal models (usual dose ~10–20 mg/kg). Adenoviral vectors delivering a suicide gene under the control of a prostate specific promoter have moved to human clinical trials [34,35,43]. The adenoviral vector used in this study is “conditionally” replication competent, and under the control of a restricted prostate cancer specific promoter. Minimal side effects are observed in animal and human studies, with this virus, due to the restriction governed by the tissue and tumor specific promoter [34–36,38].
In this study, a treatment effect was evident, being a clinically relevant reduction in serum PSA (and thus tumor volume) in both the osseous pre-treatment model and the “triple therapy” systemic administration group. However, in the systemic therapy model the difference between the triple treatment group and the double treatment groups was not as pronounced as might have been expected. Although average PSA values were lower for the triple treatment compared to single/double treatment or control groups, they lacked statistical significance. The lack of such significance could possibly be attributed to several factors. Systemic administration of the oligonucleotides by IP injection may not be an optimal method of delivery. Further research investigating the bioavailability and efficiency of this route as compared to other routes would help to clarify this point. In addition, vector backbone research may unveil more effective vehicles. For example, a TEI inhibitor that has just entered clinical trials for chronic lymphocytic leukemia (CLL), GRN 163L (Geron Corp.), has a different backbone chemistry including a lipid moiety that appears to enhance cellular uptake, and bio-distribution [47]. Another potential issue would be the duration of the telomerase inhibition. In groups treated with match and mismatch TEI, these agents were administered 3 times per week on weeks 3–8 (6 weeks) following an initial treatment with cytotoxic agent (adenoviral agent and/or docetaxel) over the first 2 weeks. Had there been a longer treatment, a more pronounced long-term effect of telomerase inhibition might have been evident. One postulated mechanism of action of telomerase inhibitors involves gradual shortening of the telomeres before an actual growth arrest can occur, raising the concern about whether the 6-week treatment was long enough to show the maximum therapeutic effect in vivo. A longer treatment period might show an increased tumor cell benefit without increasing the side effects of such agents. In addition, the cytotoxic effect of the lytic adenovirus and docetaxel tends to be more pronounced and acute in onset, when compared to telomerase inhibition. This can be explained by the different mechanisms of actions by which these agents are believed to work. It is possible that had the TEI treatment been started concomitantly, or before cytotoxic drugs were administered, there might have been a more significant difference in the PSA response to the different treatments. Lastly, the viral tumor toxicity is significant [28], and may have masked smaller differences between groups due to TEI therapy. In any event, the treatment regime used may be considered one “cycle,” and could be repeated for added effect.
Recent research at inhibiting tankyrase, an enzyme involved in recruiting and activating telomerase, has revealed that it allows for a decreased requirement of TEI dose [48]. Seimiya et al. described that, at telomerase inhibitor concentration of 2 μM, telomeres shortened to a length sufficient to cause cell crisis. At a dose of 0.75 μM however, telomere length was shortened from 5 kb and stabilized at 4 kb. The addition of tankyrase inhibitors to the later group had allowed for a further decrease of telomeres to a length short enough to cause cell crisis. Such a synergistic effect might allow a further reduction in the dose of the telomerase inhibitor, decreasing the potential side effects and allowing for higher efficiency of tumor control.
Furthermore, the combination of a chemotherapeutic agent and TEI showed a synergistic effect in the control of malignant glioma. Kondo et al. [49] have shown a synergistic effect between cisplatin and 2-5A-antisense telomerase RNA. Komata et al. [50] used a recombinant adenovirus carrying a p53 tumor suppressor in conjunction with a TEI. All of the five different glioma cell lines were highly sensitive to the combination of adenoviral vector and TEI therapy. In our study, tumor control, as evidenced from the data using the very sensitive PSA level as a surrogate marker, was significantly better with the combination therapy. PSA expression in C4-2B cells is directly proportional to viable tumor cell volume.
This study has provided valuable guidance to the future role of telomerase inhibition in the management of hormone refractory prostate cancer. As this is the only study evaluating the role of TEI in a PSA producing, intraosseous HRPC model, a few questions still remain to be answered. Similar experiments using different treatment groups, doses, routes of administration, or antineoplastic agents may provide more information about the role that TEI could play as a novel therapy for HRPC. Combination therapy in this group of animals was safe and well tolerated, and confirms previous in vitro and in vivo work [25,28]. TEI and cytotoxic combination therapy may allow for a decrease of dosage of administered chemotherapeutic agent, potentially offering a safer profile. Clinical trails are warranted using novel small-molecule telomerase enzyme template inhibitors with cytotoxic drug(s) in men with prostate cancer.
CONCLUSION
Telomerase activity has been found in most types of human tumors, including prostate cancer cells. Synthetic small-molecule telomerase template enzyme inhibitors of the antisense oligonucleotide family, acting as an enzyme inhibitor, can suppress the tumor in pretreated cells then grow in vivo in the subcutaneous and osseous space. When combined with cytotoxic therapy, systemic telomerase inhibitor treatment is safe in osseous tumor xenografts growing in immunodeficient mice, and minimizes tumor viability, based on serum PSA and analysis of bone histology. To better evaluate their utility as single or combination potential therapy in humans, further large scale preclinical, and clinical studies are warranted.
Acknowledgments
The authors are thankful to Isis pharmaceuticals for providing telomerse inhibitors, to Wooding E. Wright, Ph.D. for providing insight into inhibitor and telomere biology, to University of Texas Southwestern Urology Department for providing administrative support (John D. McConnell, MD, an Claus G. Roerhborn, MD), and to Leland W.C. Chung, PhD, and Haiyen Zhau, PhD. for providing stimulus, interest and education on prostate cancer and cell line models. This work was supported by the Department of Defense (DOD), CDMRP Program, Young Investigator Award, Grant # DAMD 17-01-1-0107, and DAMD 17-02-1-0148 (K.S.K.), and DOD PCRP-02 Consortium Grant (K.S.K.), and the Minnesota Medical Foundation: Center for Prostate Cancer, and Doughery Family Endowment funds (K.S.K.).
Grant sponsor: Department of Defense (DOD), CDMRP Program, Young Investigator Award; Grant numbers: DAMD 17-01-1-0107, DAMD 17-02-1-0148; Grant sponsor: PCRP-02 Consortium Grant; Grant sponsor: Minnesota Medical Foundation: Center for Prostate Cancer, and Doughery Family Endowment funds.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59(4):228. [Google Scholar]
- 2.Miller DC, Hafez KS, Stewart A, Montie JE, Wei JT. Prostate carcinoma presentation, diagnosis, and staging: An update from the National Cancer Data Base. Cancer. 2003;98(6):1169–1178. doi: 10.1002/cncr.11635. [DOI] [PubMed] [Google Scholar]
- 3.Roehl KA, Han M, Ramos CG, Antenor JA, Catalona WJ. Cancer progression and survival rates following anatomical radical retropubic prostatectomy in 3,478 consecutive patients: Long-term results. J Urol. 2004;172(3):910–914. doi: 10.1097/01.ju.0000134888.22332.bb. [DOI] [PubMed] [Google Scholar]
- 4.Han M, Partin AW, Pound CR, Epstein JI, Walsh PC. Long-term biochemical disease-free and cancer-specific survival following anatomic radical retropubic prostatectomy. The 15-year Johns Hopkins experience. Urol Clin North Am. 2001;28(3):555–565. doi: 10.1016/s0094-0143(05)70163-4. [DOI] [PubMed] [Google Scholar]
- 5.Perez CA, Michalski JM, Lockett MA. Chemical disease-free survival in localized carcinoma of prostate treated with external beam irradiation: Comparison of American Society of Therapeutic Radiology and Oncology Consensus or 1 ng/mL as endpoint. Int J Radiat Oncol Biol Phys. 2001;49(5):1287–1296. doi: 10.1016/s0360-3016(00)01492-9. [DOI] [PubMed] [Google Scholar]
- 6.Kuban DA, Thames HD, Levy LB, Horwitz EM, Kupelian PA, Martinez AA, Michalski JM, Pisansky TM, Sandler HM, Shipley WU, Zelefsky MJ, Zietman AL. Long-term multi-institutional analysis of stage T1-T2 prostate cancer treated with radiotherapy in the PSA era. Int J Radiat Oncol Biol Phys. 2003;57(4):915–928. doi: 10.1016/s0360-3016(03)00632-1. [DOI] [PubMed] [Google Scholar]
- 7.Robson M, Dawson N. How is androgen-dependent metastatic prostate cancer best treated? Hematol Oncol Clin North Am. 1996;10(3):727–747. doi: 10.1016/s0889-8588(05)70364-6. [DOI] [PubMed] [Google Scholar]
- 8.Smaletz O, Scher HI, Small EJ, Verbel DA, McMillan A, Regan K, Kelly WK, Kattan MW. Nomogram for overall survival of patients with progressive metastatic prostate cancer after castration. J Clin Oncol. 2002;20(19):3972–3982. doi: 10.1200/JCO.2002.11.021. [DOI] [PubMed] [Google Scholar]
- 9.Koeneman KS, Pan CX, Jin JK, Pyle JM, III, Flanigan RC, Shankey TV, Diaz MO. Telomerase activity, telomere length, and DNA ploidy in prostatic intraepithelial neoplasia (PIN) J Urol. 1998;160(4):1533–1539. [PubMed] [Google Scholar]
- 10.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266(5193):2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 11.Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33(5):787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 12.Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD, Ziaugra L, Beijersbergen RL, Davidoff MJ, Liu Q, Bacchetti S, Haber DA, Weinberg RA. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90(4):785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB, Cech TR. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277(5328):955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- 14.Harrington L, Zhou W, McPhail T, Oulton R, Yeung DS, Mar V, Bass MB, Robinson MO. Human telomerase contains evolutionarily conserved catalytic and structural subunits. Genes Dev. 1997;11(23):3109–3115. doi: 10.1101/gad.11.23.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng J, Funk WD, Wang SS, Weinrich SS, Avilion AA, Chiu CP, Adams RR, Chang E, Allsopp RC, Yu J, Le S, West MD, Harley CB, Andrews WH, Greider CW, Villeponteau B. The RNA component of human telomerase. Science (Washington, DC) 1995;269:1236–1241. doi: 10.1126/science.7544491. [DOI] [PubMed] [Google Scholar]
- 16.Lin Y, Uemura H, Fujinami K, Hosaka M, Iwasaki Y, Kitamura H, Harada M, Kubota Y. Detection of telomerase activity in prostate needle-biopsy samples. Prostate. 1998;36:121–128. doi: 10.1002/(sici)1097-0045(19980701)36:2<121::aid-pros7>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 17.Zhang W, Kapusta LR, Slingerland JM, Klotz LH. Telomerase activity in prostate cancer, prostatic intraepithelial neoplasia, and benign prostatic epithelium. Cancer Res. 1998;58:619–622. [PubMed] [Google Scholar]
- 18.Sommerfeld HJ, Meeker AK, Piatyszek MA, Bova GS, Shay JW, Coffey DS. Telomerase activity: A prevalent marker of malignant human prostate tissue. Cancer Res. 1996;56:218–222. [PubMed] [Google Scholar]
- 19.Sigala S, Faraoni I, Botticini D, Paez-Pereda M, Missale C, Bonmassar E, Spano P. Suppression of telomerase, reexpression of KAI1, and abrogation of tumorigenicity by nerve growth factor in prostate cancer cell lines. Clin Cancer Res. 1999;5(5):1211–1218. [PubMed] [Google Scholar]
- 20.Incles CM, Schultes CM, Kempski H, Koehler H, Kelland LR, Neidle S. A G-quadruplex telomere targeting agent produces p16-associated senescence and chromosomal fusions in human prostate cancer cells. Mol Cancer Ther. 2004;3(10):1201–1206. [PubMed] [Google Scholar]
- 21.Damm K, Hemmann U, Garin-Chesa P, Hauel N, Kauffmann I, Priepke H, Niestroj C, Daiber C, Enenkel B, Guilliard B, Lauritsch I, Müller E, Pascolo E, Sauter G, Pantic M, Martens UM, Wenz C, Lingner J, Kraut N, Rettig WJ, Schnapp A. A highly selective telomerase inhibitor limiting human cancer cell proliferation. EMBO J. 2001;20(24):6958–6968. doi: 10.1093/emboj/20.24.6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ouchi H, Ishiguro H, Ikeda N, Hori M, Kubota Y, Uemura H. Genistein induces cell growth inhibition in prostate cancer through the suppression of telomerase activity. Int J Urol. 2005;12(1):73–80. doi: 10.1111/j.1442-2042.2004.00973.x. [DOI] [PubMed] [Google Scholar]
- 23.Swami S, Krishnan AV, Peehl DM, Feldman D. Genistein potentiates the growth inhibitory effects of 1,25-dihydroxyvitamin D3 in DU145 human prostate cancer cells: Role of the direct inhibition of CYP24 enzyme activity. Mol Cell Endocrinol. 2005;241(1–2):49–61. doi: 10.1016/j.mce.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 24.Ouchi H, Ishiguro H, Ikeda N, Hori M, Kubota Y, Uemura H. Genistein induces cell growth inhibition in prostate cancer through the suppression of telomerase activity. Int J Urol. 2005;12(1):73–80. doi: 10.1111/j.1442-2042.2004.00973.x. [DOI] [PubMed] [Google Scholar]
- 25.Chen Z, Koeneman KS, Corey DR. Consequences of telomerase inhibition and combination treatments for the proliferation of cancer cells. Cancer Res. 2003;63(18):5917–5925. [PubMed] [Google Scholar]
- 26.Asai A, Oshima Y, Yamamoto Y, Uochi TA, Kusaka H, Akinaga S, Yamashita Y, Pongracz K, Pruzan R, Wunder E, Piatyszek M, Li S, Chin AC, Harley CB, Gryaznov S. A novel telomerase template antagonist (GRN163) as a potential anticancer agent. Cancer Res. 2003;63(14):3931–3939. [PubMed] [Google Scholar]
- 27.Elayadi AN, Demieville A, Wancewicz EV, Monia BP, Corey DR. Inhibition of telomerase by 2′-O-(2-methoxyethyl) RNA oligomers: Effect of length, phosphorothioate substitution and time inside cells. 2001;29(8):1683–1689. doi: 10.1093/nar/29.8.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Canales BK, Li Y, Thompson MG, Gleason JM, Chen Z, Malaeb B, Corey DR, Herbert BS, Shay JW, Koeneman KS. Small molecule, oligonucleotide-based telomerase template inhibition in combination with cytolytic therapy in an in vitro androgen-independent prostate cancer model. Urol Oncol. 2006;24(2):141–151. doi: 10.1016/j.urolonc.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 29.Chen Z, Monia BP, Corey DR. Telomerase Inhibition, telomere shortening, and decreased cell proliferation by cell permeable 2′-O-methoxyethyl oligonucleotides. J Med Chem. 2002;45:5423–5425. doi: 10.1021/jm025563v. [DOI] [PubMed] [Google Scholar]
- 30.Gravis G, Bladou F, Salem N, Macquart-Moulin G, Serment G, Camerlo J, Genre D, Bardou VJ, Maraninchi D, Viens P. Weekly administration of docetaxel for symptomatic metastatic hormone-refractory prostate carcinoma. Cancer. 2003;98(8):1627–1634. doi: 10.1002/cncr.11687. [DOI] [PubMed] [Google Scholar]
- 31.Friedland D, Cohen J, Miller R, Jr, Voloshin M, Gluckman R, Lembersky B, Zidar B, Keating M, Reilly N, Dimitt B. A phase II trial of docetaxel (Taxotere) in hormone-refractory prostate cancer: Correlation of antitumor effect to phosphorylation of Bcl-2. Semin Oncol. 1999;26(5 Suppl 17):19–23. [PubMed] [Google Scholar]
- 32.Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr, Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, Benson MC, Small EJ, Raghavan D, Crawford ED. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351(15):1513–1520. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- 33.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Théodore C, James ND, Turesson I, Rosenthal MA Eisenberger MA; TAX 327 Investigators. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351(15):1502–1512. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 34.Kubo H, Gardner TA, Wada Y, Koeneman KS, Gotoh A, Yang L, Kao C, Lim SD, Amin MB, Yang H, Black ME, Matsubara S, Nakagawa M, Gillenwater JY, Zhau HE, Chung LW. Phase I dose escalation clinical trial of adenovirus vector carrying osteocalcin promoter-driven herpes simplex virus thymidine kinase in localized and metastatic hormone-refractory prostate cancer. Hum Gene Ther. 2003;14(3):227–241. doi: 10.1089/10430340360535788. [DOI] [PubMed] [Google Scholar]
- 35.Koeneman KS, Kao C, Ko SC, Yang L, Wada Y, Kallmes DF, Gillenwater JY, Zhau HE, Chung LW, Gardner TA. Osteocalcin-directed gene therapy for prostate-cancer bone metastasis. World J Urol. 2000;18(2):102–110. doi: 10.1007/s003450050181. [DOI] [PubMed] [Google Scholar]
- 36.Matsubara S, Wada Y, Gardner TA, Egawa M, Park MS, Hsieh CL, Zhau HE, Kao C, Kamidono S, Gillenwater JY, Chung LW. A conditional replication-competent adenoviral vector, Ad-OC-E1a, to cotarget prostate cancer and bone stroma in an experimental model of androgen-independent prostate cancer bone metastasis. Cancer Res. 2001;61(16):6012–6019. [PubMed] [Google Scholar]
- 37.Nakamura M, Masutomi K, Kyo S, Hashimoto M, Maida Y, Kanaya T, Tanaka M, Hahn WC, Inoue M. Efficient inhibition of human telomerase reverse transcriptase expression by RNA interference sensitizes cancer cells to ionizing radiation and chemotherapy. Hum Gene Ther. 2005;16(7):859–868. doi: 10.1089/hum.2005.16.859. [DOI] [PubMed] [Google Scholar]
- 38.Fujiwara T, Kagawa S, Kishimoto H, Endo Y, Hioki M, Ikeda Y, Sakai R, Urata Y, Tanaka N, Fujiwara T. Enhanced antitumor efficacy of telomerase-selective oncolytic adenoviral agent OBP-401 with docetaxel: Preclinical evaluation of chemovirotherapy. Int J Cancer. 2006;119(2):432–440. doi: 10.1002/ijc.21846. [DOI] [PubMed] [Google Scholar]
- 39.Wu TT, Sikes RA, Cui Q, Thalmann GN, Kao C, Murphy CF, Yang H, Zhau HE, Balian G, Chung LW. Establishing human prostate cancer cell xenografts in bone: Induction of osteoblastic reaction by prostate-specific antigen-producing tumors in athymic and SCID/bg mice using LNCaP and lineage-derived metastatic sublines. Int J Cancer. 1998;77(6):887–894. doi: 10.1002/(sici)1097-0215(19980911)77:6<887::aid-ijc15>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 40.Thalmann GN, Sikes RA, Wu TT, Degeorges A, Chang SM, Ozen M, Pathak S, Chung LW. LNCaP progression model of human prostate cancer: Androgen-independence and osseous metastasis. Prostate. 2000;44(2):91–103. doi: 10.1002/1097-0045(20000701)44:2<91::aid-pros1>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 41.Koeneman KS, Yeung F, Chung LW. Osteomimetic properties of prostate cancer cells: A hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. Prostate. 1999;39(4):246–261. doi: 10.1002/(sici)1097-0045(19990601)39:4<246::aid-pros5>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 42.Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LW. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: Role of bone stromal cells. Int J Cancer. 1994;57(3):406–412. doi: 10.1002/ijc.2910570319. [DOI] [PubMed] [Google Scholar]
- 43.Chung LW, Hsieh CL, Law A, Sung SY, Gardner TA, Egawa M, Matsubara S, Zhau HE. New targets for therapy in prostate cancer: Modulation of stromal-epithelial interactions. Urology. 2003;62(5 Suppl 1):44–54. doi: 10.1016/s0090-4295(03)00796-9. [DOI] [PubMed] [Google Scholar]
- 44.Wong KK, Chang S, Weiler SR, Ganesan S, Chaudhuri J, Zhu C, Artandi SE, Rudolph KL, Gottlieb GJ, Chin L, Alt FW, DePinho RA. Telomere dysfunction impairs DNA repair and enhances sensitivity to ionizing radiation. Nat Genet. 2000;26(1):85–88. doi: 10.1038/79232. [DOI] [PubMed] [Google Scholar]
- 45.Tauchi T, Nakajima A, Sashida G, Shimamoto T, Ohyashiki JH, Abe K, Yamamoto K, Ohyashiki K. Inhibition of human telomerase enhances the effect of the tyrosine kinase inhibitor, imatinib, in BCR-ABL-positive leukemia cells. Clin Cancer Res. 2002;8(11):3341–3347. [PubMed] [Google Scholar]
- 46.Ward RJ, Autexier C. Pharmacological telomerase inhibition can sensitize drug-resistant and drug-sensitive cells to chemotherapeutic treatment. Mol Pharmacol. 2005;68(3):779–786. doi: 10.1124/mol.105.011494. [DOI] [PubMed] [Google Scholar]
- 47.Dikmen ZG, Gellert GC, Jackson S, Gryaznov S, Tressler R, Dogan P, Wright WE, Shay JW. In vivo inhibition of lung cancer by GRN163L: A novel human telomerase inhibitor. Cancer Res. 2005;65(17):7866–7873. doi: 10.1158/0008-5472.CAN-05-1215. [DOI] [PubMed] [Google Scholar]
- 48.Seimiya H, Muramatsu Y, Ohishi T, Tsuruo T. Tankyrase 1 as a target for telomere-directed molecular cancer therapeutics. Cancer Cell. 2005;7(1):25–37. doi: 10.1016/j.ccr.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 49.Kondo Y, Komata T, Kondo S. Combination therapy of 2-5A antisense against telomerase RNA and cisplatin for malignant gliomas. Int J Oncol. 2001;18(6):1287–1292. doi: 10.3892/ijo.18.6.1287. [DOI] [PubMed] [Google Scholar]
- 50.Komata T, Kondo Y, Koga S, Ko SC, Chung LW, Kondo S. Combination therapy of malignant glioma cells with 2-5A-antisense telomerase RNA and recombinant adenovirus p53. Gene Ther. 2000;7(24):2071–2079. doi: 10.1038/sj.gt.3301327. [DOI] [PubMed] [Google Scholar]