

Abstract
Long term potentiation and long term depression of synaptic responses in the hippocampus are thought to be critical for certain forms of learning and memory, although until recently it has been difficult to demonstrate that long term potentiation or long term depression occurs during hippocampus-dependent learning. Induction of long term potentiation or long term depression in hippocampal slices in vitro modulates phosphorylation of the α-amino-3-hydrozy-5-methylisoxazole-4-propionic acid subtype of glutamate receptor subunit GluR1 at distinct phosphorylation sites. In long term potentiation, GluR1 phosphorylation is increased at the Ca2+/calmodulin-dependent protein kinase and protein kinase C site serine 831, whereas in long term depression, phosphorylation of the protein kinase A site serine 845 is decreased. Indeed, phosphorylation of one or both of these sites is required for long term synaptic plasticity and for certain forms of learning and memory. Here we demonstrate that training in a hippocampus-dependent learning task, contextual fear conditioning is associated with increased phosphorylation of GluR1 at serine 831 in the hippocampal formation. This increased phosphorylation is specific to learning, has a similar time course to that in long term potentiation, and like memory and long term potentiation, is dependent on N-methyl-D-aspartate receptor activation during training. Furthermore, the learning-induced increase in serine 831 phosphorylation is present at synapses and is in heteromeric complexes with the glutamate receptor subunit GluR2. These data indicate that a biochemical correlate of long term potentiation occurs at synapses in receptor complexes in a final, downstream, postsynaptic effector of long term potentiation during learning in vivo, further strengthening the link between long term potentiation and memory.
Bidirectional synaptic plasticity, manifested experimentally as long term potentiation (LTP)2 and long term depression (LTD), is thought to be critical for certain forms of learning and memory (1–4). Prevention of LTP in the hippocampus pharmacologically (4, 17), electrophysiologically (18–20), and genetically (4, 21–25) has been shown in many cases to prevent hippocampus-dependent forms of learning and memory including contextual fear conditioning and spatial memory in a Morris water maze task with some exceptions (4, 10). Until recently, however, it has been difficult to measure lasting potentiation of synaptic responses in the hippocampus during learning (4–12).
Much effort has been directed at understanding the cellular and molecular mechanisms of LTP and LTD in the hippocampus. In particular, activation of protein kinases such as Ca2+/calmodulin-dependent protein kinase, protein kinase C, and others are thought to play a major role in induction and early phases of hippocampal LTP (14, 26–33), whereas protein phosphatases may play a corresponding role in hippocampal LTD (34–39). One important substrate for these kinases and phosphatases is thought to be the GluR1 subunit of the α-amino-3-hydrozy-5-methylisoxazole-4-propionic acid (AMPA) subtype of glutamate receptors, the primary mediator of excitatory postsynaptic currents (15, 40–43). Phosphorylation of GluR1 at serine 831 by protein kinase C or calmodulin-dependent protein kinase is known to increase AMPA receptor-mediated currents, although a recent study suggests that this increase is observed only in GluR1 homomeric channels and not in GluR1/GluR2 heteromeric AMPA receptors (44). Similarly, phosphorylation at serine 845 by protein kinase A can increase AMPA receptor-mediated currents (45–48). Phosphorylation of Ser-845 may also be important for insertion of additional AMPA subtype of glutamate receptor into the postsynaptic membrane (42, 49–51).
Recent work has characterized the pattern of phosphorylation of GluR1 at these two sites during LTP and LTD as well as during reversal of LTP (de-potentiation) and LTD (de-depression) (13, 15). For at least one hour after induction of LTP, phosphorylation of GluR1 is increased at Ser-831, whereas during LTD, dephosphorylation of Ser-845 predominates (15). When previously potentiated synapses are depotentiated, Ser-831 phosphorylation is decreased (15). When depressed synapses are de-depressed, Ser-845 phosphorylation is increased (15). These two GluR1 phosphorylation sites have been shown to be required for certain forms of learning and memory in vivo (16).
In an effort to understand whether biochemical correlates of LTP or LTD occur in the hippocampus during learning, we have determined the predominant GluR1 phosphorylation pattern in the hippocampus after a temporally discrete, hippocampus-dependent learning task. Our data reveal a learning-specific increase in Ser-831 GluR1 phosphorylation reminiscent of that seen during LTP of naïve synapses. This increased phosphorylation occurs in heteromeric complexes of GluR1 with GluR2 and is localized to the postsynaptic density (PSD). These data provide additional evidence for LTP-like biochemical changes in the hippocampus occurring at excitatory synapses during learning, providing additional data linking LTP and memory formation in the brain.
EXPERIMENTAL PROCEDURES
Behavior
6–8-Week-old male Sprague-Dawley rats (Charles River) housed 4 per cage were handled daily for 5 days in a stereotyped manner before all experiments. Contextual fear conditioning was performed in a manner similar to that previously described (52–54) in MedAssociates (ENV-008-FPU) fear conditioning chambers with a rat shock grid floor (ENV-005-FPU-R). After a 2-min habituation period, rats were subjected to three 2-s, 1.5-mA footshocks 1 min apart followed by a 1-min rest period before removal to the home cage. In a separate set of preliminary experiments, freezing behavior (motionless except respirations) was measured by observation every 10 s, and the training protocol was optimized to induce maximal contextual fear memory measured 24 h after training (not shown). In all experiments, sham-trained control rats were subjected to the identical training protocol simultaneously with their randomly chosen, paired, cage-mate but did not receive footshock. For “immediate shock” experiments, rats were placed into the conditioning chambers from their home cage, shocked for 6 s at 1.5 mA, then immediately returned to their home cage. For “latent inhibition” experiments, rats were housed in the conditioning chambers for 16 h with ad libitum access to food and water. One hour before training, food and water were removed for 1 h followed by a standard contextual fear conditioning training protocol described above. Paired, cage-mate, sham control rats were simultaneously treated identically except they received no footshock. In MK-801 experiments, rats were injected intraperitoneally with 0.4 mg/kg MK-801 dissolved in sterile saline (0.4 mg/ml stock) or vehicle alone 30 min before contextual fear conditioning.
Sample Preparation
Tissue was collected and processed essentially as described (15). Rats were euthanized at the indicated time points ± 2–3 min after training by live decapitation. The brain was removed rapidly and submersed immediately into ice-cold dissecting buffer (15). The left hippocampus was rapidly dissected out, frozen on dry ice, and stored for later use at −80 °C. Whole hippocampi were homogenized in 5 ml of ice-cold homogenization buffer (0.1 M sodium phosphate, 0.1 M NaCl, 5 mM EDTA, 5 mM EGTA, 50 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, 1/100 dilution Protease Inhibitor Mixture for Mammalian Tissue (Sigma P8340), 10 mM sodium pyrophosphate, 0.5 μM okadaic acid) via sonication with brief pulses for 20–30 s. Homogenates were centrifuged at 14,000 × g for 10 min at 4 °C. Crude membrane fractions were resuspended in 750 μl of ice-cold 1% SDS with 50 mM NaF and placed at 100 °C for 5 min followed by vortexing and brief sonication. Protein concentration was determined by Bio-Rad DC protein assay, SDS sample buffer was added, and samples were heated to 100 °C for 5 min, and loaded onto SDS-PAGE gels (4–15% gradient). Samples were paired based on simultaneous experimental and sham control treatment in cage-mate pairs.
Immunoblot Analysis
Phosphorylation site-specific GluR1 antibodies were a gift of Dr. Richard Huganir and were generated and purified as described (55). Immunoblot analysis was performed essentially as described (15), except secondary antibody was used at 1:20,000, membranes were scanned while wet, and total GluR1 levels were quantified separately by normalizing to actin levels using the bottom half of the same blot with a mouse anti-actin monoclonal antibody (Clone C4, MP Biomedicals, Aurora, OH). Enhanced chemifluorescent detection afforded a very broad linear range, and all immunoblots were within the predetermined linear range for each antibody (not shown).
Postsynaptic Density Preparations
PSD preparations were prepared essentially as described (56). Bilateral hippocampi were rapidly dissected and homogenized in 5 ml of ice-cold buffer A (5 mM HEPES, pH 7.4, 1 mM MgCl2, 0.5 mM CaCl2, 1 mM NaF, 0.1 mM phenylmethylsulfonyl fluoride, 500 μl of protease inhibitor mixture (Sigma p8340) in 250 ml). All remaining steps were performed at 4 °C. Homogenates were centrifuged at 1400 × g for 10 min, and the supernatant (S1) was saved. The pellet was resuspended in 5 ml of buffer A and centrifuged 10 min at 700 × g, and supernatant (S1′) was saved and combined with S1. The supernatants were centrifuged at 13,750 ×g for 10 min to obtain pellet (P2). P2 was resuspended in 1 ml of buffer B (0.321 mM sucrose, 6 mM Tris, pH 8.0, 0.1 mM phenylmethylsulfonyl fluoride, 100 ml protease inhibitor mixture in 50 ml) with 5 strokes of a Teflon/glass homogenizer. The resulting P2 fraction was loaded onto a discontinuous sucrose gradient (1 ml each of 0.85, 1.0, and 1.15 M sucrose in 6 mM Tris, pH 8.0) that was centrifuged at 82,500 ×g for 2 h. The synaptosome fraction (between 1 and 1.15 M sucrose) was collected, the volume was adjusted to 2 ml with buffer B, and 2 ml of buffer C (6 mM Tris, pH 8.0, 1% Triton-X) was added and rocked gently in a cold room for 15 min. The synaptosome fraction was then centrifuged at 32,800 × g for 20 min, the supernatant was discarded, and pellets were saved as PSD1. PSD1 was suspended in 4 ml of buffer D (6 mM Tris, pH 8.0, 0.5% Triton-X) and gently rocked in a cold room for 15 min. PSD1 was then centrifuged at 201,800 × g for 1 h. The resulting pellets were suspended in 70 ml of 1% SDS buffer, subjected to protein assay, and immuno-blotting as described above. Using this preparation, the PSD protein PSD-95 was progressively enriched from homogenates to synaptosome to PSD fraction (not shown).
Immunoprecipitation
Immunoprecipitation of GluR2 was performed essentially as described (57). Bilateral hippocampi were rapidly dissected and homogenized in 1 ml ice-cold lysis buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 1 mM EDTA) containing 1 mM NaF, 1 mM sodium orthovanadate, Complete inhibitor tablet (Roche Applied Science 11 836 170 001), 100 nM okadaic acid, and 1 mM phenylmethylsulfonyl fluoride. The detergent Triton X-100 was then added to the final concentration of 1%, and homogenate was incubated at 4 °C with end-over-end incubation for 1 h. Samples were then centrifuged for 20 min at 4 °C, and supernatant was collected and subjected to protein assay. 2 mg of each sample was incubated with agarose beads at 4 °C for 1 h, then centrifuged for 1 min at 3000 × g. Supernatant was removed, and samples were subjected to immunoprecipitation. 5 μg of mouse anti-GluR2 antibody (Zymed Laboratories Inc. 32-0300) was added to the sample and incubated at 4 °C for 1 h. 30 μl of agarose beads were then added to samples and incubated for 2 h at 4 °C with end-over-end rotation. Samples were then centrifuged for 1 min at 3000 × g, supernatant was removed, and beads were washed with 1 ml of ice-cold lysis buffer containing 0.5% Triton X-100 3 times. After the final wash, 60 μl of SDS-PAGE sample buffer was added, and samples were boiled for 5 min. Samples were then loaded on 4–15% SDS-PAGE gels with the necessary controls, run for 1.5 h at 100 V, and transferred to polyvinylidene difluoride membranes (Millipore IPVh00010) for 50 min at 100 V. Blots were blocked with 1% bovine serum albumin in 1× Tris-buffered saline containing 0.1% Tween 20 for 1 h. Blots were then incubated with primary antibodies rabbit phospho-Ser-831 (Chemicon AB5847) and rabbit phospho-Ser-845 (Chemicon AB5849) and actin as above overnight at 4 °C. Blots were washed 5 times with 1× Tris-buffered saline-Tween 20 (0.1%) for 5 min and incubated with alkaline phosphatase-conjugated goat anti-rabbit and goat anti-mouse antibodies for 1 h at room temperature. Blot were then incubated in enhanced chemifluorescence substrate (Amersham Biosciences RPN5785) for 5 min and imaged by molecular imager (Bio-Rad molecular imager FX). Immunoblot analysis was performed as described above. Blots were then stripped, washed, blocked, and re-analyzed using rabbit total GluR1 (Chemicon AB1504), mouse total GluR2(Zymed Laboratories Inc. 32-0300) antibodies. Control immunoprecipitations performed without antibody or without beads did not pull down GluR2 or GluR1 (not shown).
RESULTS
We determined the predominant pattern of GluR1 phosphorylation in rat hippocampus induced during contextual fear conditioning via quantitative immunoblot analysis with phospho-specific GluR1 antibodies. One hour after training in contextual fear conditioning, we observed a statistically significant increase in the ratio of Ser-831 phosphorylation to total GluR1 but no detectable change in the ratio of Ser-845 phosphorylation to total, as compared with sham-trained, paired controls (Fig. 1, A–D). Total levels of GluR1, normalized to actin on the same blots to control for gel loading, were unchanged (Fig. 1, E–H). This pattern of GluR1 phosphorylation 1 h after contextual fear-conditioning training is equivalent to that observed by Lee et al. (15) 1 h after induction of LTP in area CA1 of the hippocampus. Thus, the predominant GluR1 phosphorylation pattern in the hippocampus 1 h after fear conditioning resembles that induced during LTP.
FIGURE 1. Fear-conditioning training increases phosphorylation of GluR1 at the calmodulin-dependent protein kinase/protein kinase C site Ser-831.

A, ratio of GluR1 phosphorylated at Ser-831 to total GluR1 determined on the same immunoblots and normalized to simultaneously trained, cage-mate, sham control is increased 1 h after contextual fear-conditioning (FC) training (n = 8; *, p < 0.05). B, ratio of GluR1 phosphorylated at Ser-845 to total GluR1 is unchanged 1 h after training (n = 8). C, example immunoblot for data summarized in A using antibody against phosphorylated serine 831 (calmodulin-dependent protein kinase/protein kinase C site, phospho-Ser-831 (P-S831)). The blot was stripped and re-probed with C-terminal GluR1 antibody (Total GluR1). D, example immunoblot for data summarized in B using antibody against phosphorylated serine 845 (protein kinase A site, P-S845). Blots were stripped and re-probed with Total GluR1 antibody as in C. E and F, ratio of total GluR1 to actin immunoreactivity from the same blot is unchanged 1 h after contextual fear-conditioning training. E and F show data summaries from blots stripped and re-probed for total GluR1 after phospho-Ser-831 antibody and phospho-Ser-845 antibody, respectively (n = 8). G and H, example immunoblots showing total GluR1 and actin antibodies after phospho-Ser-831 and phospho-Ser-845 antibodies in G and H, respectively.
Although these LTP-like changes in GluR1 phosphorylation are associated with stimuli that induce contextual fear-conditioning learning, it is possible that they are not an effect of learning per se but, rather, are induced by the stress of the unconditioned stimulus, footshock. To control for this, we performed experiments in which animals are subjected to footshock but do not exhibit contextual fear conditioning behaviorally.
In the first control experiment (Shock/Remove), animals receive the same unconditioned stimulus (footshock) but do not have time to associate the unconditioned stimulus with the conditioned stimulus (context). Rats were placed in the conditioning chamber and immediately subjected to a footshock of the same total duration and amplitude used to induce contextual fear conditioning followed by immediate removal to their home cage. When contextual memory was tested 24 h after training, Shock/Remove rats do not exhibit significant memory recall compared with traditionally trained, control rats (Fig. 2A). Indeed, the behavior of the Shock/Remove group resembles that of sham-trained rats that have not been shocked at all during training (Fig. 2A). Thus, Shock/Remove training provides a group of animals subjected to footshock without learning the association between the context and the unconditioned stimulus (footshock), although it is likely that some other form of learning takes place in these animals.
FIGURE 2. Three behavioral experiments in which rats are shocked but do not exhibit contextual learning.
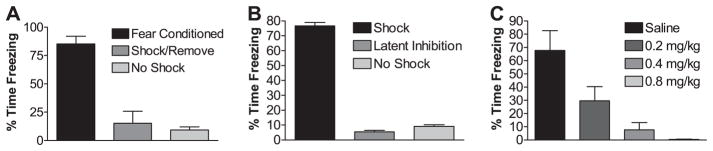
A, rats placed into a contextual training chamber, shocked immediately, and removed to the home cage immediately thereafter (Shock/Remove, n = 8) show dramatically reduced contextual fear memory measured 24 h after training compared with traditionally fear-conditioned rats (Fear Conditioned, n = 6). Contextual fear memory of the Shock/Remove group was not significantly different from sham-trained rats that did not receive footshock during training (No Shock, n = 8). B, rats pre-exposed to the training chamber for 16 h before training (Latent Inhibition, n = 7) showed dramatically reduced contextual fear memory measured 24 h after training compared with traditionally fear-conditioned rats that were pre-exposed to a completely different novel context (Shock, n = 8). Contextual fear memory of the Latent Inhibition group was similar in magnitude to sham-trained animals that did not receive footshock (No Shock, n = 8). C, NMDA receptor inhibition with MK-801 30 min before training significantly decreased contextual fear memory measured 24 h after training in a dose-dependent manner.
In the second control experiment (Latent Inhibition), rats were pre-exposed to the fear-conditioning chamber for many hours before traditional contextual fear conditioning training. Prior exposure to the context is known to inhibit subsequent associative learning to that particular context, a phenomenon known as latent inhibition. When contextual memory was tested 24 h after training in pre-exposed animals, this group did not exhibit significant memory (Fig. 2B). Control rats pre-exposed to a completely different context then trained in traditional fear conditioning in the fear-conditioning chambers exhibited robust learning (Fig. 2B, Shock). The behavior of the Latent Inhibition group resembled sham-trained rats that were not shocked during training (Fig. 2B). Thus, prior exposure to the context (conditioning chamber) prevents a subsequent aversive conditioning to the context. This allows for yet another group of animals subjected to footshock that do not learn to associate the context with the unconditioned stimulus.
Both LTP and contextual fear-conditioning learning are known to be blocked by N-methyl-D-aspartate (NMDA) receptor antagonists. We have replicated the NMDA receptor dependence of contextual fear conditioning to determine whether the learning-induced alterations in GluR1 phosphorylation were dependent on NMDA receptor activation similar to the GluR1 phosphorylation during LTP. The NMDA receptor antagonist MK-801 blocked contextual fear conditioning in a dose-dependent manner as has been previously demonstrated (Fig. 2C) (58, 59).
Footshock alone does not induce GluR1 phosphorylation. Shock/Remove rats were compared with similarly handled, unshocked controls 1 h after training, and no significant differences in phosphorylated or total GluR1 were observed (Fig. 3, A and B). Similarly, no significant differences in phosphorylation of GluR1 were seen in the Latent Inhibition group compared with similarly handled, unshocked controls (Fig. 3, C and D). Thus, footshock alone does not lead to the LTP-like GluR1 phosphorylation changes unless significant learning occurs.
FIGURE 3. Footshock alone is not sufficient to increase GluR1 phosphorylation, and the increase in GluR1 Ser-831 phosphorylation is NMDA receptor-dependent.

A and B, rats immediately shocked and removed (Shock/Remove, n = 8) showed no change in Ser-831 or Ser-845 phosphorylation compared with similarly handled, unshocked controls (Sham Control, n = 8) measured 1 h after training. C and D, rats trained after latent inhibition (Latent Inhibition, n = 8) showed no change in Ser-831 or Ser-845 phosphorylation compared with similarly treated, unshocked controls (Sham Control, n = 8) measured 1 h after training. E and F, rats trained in the presence of 0.4 mg/kg MK-801 (MK-801 Trained, n = 8) showed no change in Ser-831 or Ser-845 phosphorylation compared with similarly treated, unshocked controls (MK-801 Sham, n = 8) measured 1 h after training.
Learning-induced changes in GluR1 phosphorylation are dependent on NMDA receptor activation as they are in LTP. Fear conditioning training in the presence of the NMDA receptor antagonist MK-801 (0.4 mg/kg intraperitoneal 30 min before training) completely blocked the learning-induced increase in GluR1 phosphorylation at Ser-831 (Fig. 3, E and F).
We next wanted to better understand the onset and duration of GluR1 phosphorylation during learning. Phosphorylation of GluR1 at Ser-831 was dramatically and significantly increased 5 min after training (Fig. 4, A and C), whereas phosphorylation at Ser-845 was increased at 5 min but not significantly (Fig. 4, B and D). The increase in Ser-831 phosphorylation 5 min after training was also NMDA receptor-dependent as shown by preventing the increase by training in the presence of MK-801 (not shown, n = 10, p = 0.20). By 2 h after training in contextual fear conditioning, no changes in GluR1 phosphorylation were observed (Fig. 4, E–H). No change in total GluR1 levels was observed at either time point (not shown).
FIGURE 4. Learning-induced increase in Ser-831 phosphorylation is present as early as 5 min after training but is transient.

A, ratio of GluR1 phosphorylated at Ser-831 (S831) to total GluR1 is significantly increased 5 min after contextual fear conditioning training (FC; n = 8; *, p < 0.05). B, ratio of GluR1 phosphorylated at Ser-845 to total GluR1 is increased, although not significantly (n.s.), 5 min after contextual fear conditioning training (n = 11, p = 0.21). C and D, example immunoblots for A and B. E, GluR1 phosphorylation at Ser-831 (P-S831) is back to control levels 2 h after contextual fear conditioning training (n = 8). F, no change in GluR1 phosphorylation at Ser-845 2 h after contextual fear conditioning training (n = 8). G and H, example blots for E and F labeled as in C and D.
In an effort to demonstrate the synaptic localization of the learning-induced increase in phosphorylation of Ser-831 GluR1, we determined whether this increase can be demonstrated in hippocampal PSD fractions from fear-conditioned animals. We first isolated PSDs from bilateral hippocampi of fear-conditioned animals and sham-trained controls using standard subcellular fractionation methods (60–68) 5 min after fear-conditioning training. We then performed quantitative immunoblots for phosphorylated Ser-831 and Ser-845 as before. A significant increase in Ser-831 phosphorylation relative to total GluR1 was observed in PSDs isolated from fear-conditioned animals compared with sham-trained controls (Fig. 5). Again, no measurable change was observed in Ser-845 phosphorylation (Fig. 5) or total GluR1 phosphorylation (normalized to actin, p = 0.69, n = 12; normalized to control total GluR1 only, p = 0.27, n = 12; not shown). Thus, the increase in GluR1 phosphorylation in the hippocampus during learning occurs at synaptic GluR1 subunits.
FIGURE 5. Learning-induced increase in Ser-831 phosphorylation is present at synapses (postsynaptic density preparations).

A, ratio of GluR1 phosphorylated at Ser-831 (S831) to total GluR1 is significantly elevated in hippocampal PSDs made from fear-conditioned (FC) animals compared with PSDs from sham trained animals (n = 12; *, p < 0.05). B, no change in Ser-845 GluR1 phosphorylation (P-S845) in PSDs from fear-conditioned animals (n = 12, p = 0.94). C and D, example immunoblots for A and B.
GluR1-containing AMPA-type glutamate receptors in the hippocampus are thought to predominantly exist in heteromeric complexes with GluR2. Thus, we asked whether the increase in GluR1 phosphorylation occurs on GluR1 subunits after immunoprecipitation with GluR2 antibodies. We immunoprecipitated GluR2 from hippocampal homogenates of fear-conditioned and sham-trained controls 5 min after fear-conditioning training. Quantitative immunoblots of the resulting GluR2 immunoprecipitates were then performed with phosphorylated GluR1 Ser-831 and Ser-845 antibodies as before. A significant increase in Ser-831 relative to total GluR1 was observed in GluR2 immunoprecipitates isolated from fear-conditioned animals compared with controls (Fig. 6). No measurable change was observed in Ser-845 phosphorylation (Fig. 6) or total GluR1 (normalized to control total GluR1 only, p = 0.54, n = 18; not shown). Thus, the increase in GluR1 phosphorylation during learning occurs in heteromeric complexes with GluR2.
FIGURE 6. Increase in Ser-831 phosphorylation present in heteromeric complexes with GluR2.

A, ratio of GluR1 phosphorylated at Ser-831 to total GluR1 is significantly elevated in GluR2 immunoprecipitations from hippocampi from fear-conditioned animals compared with PSDs from sham trained controls (n = 18; *, p < 0.05). B, no significant change in Ser-845 GluR1 phosphorylation in GluR2 immunoprecipitations from fear-conditioned animals (n = 18, p = 0.25). C and D, example immunoblots for A and B. E, GluR2 immunoprecipitation (IP) from control hippocampus blotted with total GluR2, total GluR1, phospho (P)-Ser-831, and phospho-Ser-845 antibodies. Immunoprecipitation lanes represent total GluR2 immunoprecipitate from 1000 μg of starting hippocampal lysate. Homog lanes represent 200 μg of total protein from starting material lysate (20% of that used for immunoprecipitation).
DISCUSSION
Our data reveal an increase in Ser-831 GluR1 phosphorylation during learning reminiscent of that seen during LTP of naïve synapses (15). These changes occur at the PSD and in heteromeric GluR1/2 complexes. These data provide evidence for LTP-like biochemical changes at postsynaptic glutamate receptors in the hippocampus occurring during contextual fear learning, providing additional data linking LTP and memory formation in the brain.
Although LTP-like changes in GluR1 phosphorylation are correlated with learning, it is possible that they are induced by footshock alone. We have performed control experiments in which animals are subjected to footshock but do not exhibit fear conditioning. Animals subjected to immediate shock/removal and latent inhibition were compared with paired, unshocked controls, and no significant differences in phosphorylated or total GluR1 were observed. Thus, footshock alone does not alter GluR1 phosphorylation.
The change in GluR1 phosphorylation has a time course and magnitude similar to that observed after LTP induction in hippocampal slices (15). Our results cannot determine whether this represents a small increase in many hippocampal neurons or a large increase in a smaller number of neurons. The similar time course suggests that the LTP and learning-induced GluR1 phosphorylation may be functionally related, although this remains speculative.
Additionally, like LTP and behaviorally measured memory, these biochemical changes are dependent on NMDA receptor activation. It is certainly possible that blockade of NMDA receptors blocks learning by a mechanism other than blocking LTP; however, the NMDA receptor dependence of LTP, behavioral learning, and of the changes in GluR1 phosphorylation is compelling.
Although the studies linking LTP and memory in the literature are numerous, several studies have found instances in which changes in LTP do not result in the expected alterations in learning and memory(for an excellent review, see Ref. 4). The most compelling of these are instances where LTP is decreased and memory is increased (69). Other studies show decreased LTP with no effect on memory (70–78). Indeed there are many studies in which LTP is increased while memory is decreased or unchanged (69, 79–83). These studies make clear that a one-to-one correlation between memory and LTP is unlikely, although LTP does seem to be one important component of learning and memory.
Our data do not exclude a learning-associated change in Ser-845 phosphorylation in hippocampal synapses during contextual fear conditioning. There is considerable evidence suggesting elevations of cAMP and protein kinase A activity are involved with both LTP and learning (84–92). Although we provide evidence that LTP-like biochemical changes occur in the hippocampus during learning, our data cannot rule out the possibility that simultaneous LTD (Ser-845 de-phosphorylation), de-potentiation (Ser-831 dephosphorylation), and de-depression-induced (Ser-845 phosphorylation) biochemical changes occur. The present experiments merely examine the predominant, net GluR1 phosphorylation changes in the entire hippocampal volume. For example, if equal amounts of LTD and de-depression are occurring, we may see no net change in Ser-845 phosphorylation, although such changes may be occur at different synapses in the volume of tissue simultaneously. Likewise, if a significant amount of de-potentiation is occurring, favoring decreases in Ser-831 phosphorylation, we may be masking an even greater amount of GluR1 phosphorylation at different synapses or in different neurons. Finally, we may miss more subtle changes in GluR1 phosphorylation (e.g. at Ser-845) occurring in too few neurons or synapses to detect in the volume of hippocampal tissue. Thus, our data do not rule out changes in Ser-845 phosphorylation during learning. They merely highlight a net increase in Ser-831 phosphorylation.
The lack of longer-lasting increases in Ser-831 phosphorylation during learning and LTP can be explained in various ways. The number of potentiated synapses with phosphorylated GluR1 may decrease over time below the limits of biochemical detection in the volume of tissue. Alternatively, the GluR1 phosphorylation may diminish over time, allowing different synaptic plasticity mechanisms to predominate after the first 1–2 h.
Ours is not the first study to observe LTP-like biochemical changes in the hippocampus after training in contextual fear conditioning (53, 54, 93, 94). This study, however, is the first to demonstrate such changes in a final, downstream, postsynaptic effector of LTP such as GluR1 (40, 42, 95) in contextual fear conditioning and the first to show such changes occur in the PSD fraction and in heteromeric complexes with GluR2 subunits.
Our findings are also consistent with previous studies correlating biochemical changes with inhibitory avoidance training, another form of aversively motivated learning. Cammarota et al. (96) found that inhibitory avoidance training leads to increased Ca2+-independent and total calmodulin-dependent protein kinase activity, measured in post hoc kinase assays, immediately after training. The same study reported a delayed increase in synaptosomal GluR1 phosphorylation 120 min after training (96). This experiment was performed in a post hoc back phosphorylation kinase assay in which endogenous calmodulin-dependent protein kinase was activated artificially with Ca2+/calmodulin in synaptosomal homogenates and, therefore, does not reflect the in vivo phosphorylation state of GluR1 (96). A major strength of the Cammarota study (96) was the use of shocked controls that were subjected to similar foot-shock but did not acquire inhibitory avoidance learning. In a subsequent study, Bevilaqua et al. (97) directly demonstrated an increase in Ser-831 phosphorylation of GluR1, with no change in Ser-845 phosphorylation, in isolated PSDs 30 and 90 min after inhibitory avoidance training when compared with naïve unshocked, unhandled, home cage controls. This study did not, however, perform controls to rule out a nonspecific effect of footshock nor an effect of novel context exposure, although they did demonstrate that their effects were NMDA receptor-dependent (97). A recently published study by Whitlock et al. (12) independently demonstrated an increase in Ser-831 GluR1 phosphorylation during inhibitory avoidance learning that is strikingly similar in magnitude and time course to the present findings. Comparing inhibitory avoidance-trained animals to unshocked, “walk-through” controls, they showed a significant increase in Ser-831, but not Ser-845 phosphorylation at 5 and 30 min after training. As a control for footshock alone, they compared a group of shock only animals to unshocked, unhandled, naïve controls and found no difference. As in the present study, NMDA receptor antagonists during training prevented the increase in Ser-831 phosphorylation. They also demonstrated that this increase in Ser-831 phosphorylation occurred in synaptoneurosome preparations. However, synaptoneurosome preparations only partially enrich for synaptic proteins. Thus, this study was not able to confirm the synaptic localization of the increased GluR1 phosphorylation. Nor did they determine whether the increase in GluR1 occurred in heteromeric complexes with GluR2, the functional receptor complex thought to be present at synapses.
Overall, our data both confirm and extend previous findings of increased Ser-831 phosphorylation in the hippocampus after an aversive memory task. The contextual fear conditioning-induced increase in Ser-831 GluR1 phosphorylation is specifically associated with learning rather than a nonspecific effect of footshock or novel context exposure, as shown with two separate controls (shock/remove and latent inhibition), and can be observed in whole hippocampal homogenates. Interestingly, the increase in Ser-831 GluR1 phosphorylation during fear-conditioning consolidation in whole hippocampal homogenates is similar in magnitude and time course to that induced by inhibitory avoidance and by LTP (12, 15, 97).
Glutamate receptors may be found at the synapse or in various non-synaptic compartments including extrasynaptic plasma membrane or endosomal compartments. By demonstrating the learning-induced increase in Ser-831 phosphorylation in PSD preparations, we have further localized these changes to synaptic glutamate receptors. Using immunoprecipitation of GluR2, we have demonstrated directly that the increased Ser-831 phosphorylation during learning occurs in heteromeric complexes with GluR2. Thus, it is likely that our observed alterations in GluR1 phosphorylation during contextual fear conditioning occur at functional, synaptic receptors.
Although it is clear that the phosphorylation state of Ser-831 and Ser-845 is important for synaptic plasticity and learning and memory (16), we have now demonstrated for the first time that one of these sites is actively regulated at synapses during contextual fear conditioning in vivo. The pattern of GluR1 phosphorylation resembles that of LTP and appears to occur in heteromeric complexes with GluR2 at the PSD, providing an additional biochemical correlate suggesting that LTP occurs in the hippocampus during learning.
Acknowledgments
We thank Drs. Richard Huganir and Hey-Kyoung Lee for antibodies and protocols. We thank Divya Balasubramanian and Phillip Williams for technical assistance with behavioral experiments.
Footnotes
This work was supported by National Institutes of Health Grant MH65975 and by National Alliance for Research on Schizophrenia and Depression Young Investigator Award 2003 Leiber Investigator (to C. M. P.).
The abbreviations used are: LTP, long term potentiation; LTD, long term depression; AMPA, α-amino-3-hydrozy-5-methylisoxazole-4-propionic acid; NMDA, N-methyl-D-aspartate; PSD, postsynaptic density.
References
- 1.Matynia A, Kushner SA, Silva AJ. Annu Rev Genet. 2002;36:687–720. doi: 10.1146/annurev.genet.36.062802.091007. [DOI] [PubMed] [Google Scholar]
- 2.Shapiro ML, Eichenbaum H. Hippocampus. 1999;9:365–384. doi: 10.1002/(SICI)1098-1063(1999)9:4<365::AID-HIPO4>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 3.Kandel ER. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- 4.Martin SJ, Grimwood PD, Morris RG. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- 5.Green EJ, McNaughton BL, Barnes CA. J Neurosci. 1990;10:1455–1471. doi: 10.1523/JNEUROSCI.10-05-01455.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharp PE, McNaughton BL, Barnes CA. Brain Res. 1985;339:361–365. doi: 10.1016/0006-8993(85)90105-2. [DOI] [PubMed] [Google Scholar]
- 7.Moser E, Mathiesen I, Andersen P. Science. 1993;259:1324–1326. doi: 10.1126/science.8446900. [DOI] [PubMed] [Google Scholar]
- 8.Mitsuno K, Sasa M, Ishihara K, Ishikawa M, Kikuchi H. Physiol Behav. 1994;55:633–638. doi: 10.1016/0031-9384(94)90037-x. [DOI] [PubMed] [Google Scholar]
- 9.Green EJ, Greenough WT. J Neurophysiol. 1986;55:739–750. doi: 10.1152/jn.1986.55.4.739. [DOI] [PubMed] [Google Scholar]
- 10.Sweatt JD. Mechanisms of Memory. Academic Press, Inc; New York: 2003. [Google Scholar]
- 11.Andersen P, Moser E, Moser MB, Trommald M. J Physiol Paris. 1996;90:349. doi: 10.1016/s0928-4257(97)87917-x. [DOI] [PubMed] [Google Scholar]
- 12.Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- 13.Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 14.Soderling TR, Derkach VA. Trends Neurosci. 2000;23:75–80. doi: 10.1016/s0166-2236(99)01490-3. [DOI] [PubMed] [Google Scholar]
- 15.Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- 16.Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL. Cell. 2003;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- 17.Morris RG, Anderson E, Lynch GS, Baudry M. Nature. 1986;319:774–776. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- 18.Moser EI, Krobert KA, Moser MB, Morris RG. Science. 1998;281:2038–2042. doi: 10.1126/science.281.5385.2038. [DOI] [PubMed] [Google Scholar]
- 19.McNaughton BL, Barnes CA, Rao G, Baldwin J, Rasmussen M. J Neurosci. 1986;6:563–571. doi: 10.1523/JNEUROSCI.06-02-00563.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castro CA, Silbert LH, McNaughton BL, Barnes CA. Nature. 1989;342:545–548. doi: 10.1038/342545a0. [DOI] [PubMed] [Google Scholar]
- 21.Silva AJ, Paylor R, Wehner JM, Tonegawa S. Science. 1992;257:206–211. doi: 10.1126/science.1321493. [DOI] [PubMed] [Google Scholar]
- 22.Silva AJ, Wang Y, Paylor R, Wehner JM, Stevens CF, Tonegawa S. Cold Spring Harbor Symp Quant Biol. 1992;57:527–539. doi: 10.1101/sqb.1992.057.01.058. [DOI] [PubMed] [Google Scholar]
- 23.Grant SG, O’Dell TJ, Karl KA, Stein PL, Soriano P, Kandel ER. Science. 1992;258:1903–1910. doi: 10.1126/science.1361685. [DOI] [PubMed] [Google Scholar]
- 24.Tonegawa S, Tsien JZ, McHugh TJ, Huerta P, Blum KI, Wilson MA. Cold Spring Harbor Symp Quant Biol. 1996;61:225–238. [PubMed] [Google Scholar]
- 25.Mayford M, Mansuy IM, Muller RU, Kandel ER. Curr Biol. 1997;7:580–589. doi: 10.1016/s0960-9822(06)00287-9. [DOI] [PubMed] [Google Scholar]
- 26.Sweatt JD. Curr Biol. 2001;11:391–394. doi: 10.1016/s0960-9822(01)00216-0. [DOI] [PubMed] [Google Scholar]
- 27.Sweatt JD. Learn Mem. 1999;6:399–416. doi: 10.1101/lm.6.5.399. [DOI] [PubMed] [Google Scholar]
- 28.Roberson ED, English JD, Sweatt JD. Learn Mem. 1996;3:1–24. doi: 10.1101/lm.3.1.1. [DOI] [PubMed] [Google Scholar]
- 29.Ling DS, Benardo LS, Serrano PA, Blace N, Kelly MT, Crary JF, Sacktor TC. Nat Neurosci. 2002;5:295–296. doi: 10.1038/nn829. [DOI] [PubMed] [Google Scholar]
- 30.Pettit DL, Perlman S, Malinow R. Science. 1994;266:1881–1885. doi: 10.1126/science.7997883. [DOI] [PubMed] [Google Scholar]
- 31.Malinow R, Schulman H, Tsien RW. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 32.Malinow R, Madison DV, Tsien RW. Nature. 1988;335:820–824. doi: 10.1038/335820a0. [DOI] [PubMed] [Google Scholar]
- 33.Powell CM, Johnston D, Sweatt JD. J Biol Chem. 1994;269:27958–27963. [PMC free article] [PubMed] [Google Scholar]
- 34.Morishita W, Connor JH, Xia H, Quinlan EM, Shenolikar S, Malenka RC. Neuron. 2001;32:1133–1148. doi: 10.1016/s0896-6273(01)00554-2. [DOI] [PubMed] [Google Scholar]
- 35.Mulkey RM, Endo S, Shenolikar S, Malenka RC. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- 36.Mulkey RM, Herron CE, Malenka RC. Science. 1993;261:1051–1055. doi: 10.1126/science.8394601. [DOI] [PubMed] [Google Scholar]
- 37.Thiels E, Kanterewicz BI, Knapp LT, Barrionuevo G, Klann E. J Neurosci. 2000;20:7199–7207. doi: 10.1523/JNEUROSCI.20-19-07199.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Norman ED, Thiels E, Barrionuevo G, Klann E. J Neurochem. 2000;74:192–198. doi: 10.1046/j.1471-4159.2000.0740192.x. [DOI] [PubMed] [Google Scholar]
- 39.Jouvenceau A, Billard JM, Haditsch U, Mansuy IM, Dutar P. Eur J Neurosci. 2003;18:1279–1285. doi: 10.1046/j.1460-9568.2003.02831.x. [DOI] [PubMed] [Google Scholar]
- 40.Song I, Huganir RL. Trends Neurosci. 2002;25:578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- 41.Roche KW, Tingley WG, Huganir RL. Curr Opin Neurobiol. 1994;4:383–388. doi: 10.1016/0959-4388(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 42.Malinow R. Philos Trans R Soc Lond B Biol Sci. 2003;358:707–714. doi: 10.1098/rstb.2002.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soderling TR. Neurochem Int. 1996;28:359–361. doi: 10.1016/0197-0186(95)00098-4. [DOI] [PubMed] [Google Scholar]
- 44.Oh MC, Derkach VA. Nat Neurosci. 2005;8:853–854. doi: 10.1038/nn1476. [DOI] [PubMed] [Google Scholar]
- 45.Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 46.Barria A, Derkach V, Soderling T. J Biol Chem. 1997;272:32727–32730. doi: 10.1074/jbc.272.52.32727. [DOI] [PubMed] [Google Scholar]
- 47.Derkach V, Barria A, Soderling TR. Proc Natl Acad Sci U S A. 1999;96:3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. J Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boehm J, Malinow R. Biochem Soc Trans. 2005;33:1354–1356. doi: 10.1042/BST0331354. [DOI] [PubMed] [Google Scholar]
- 50.Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- 51.Oh MC, Derkach VA, Guire ES, Soderling TR. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- 52.Powell CM, Schoch S, Monteggia L, Barrot M, Matos MF, Feldmann N, Sudhof TC, Nestler EJ. Neuron. 2004;42:143–153. doi: 10.1016/s0896-6273(04)00146-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR. Nat Neurosci. 1998;1:595–601. doi: 10.1038/2830. [DOI] [PubMed] [Google Scholar]
- 54.Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- 55.Mammen AL, Kameyama K, Roche KW, Huganir RL. J Biol Chem. 1997;272:32528–32533. doi: 10.1074/jbc.272.51.32528. [DOI] [PubMed] [Google Scholar]
- 56.Carlin RK, Grab DJ, Cohen RS, Siekevitz P. J Cell Biol. 1980;86:831–845. doi: 10.1083/jcb.86.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kjoller C, Diemer NH. Neurochem Int. 2000;37:7–15. doi: 10.1016/s0197-0186(00)00008-5. [DOI] [PubMed] [Google Scholar]
- 58.Kim JJ, DeCola JP, Landeira-Fernandez J, Fanselow MS. Behav Neurosci. 1991;105:126–133. doi: 10.1037//0735-7044.105.1.126. [DOI] [PubMed] [Google Scholar]
- 59.Bordi F, Marcon C, Chiamulera C, Reggiani A. Neuropharmacology. 1996;35:1557–1565. doi: 10.1016/s0028-3908(96)00101-3. [DOI] [PubMed] [Google Scholar]
- 60.Walikonis RS, Jensen ON, Mann M, Provance DW, Jr, Mercer JA, Kennedy MB. J Neurosci. 2000;20:4069–4080. doi: 10.1523/JNEUROSCI.20-11-04069.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kennedy MB. Trends Neurosci. 1997;20:264–268. doi: 10.1016/s0166-2236(96)01033-8. [DOI] [PubMed] [Google Scholar]
- 62.Hunt CA, Schenker LJ, Kennedy MB. J Neurosci. 1996;16:1380–1388. doi: 10.1523/JNEUROSCI.16-04-01380.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kennedy MB. Curr Opin Neurobiol. 1993;3:732–737. doi: 10.1016/0959-4388(93)90145-o. [DOI] [PubMed] [Google Scholar]
- 64.Cho KO, Hunt CA, Kennedy MB. Neuron. 1992;9:929–942. doi: 10.1016/0896-6273(92)90245-9. [DOI] [PubMed] [Google Scholar]
- 65.Kennedy MB, Bennett MK, Erondu NE. Proc Natl Acad Sci U S A. 1983;80:7357–7361. doi: 10.1073/pnas.80.23.7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheng D, Hoogenraad CC, Rush J, Ramm E, Schlager MA, Duong DM, Xu P, Wijayawardana SR, Hanfelt J, Nakagawa T, Sheng M, Peng J. Mol Cell Proteomics. 2006;5:1158–1170. doi: 10.1074/mcp.D500009-MCP200. [DOI] [PubMed] [Google Scholar]
- 67.Chen X, Vinade L, Leapman RD, Petersen JD, Nakagawa T, Phillips TM, Sheng M, Reese TS. Proc Natl Acad Sci U S A. 2005;102:11551–11556. doi: 10.1073/pnas.0505359102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peng J, Kim MJ, Cheng D, Duong DM, Gygi SP, Sheng M. J Biol Chem. 2004;279:21003–21011. doi: 10.1074/jbc.M400103200. [DOI] [PubMed] [Google Scholar]
- 69.Pavlov I, Voikar V, Kaksonen M, Lauri SE, Hienola A, Taira T, Rauvala H. Mol Cell Neurosci. 2002;20:330–342. doi: 10.1006/mcne.2002.1104. [DOI] [PubMed] [Google Scholar]
- 70.Meiri N, Sun MK, Segal Z, Alkon DL. Proc Natl Acad Sci U S A. 1998;95:15037–15042. doi: 10.1073/pnas.95.25.15037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bannerman DM, Chapman PF, Kelly PA, Butcher SP, Morris RG. J Neurosci. 1994;14:7404–7414. doi: 10.1523/JNEUROSCI.14-12-07404.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC, Kandel ER. Cell. 1996;87:1015–1023. doi: 10.1016/s0092-8674(00)81796-1. [DOI] [PubMed] [Google Scholar]
- 73.Huang YY, Bach ME, Lipp HP, Zhuo M, Wolfer DP, Hawkins RD, Schoonjans L, Kandel ER, Godfraind JM, Mulligan R, Collen D, Carmeliet P. Proc Natl Acad Sci U S A. 1996;93:8699–8704. doi: 10.1073/pnas.93.16.8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Minichiello L, Korte M, Wolfer D, Kuhn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- 75.Ho N, Liauw JA, Blaeser F, Wei F, Hanissian S, Muglia LM, Wozniak DF, Nardi A, Arvin KL, Holtzman DM, Linden DJ, Zhuo M, Muglia LJ, Chatila TA. J Neurosci. 2000;20:6459–6472. doi: 10.1523/JNEUROSCI.20-17-06459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Allen PB, Hvalby O, Jensen V, Errington ML, Ramsay M, Chaudhry FA, Bliss TV, Storm-Mathisen J, Morris RG, Andersen P, Greengard P. J Neurosci. 2000;20:3537–3543. doi: 10.1523/JNEUROSCI.20-10-03537.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nosten-Bertrand M, Errington ML, Murphy KP, Tokugawa Y, Barboni E, Kozlova E, Michalovich D, Morris RG, Silver J, Stewart CL, Bliss TV, Morris RJ. Nature. 1996;379:826–829. doi: 10.1038/379826a0. [DOI] [PubMed] [Google Scholar]
- 78.Zamanillo D, Sprengel R, Hvalby O, Jensen V, Burnashev N, Rozov A, Kaiser KM, Koster HJ, Borchardt T, Worley P, Lubke J, Frotscher M, Kelly PH, Sommer B, Andersen P, Seeburg PH, Sakmann B. Science. 1999;284:1805–1811. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]
- 79.Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z. Neuron. 2002;35:121–133. doi: 10.1016/s0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- 80.Gu Y, McIlwain KL, Weeber EJ, Yamagata T, Xu B, Antalffy BA, Reyes C, Yuva-Paylor L, Armstrong D, Zoghbi H, Sweatt JD, Paylor R, Nelson DL. J Neurosci. 2002;22:2753–2763. doi: 10.1523/JNEUROSCI.22-07-02753.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O’Dell TJ, Grant SG. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- 82.Walther T, Balschun D, Voigt JP, Fink H, Zuschratter W, Birchmeier C, Ganten D, Bader M. J Biol Chem. 1998;273:11867–11873. doi: 10.1074/jbc.273.19.11867. [DOI] [PubMed] [Google Scholar]
- 83.Jun K, Choi G, Yang SG, Choi KY, Kim H, Chan GC, Storm DR, Albert C, Mayr GW, Lee CJ, Shin HS. Learn Mem. 1998;5:317–330. [PMC free article] [PubMed] [Google Scholar]
- 84.Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- 85.Frey U, Huang YY, Kandel ER. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- 86.Huang YY, Kandel ER. Learn Mem. 1994;1:74–82. [PubMed] [Google Scholar]
- 87.Huang YY, Zakharenko SS, Schoch S, Kaeser PS, Janz R, Sudhof TC, Siegelbaum SA, Kandel ER. Proc Natl Acad Sci U S A. 2005;102:9365–9370. doi: 10.1073/pnas.0503777102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Impey S, Mark M, Villacres EC, Poser S, Chavkin C, Storm DR. Neuron. 1996;16:973–982. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- 89.Matthies H, Reymann KG. Neuroreport. 1993;4:712–714. doi: 10.1097/00001756-199306000-00028. [DOI] [PubMed] [Google Scholar]
- 90.Otmakhova NA, Otmakhov N, Mortenson LH, Lisman JE. J Neurosci. 2000;20:4446–4451. doi: 10.1523/JNEUROSCI.20-12-04446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Roberson ED, Sweatt JD. J Biol Chem. 1996;271:30436–30441. doi: 10.1074/jbc.271.48.30436. [DOI] [PubMed] [Google Scholar]
- 92.Villacres EC, Wong ST, Chavkin C, Storm DR. J Neurosci. 1998;18:3186–3194. doi: 10.1523/JNEUROSCI.18-09-03186.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Levenson JM, O’Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- 94.Hall J, Thomas KL, Everitt BJ. Nat Neurosci. 2000;3:533–535. doi: 10.1038/75698. [DOI] [PubMed] [Google Scholar]
- 95.Malinow R, Malenka RC. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 96.Cammarota M, Bernabeu R, Levi De Stein M, Izquierdo I, Medina JH. Eur J Neurosci. 1998;10:2669–2676. doi: 10.1046/j.1460-9568.1998.00254.x. [DOI] [PubMed] [Google Scholar]
- 97.Bevilaqua LR, Medina JH, Izquierdo I, Cammarota M. Neuroscience. 2005;136:397–403. doi: 10.1016/j.neuroscience.2005.08.007. [DOI] [PubMed] [Google Scholar]