

Abstract
Metallo-β-lactamases (MBLs) are a growing threat to the use of almost all clinically used β-lactam antibiotics. The identification of broad-spectrum MBL inhibitors is hampered by the lack of a suitable screening platform, consisting of appropriate substrates and a set of clinically relevant MBLs. We report procedures for the preparation of a set of clinically relevant metallo-β-lactamases (i.e., NDM-1 (New Delhi MBL), IMP-1 (Imipenemase), SPM-1 (São Paulo MBL), and VIM-2 (Verona integron-encoded MBL)) and the identification of suitable fluorogenic substrates (umbelliferone-derived cephalosporins). The fluorogenic substrates were compared to chromogenic substrates (CENTA, nitrocefin, and imipenem), showing improved sensitivity and kinetic parameters. The efficiency of the fluorogenic substrates was exemplified by inhibitor screening, identifying 4-chloroisoquinolinols as potential pan MBL inhibitors.
Introduction
Selection pressure caused by frequent use of β-lactam antibiotics, including penicillins, cephalosporins, carbapenems, and monobactams, has resulted in the emergence and dissemination of highly efficient resistance mechanisms in clinically relevant bacterial pathogens. This rapid adaptation of bacterial strains to commonly used antibiotics is an increasing global threat to public health care.1,2 An important mechanism of resistance to β-lactam antibiotics, including clinically challenging Gram-negative organisms, involves β-lactamase catalysis. β-Lactamases can be classified into those employing a nucleophilic serinyl residue at their active site (serine-β-lactamases, SBLs, classes A, C, and D) and those employing one or two zinc ions to promote hydrolysis (metallo-β-lactamases, MBLs, class B).3 The combination of a penicillin antibiotic and an SBL inhibitor has substantially extended the utility of the former, as, for example, in the case of amoxicillin and clavulanic acid (Augmentin).4 In addition to clavulanic acid, two other class A (penicillinase) β-lactamase inhibitors are used, tazobactam and sulbactam; all three inhibitors are themselves β-lactams, though in contrast to penicillins they react with SBLs to form relatively stable acyl-enzyme complexes.5 These SBL inhibitors, however, appear to have no effect on MBLs.6 β-Lactam antibiotics have been developed that possess resistance to SBLs, or as in the case of carbapenems, they have been developed to be inhibitors or relatively poor substrates.7 However, since the pioneering introduction of the SBL-targeting inhibitors, progress in the development of clinically useful, β-lactamase-specific inhibitors has been limited. In the case of SBLs, at least one compound, Avibactam, is currently in clinical trials as a combined class A and class C SBL inhibitor.8
MBLs, which, unlike the SBLs, are not structurally or mechanistically related to the penicillin-binding proteins (PBPs), were first identified approximately 50 years ago.9 However, as a result of their apparently restricted distribution to the chromosomes of less pathogenic species, they were long not considered to represent a significant threat to the clinical effectiveness of β-lactams. The emergence of horizontally acquired MBLs, where horizontal gene transfer permits genetic material to be transferred between individual bacteria of the same species or between different species, has led to their extensive dissemination across geographical and species boundaries and now threatens the effectiveness of almost all β-lactams, including carbapenems (i.e., “last resort” antibiotics);10 the monobactam aztreonam is a current exception. MBLs identified on transferrable plasmids (e.g., the NDM-, IMP-, VIM-, GIM-, and SPM-type enzymes) are considered a prevalent and immediate clinical problem.11 Various structurally diverse types of MBL inhibitors have been described, including carboxylic/succinic acids,12 triazoles/tetrazoles,13 thiols,14 trifluoromethyl ketones,15 and others.16 To date, no clinically useful MBL inhibitor has been reported. This, in part, may reflect the technical and scientific challenges in the development of an MBL−β-lactam-based combination therapy. There is also extensive structural diversity in the MBL family, particularly in the proposed mobile loop regions around the active site.17 For an MBL inhibitor to be clinically useful, it is possible that more than one MBL will need to be inhibited. In an effort to help enable work that will lead to the development of useful MBL inhibitors, we here describe an assay platform for clinically relevant MBLs, including protein production procedures and assay conditions using both chromogenic and fluorogenic substrates.
Results and Discussion
Several screening methods for the in vitro and in vivo detection of β-lactamases have been reported.18 Representative substrates currently in use for β-lactamases include chromogenic cephalosporin-based substrates, such as CENTA,19 PADAC,20 and nitrocefin,21 cephalosporin-based fluorogenic substrates,22 bioluminescent probes,23 and fluorescence resonance energy transfer (FRET)-based substrates.24 These substrates have mainly been applied in research centered around SBLs and only in one case on MBLs (L1 and CcrA).22c The large-scale application of these type of compounds in automated high-throughput screening (HTS) for MBL inhibitors is, however, hampered by their lengthy and often difficult synthesis and the high costs associated with these substrates. More importantly, these substrates suffer from poor substrate recognition by some MBLs as a result of the high diversity of this enzyme family whose members vary significantly in sequence, structure, and substrate specificity, thus making it hard to use a single substrate for broad MBL activity screening. The development of new assays for broad-range MBL activity screening, based on hydrolysis of chromogenic or fluorogenic β-lactams, would, however, significantly facilitate inhibitor identification. Not unimportantly, large-scale inhibitor screening against different MBLs needs an inexpensive substrate, thus requiring a simple, scalable, and cost-efficient synthetic route. To develop assay procedures suitable for a range of MBLs, we targeted four clinically relevant enzymes as well as the MBL from Bacillus cereus (BcII), which has been extensively used as a model enzyme. The clinically relevant enzymes chosen were NDM-1 (New Delhi MBL), IMP-1 (Imipenemase), SPM-1 (São Paulo MBL), and VIM-2 (Verona integron-encoded MBL). To identify MBL substrates useful for efficient inhibitor screening, we prepared and tested substrates on the above-mentioned panel of MBLs. These substrates included three chromogenic substrates, i.e., imipenem, nitrocefin, and CENTA, and five coumarin-linked substrates, i.e., FC1–FC5 (Figure 1). Fluorescent substrates FC1 and FC2 release 7-mercapto-4-methylcoumarin upon hydrolysis, resulting in a decrease of fluorescence. Conversely, fluorogenic substrates FC3–FC5 liberate 7-hydroxycoumarin upon hydrolysis of the lactam ring, producing an increased fluorescence signal (Figure 2).
Figure 1.

Substrates for metallo-β-lactamase activity measurements.
Figure 2.
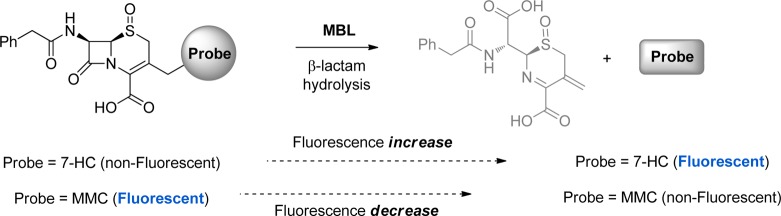
Hydrolysis of substrates by MBLs, resulting in either an increase or a decrease of fluorescence.
Protein Production
Details are provided in the Supporting Information. For the production of recombinant BcII, IMP-1, and NDM-1 MBLs in Escherichia coli, vectors that have been previously described were used, i.e., the pET9a-BcII, pET-26b IMP-1, and pOPINF NDM-1 plasmids.25 In the case of the MBLs VIM-2 and SPM-1, new pTriEx-based pOPINF vectors were constructed.26 These vectors encode the mature VIM-2 and SPM-1 sequences (i.e., with the periplasmic export sequences removed) fused to N-terminal hexahistidine tags, cleavable with 3C protease. The VIM-2 and SPM-1 genes were codon optimized for E. coli (Genscript, Piscataway, NJ) and inserted into the pOPINF vector. All the MBLs were produced using in-house-constructed E. coli BL21(DE3) pLyS cells, which were grown in 2TY medium. For the protein purifications standard procedures were used. The purity of the MBL proteins was determined to be >95% by SDS–PAGE analysis (see the Supporting Information, Figure SI_4).
Synthesis of Substrates
CENTA was prepared using a modified literature procedure.27 Cleavage of 5,5′-dithiobis(2-nitrobenzoic acid) by dithiothreitol (DTT) yielded 3-carboxyl-4-nitrothiophenol, which was subsequently used to convert commercially available cephalothin into CENTA by heating (65 °C, 3 h). Use of chromatographically purified 3-carboxyl-4-nitrothiophenol gave CENTA in high purity and satisfactory yield after a single acid/base extraction.28
Fluorogenic cephalosporin 1 (FC1) was prepared from commercially available p-methoxybenzyl (PMB) ester-protected chlorocephalosporin (CC) via substitution with 7-mercapto-4-methylcoumarin (MMC), followed by acid-mediated PMB deprotection (Scheme 1A). Treatment of CC with MMC resulted in the formation of both the Δ2- and Δ3-alkene isomers of compound 1. Subjecting the Δ2–Δ3 isomer mixture to acid-mediated PMB deprotection conditions led to complete deprotection within 1 h, generating FC1 (Δ2–Δ3 mix). To circumvent the Δ2–Δ3 isomerization issue, oxidation of the thiazine sulfur atom with mCPBA was performed prior to alkylation (Scheme 1A).29 The Δ3-alkene (S)-sulfoxide 2 was obtained in good yield (72%); subsequent alkylation with MMC in the presence of N,N′-diisopropylethylamine gave 3 as a single isolate isomer in reasonable yield (49%). Acid-mediated PMB deprotection yielded FC2 in good yield (88%). Notably, MMC appears to be nonfluorescent in its free form, whereas the thioether cephalosporin derivatives (i.e., 1 and 3) showed significant fluorescence. While this was not our objective, FC2 has potential as an “inverse fluorogenic” substrate (for spectroscopic data see the Supporting Information, Figure SI_1).
Scheme 1. Synthesis of FC1-5: (A) Thiacoumarin Cephalosporin, (B) Hydroxylcoumarin Cephalosporins.

Reagents and conditions: (a) MMC, DiPEA, DMF, rt, 2 h; (b) TFA/anisole (5:1), 0 °C, 30 min; (c) mCPBA (1 equiv), CH2Cl2, 0 °C, 1 h; (d) NaI (10 equiv), acetone, rt, 2 h, (e) 7-HC, K2CO3, MeCN, rt, 4 h; (f) mCPBA (2 equiv), CH2Cl2, 0 °C, 3 h.
With the aim of developing a fluorogenic MBL substrate, we focused on 7-hydroxycoumarin (7-HC) cephalosporin derivatives, with the knowledge that umbelliferone shows strong fluorescence in its free form while being nearly nonfluorescent when ether-derivatized.30 During the course of our investigations, Rao and co-workers reported the synthesis and application of FC3 and FC4 as SBL fluorogenic substrates (specific for BalC over TEM-1).31 Their reported procedure (with minimal Δ2–Δ3 isomerization) for the synthesis of FC3 and FC4 proved to be efficient and yielded the desired fluorogenic probes (FC3 and FC4) in satisfactory yields (Scheme 1B). Besides substrates FC3 (sulfide) and FC4 ((S)-sulfoxide), we also prepared substrate FC5 (sulfone). Treatment of compound 4 with 2 equiv of mCPBA gave clean conversion to compound 6, which upon PMB deprotection yielded substrate FC5 in good yield.
Metallo-β-lactamase Assays
Initially time-dependent changes in absorbance and/or emission spectra upon hydrolysis of the potential MBL substrates were recorded to investigate the optimal wavelengths for analysis (see the Supporting Information, Figures SI_2 and SI_3). For the frequently used chromogenic substrates, i.e., imipenem (carbapenem) and nitrocefin and CENTA (cephalosporins), this resulted in preferred readout absorption wavelengths of 300, 405, and 492 nm, respectively. For fluorogenic substrates FC3–FC5 the absorption and emission spectra of umbelliferon in HEPES assay buffer (pH 7.5) were measured and their corresponding optimal wavelengths applied in the fluorescence-based assay (λex = 380 nm and λem = 460 nm).32
We then determined kinetic parameters for different MBL–substrate combinations. The data for the chromogenic substrates (Table 1) are generally in good agreement with previously reported data,27,39 with the only substantial dissimilarity being the lower KM values obtained by us in the case of CENTA, possibly because of increased purity of the substrate. Direct quantitative comparison of the obtained data with reported values is, however, difficult as a result of variations in protein production conditions, concentration of metal ions added, etc. The improved stability of CENTA compared to imipenem renders it an attractive substrate for inhibitor identification. However, MBL inhibition by the hydrolyzed cephalosporin products (for BcII,40 CphA,41 and Sfh-I42) or the liberated thiophenol from CENTA (for IMP-1) could complicate interpretation of the results. Examination of the data in Table 1 reveals the high sensitivity of the fluorogenic MBL substrates, which enable the use of enzyme concentrations (generally ≫1 nM) well below those used with chromogenic substrates (generally 1–50 nM).
Table 1. Kinetic Data for Chromogenic and Fluorogenic Substrates on MBLs.
| literature data |
||||||||
|---|---|---|---|---|---|---|---|---|
| entry | enzyme | substrate | [E]a (pM) | KM (μM) | kcat (s–1) | kcat/KM (μM–1·s–1) | kcat/KM (μM–1·s–1) | ref |
| 1 | NDM-1 | nitrocefin | 1000 | 8.8 ± 1.1 | 25.3 | 2.9 | 4.1 | (33) |
| 2 | CENTA | 1000 | 34.6 ± 6.6 | 92.5 | 2.7 | NDb | ND | |
| 3 | imipenem | 250 | 111.2 ± 11.7 | 398.5 | 3.6 | 0.5 | (33) | |
| 4 | FC3 | 50 | 17.6 ± 1.7 | 102.6 | 5.8 | |||
| 5 | FC4 | 50 | 4.0 ± 0.8 | 125.7 | 32.0 | |||
| 6 | FC5 | 50 | 2.4 ± 0.2 | 298 | 124.1 | |||
| 7 | VIM-2 | nitrocefin | 100 | 7.2 ± 0.6 | 225.6 | 31.2 | 42.7 | (34) |
| 8 | CENTA | 1000 | 26.1 ± 3.1 | 48.2 | 1.8 | ND | ND | |
| 9 | imipenem | 1000 | 37.8 ± 11.5 | 41.6 | 1.1 | 3.8 | (34) | |
| 10 | FC4 | 50 | 6.3 ± 0.4 | 125.5 | 20.0 | |||
| 11 | FC5 | 100 | 15.2 ± 1.2 | 291.2 | 19.1 | |||
| 12 | IMP-1 | nitrocefin | 50 | 55.7 ± 4.8 | 2794 | 50.2 | 2.3 | (35) |
| 13 | CENTA | 100 | 17.1 ± 2.0 | 431.9 | 25.3 | 2.0 | (27) | |
| 14 | imipenem | 50 | 42.7 ± 6.7 | 1200 | 28.1 | 1.2 | (35) | |
| 15 | FC4 | 1 | 15.2 ± 0.7 | 12200 | 807 | |||
| 16 | FC5 | 5 | 16.8 ± 1.9 | 8706 | 517 | |||
| 17 | SPM-1 | nitrocefin | 50 × 103 | 16.0 ± 3.7c | 0.7 | 0.04 | 0.12 | (36) |
| 18 | CENTA | 10 × 103 | 25.3 ± 6.1 | 6.1 | 0.2 | ND | ND | |
| 19 | imipenem | 10 × 103 | 330.9 ± 35.5 | 37.2 | 0.1 | 1.0 | (36) | |
| 20 | FC4 | 500 | 2.5 ± 0.2 | 8.7 | 3.5 | |||
| 21 | FC5 | 1000 | 2.7 ± 0.2 | 27.38 | 10.3 | |||
| 22 | BcII | nitrocefin | 1000 | 8.1 ± 1.0 | 14.9 | 1.8 | 0.64 | (37) |
| 23 | CENTA | 1000 | 135.9 ± 16.4 | 8.9 | 0.06 | 0.15 | (27) | |
| 24 | imipenem | 1000 | >1000 | 583 | 0.4 | 0.13 | (38) | |
| 25 | FC4 | 1000 | 24.4 ± 1.7 | 17.1 | 0.7 | |||
| 26 | FC5 | 1000 | 40.4 ± 3.3 | 151.1 | 3.7 | |||
The enzyme concentrations of the purified proteins were determined using a NanoDrop spectrometer; concentrations of diluted solutions used in the assay were calculated from the original concentration.
ND = not determined.
The following apparent Ki value for substrate/product inhibition was determined: 36.6 ± 15.2.
In the case of the combination of NDM-1 and FC3–FC5, up to 5–20 times lower enzyme concentration was used as compared to enzyme concentrations required for imipenem or nitrocefin. Further testing of NDM-1 with the three fluorogenic substrates FC3–FC5, with different sulfur oxidation states, disclosed significantly different kcat/KM values (up to 20-fold), implying catalytic efficiency is altered by the oxidation state of the thiazine sulfur atom. We observed that the stability of FC3 in aqueous solution is substantially lower than that of FC4 and FC5. Autohydrolysis studies in buffer at room temperature showed substantial hydrolysis of FC3 after 3 h, while both FC4 and FC5 were found to be stable for at least 8 h. Solutions of FC4 and FC5 could be used for 1–2 days when stored on ice. Moreover, DMSO stock solutions, stored at −20 °C, could be used several months after preparation without any detectable decomposition. Storage of compounds FC4 and FC5 as solids at −20 °C enabled use of these compounds over a prolonged period of time (i.e., months). As a result, FC4 and FC5 were tested on the other MBLs.
For nearly all the clinically important MBLs, FC4 and FC5 showed higher kcat/KM values than the chromogenic substrates (with VIM-2 being the only exception). On analysis of the VIM-2 results, we found that FC5 is preferred over FC4 and performs equally well as nitrocefin, having similar kcat values though slightly different KM values (7.2 μM vs 15.2 μM, respectively) and consequently different kcat/KM values (31.2 μM–1·s–1 vs 19.1 μM–1·s–1).
The kinetic data of the five substrates for IMP-1 reveal that FC4 outperforms all other substrates mainly as a result of its high kcat value (i.e., 807 μM–1·s–1). As depicted in Figure 2A substrate concentrations up to 100 μM could be applied without perturbing the measurement. Although FC4 has a low KM compared to the other substrates tested on IMP-1, its high sensitivity allows for accurate measurement as indicted by the low interexperimental error (Figure 3A, experiment performed in triplicate).43 As a result of the high sensitivity of both FC4 and FC5, a 20-fold lower IMP-1 concentration could be used (i.e., picomolar enzyme concentration range) while retaining a KM value in the micromolar range, thus allowing for the detection of weak binding fragments or slow-binding inhibitors.44
Figure 3.

(Top) IMP-1 with FC4. (Bottom) SPM-1 with nitrocefin (substrate or product inhibition). Errors are reported as standard errors, n = 3.
In the case of SPM-1, both FC4 and FC5 outperform all the tested chromogenic substrates. Interestingly, we found that SPM-1 is inhibited by nitrocefin/nitrocefin-derived species at a concentration of 36 μM (see Figure 3B and Table 1). Although β-lactams can act as inhibitors of MBLs, as is apparent for nitrocefin and imipenem at high concentrations (e.g., nitrocefin for SPM-1, this paper, and for CphA and Sfh-I, refs (45) and (42), respectively); the high sensitivity of the here reported fluorogenic substrates (FC4 and FC5) allows for the use of extremely low substrate concentrations.
In Silico Studies
In an attempt to investigate the different substrate binding constants of nitrocefin, imipenem, CENTA, and FC4 for the different MBLs, we performed in silico docking studies. Docking of nitrocefin, imipenem, CENTA, and FC4 in IMP-1, NDM-1, and VIM-2 (PDB IDs 1JJT,12b3Q6X,46 and 1KO3,47 respectively) was performed using AutoDock 448 and SPROUT software.49 After analysis of the obtained unhydrolyzed–enzyme models (see the Supporting Information), the different docking poses, calculated by Autodock 4 and SPROUT, were scored and compared to the substrate binding constants (KM) (Table SI_1, Supporting Information). Assessment of the SPROUT docking scores generally reveals a better correlation with the kinetic parameters obtained in vitro (specifically KM values in the cases of NDM-1 and VIM-2) compared to the docking scores generated by Autodock 4. However, for all three enzymes, the highest ranking substrate in terms of the SPROUT and Autodock scores is FC4, which was also measured to have the highest binding affinity for all three enzymes. Although in-depth structural analyses are required, these modeling results suggest that the cephalosporins may bind to the active site in more than one orientation, including catalytically nonproductive ones. Overall, these results emphasize the importance of experimentally determining optimal MBL–substrate combinations and the need for more structural analyses on MBL–substrate complexes.
Inhibitor Screening
To achieve clinical utility, MBL inhibitors may need to act on more than one MBL subfamily. This is potentially challenging because of the generally low sequence similarity between MBLs of different subfamilies (B1, B2, B3). Most clinically relevant MBLs are from the B1 subfamily; however, SPM-1 presents a high similarity with both the B1 and B2 subfamilies50 and hence was used as a starting point for proof of principle inhibitor screening. To validate our screening method, we screened a series of 4-chloroisoquinolinol derivatives (7a–7g)28 against SPM-1 using FC4 as a reporter substrate (Table 2). This set of compounds was selected as a result of their previously reported inhibition of metal-binding oxygenases.51 Screening for residual activity of compounds 7a–7g, at concentrations of 200 μM and 1 mM, revealed that the 4-chloroisoquinolinols bearing a Phe-, Tyr-, or Val-based side chain were poor inhibitors. In contrast, the compounds derivatized with a Trp, Leu, Asp, or Glu residue inhibited SPM-1, with the tryptophan-functionalized chloroisoquinolinols (S)-7d and (R)-7d giving the most promising results (3% and 8% residual activity (RA) at 200 μM, respectively).
Table 2. Residual Activities (RAs) on SPM-1 at 1 mM and 200 μMa,b.
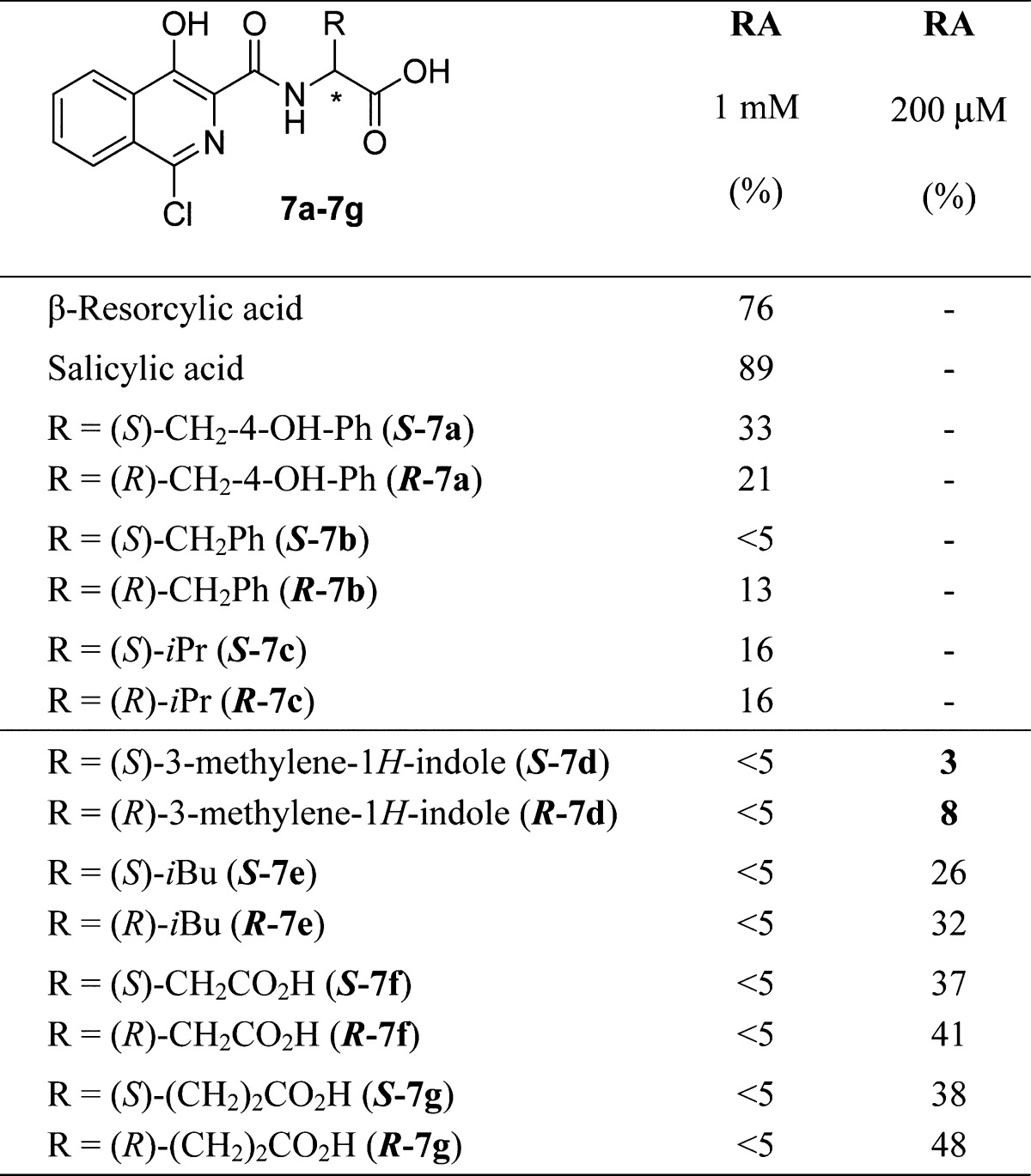
For the synthesis of compounds 7a–7g see the Supporting Information.
Following from the positive screening results obtained with the SPM-1/FC4 pair, IC50 values were determined for (S)-7d and (R)-7d against our MBL panel using FC4 and the conditions reported in Table 1; the FC4 concentrations correspond to the experimentally determined KM values (Table 1). The IC50 values (Table 3) reveal that both (S)-7d and (R)-7d are relatively weak, but broad-spectrum MBL inhibitors with IC50 values ranging from 20 to 130 μM. Since the inhibitor activity of these compounds is in the low micromolar range, they may serve as good starting points for further optimization.
Table 3. IC50 Values (μM) for a Panel of Metallo-β-lactamasesa.
| MBL | (S)-7d | (R)-7d | MBL | (S)-7d | (R)-7d |
|---|---|---|---|---|---|
| SPM-1 | 23.2 ± 1.1 | 46.6 ± 1.1 | VIM-2 | 54.7 ± 1.4 | 55.3 ± 1.3 |
| IMP-1 | 75.6 ± 1.5 | 74.1 ± 1.6 | Bc II | 61.3 ± 1.3 | 132.4 ± 1.3 |
| NDM-1 | 61.4 ± 1.3 | 47.1 ± 1.1 |
IC50 determinations were performed in duplicate over a range of 0.25–200 μM.
To further validate our assay platform, we measured the IC50 value of the reported inhibitor l-captopril52 against NDM-1. Measuring at the KM value of FC4 on NDM-1, we obtained an IC50 value of 10.0 ± 1.9 μM corresponding to a Ki of 5.0 μM for l-captopril.53 Using imipenem as the reported substrate, an IC50 of 13.2 ± 1.5 μM corresponding to a Ki of 6.6 μM were determined. Thus, under the same assay conditions, we found that the fluorogenic substrate (FC4) and the chromogenic substrate (imipenem) produced similar IC50 and Ki values. However, our values are lower than the previously reported IC50 and Ki values for l-captopril on NDM-1 using imipenem as the reported substrate (202 μM54 and 39 μM,52b respectively), possibly reflecting differences in assay conditions, protein constructs, and enzyme production procedures.
Conclusions
The development of broad-screen MBL inhibitors has been hampered by the lack of an appropriate, efficient screening platform for multiple MBLs. The development of such a screening platform has been hindered by the lack of suitable detection substrates. The results of our comparative study, employing five substrates and five MBLs, reveal striking differences in the kinetic parameters for different enzyme–substrate combinations. Fluorogenic substrates FC4 and FC5 are highly sensitive and efficient substrates for the clinically relevant MBLs, with one or both of them having kcat/KM values of >10 μM–1·s–1. The suitability of FC4 for inhibitor screening was demonstrated by testing a set of 4-chloroisoquinolinols as MBL inhibitors. This work yielded two compounds ((S)-7d and (R)-7d) that displayed inhibitory activity against our panel of MBLs, albeit at moderate inhibition concentrations (μM); nonetheless, the results reveal the feasibility of broad-spectrum MBL inhibition.
Experimental Section
The Supporting Information contains a complete general experimental section, including all procedures and equipment used. In general, chemicals were purchased from commonly used suppliers (Aldrich, Acros, Alfa Aesar, and TCI) and were used without further purification. (6R,7R)-4-Methoxybenzyl 3-(chloromethyl)-8-oxo-7-(2-phenylacetamido)-5-thia-1-aza-bicyclo[4.2.0]oct-2-ene-2-carboxylate (PMB-protected Cep-Cl) was obtained from Activate Scientific (Prien, Germany).
(6R,7R)-4-Methoxybenzyl 8-Oxo-3-(((2-oxo-2H-chromen-7-yl)oxy)methyl)-7-(2-phenylacetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate (4)
To a suspension of Cl-Cep-OPMB ester (680 mg, 1.4 mmol) in acetone (10 mL) was added NaI (2.10 g, 14.0 mmol, 10 equiv). The reaction was stirred at rt for 2 h, after which the solvent was removed in vacuo. The crude mixture was partitioned between H2O (10 mL) and EtOAc (10 mL), and the layers were separated. The H2O layer was extracted with EtOAc (2 × 15 mL), after which the combined organic layers were washed with a 5% NaS2O3 aq solution (2 × 20 mL) and brine (20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was dissolved in MeCN (15 mL), and 7-hydroxycoumarin (454 mg, 2.8 mmol, 2 equiv) and K2CO3 (4.2 mmol, 4 equiv) were added. The conversion of the reaction was monitored by TLC analysis (cHex/EtOAc, 1:1), and completion was reached after 4 h. The solvent was evaporated in vacuo, and the resulting crude mixture was partitioned between H2O (10 mL) and EtOAc (10 mL). After separation of the layers, the H2O layer was extracted with EtOAc (2 × 15 mL), after which the combined organic layers were washed with a 5% NaS2O3 aq solution (2 × 20 mL) and brine (20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography (cHex/EtOAc, 1:1) to yield compound 4 as a light brown solid (392 mg, 46%). Rf = 0.30 (cHex/EtOAc, 1:1). 1H NMR (400 MHz, DMSO-d6): δ = 9.20 (d, J = 8.0 Hz, 1H), 7.98 (d, J = 9.5 Hz, 1H), 7.59 (d, J = 8.5 Hz, 1H), 7.17 - 7.33 (m, 8H), 6.92 (d, J = 1.5 Hz, 2H), 6.87 (dd, J = 8.5, 2.5 Hz, 1H), 6.79 (d, J = 8.5 Hz, 2H), 6.30 (d, J = 9.5 Hz, 1H), 5.44 (dd, J = 7.5, 4.0 Hz, 1H), 5.11–5.19 (m, 2H), 5.01–5.10 (m, app d, 2H), 4.64–4.76 (m, app q, 2H), 3.69 (s, 3H), 3.47–3.57 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6): δ = 170.9, 167.0, 163.8, 160.9, 160.3, 159.3, 155.2, 144.3, 135.7, 130.0 (2C), 129.5, 129.1 (2C), 128.2 (2C), 126.9, 126.5, 122.4, 118.8, 113.7 (2C), 112.9, 112.7 (2C), 101.4, 69.8, 67.2, 60.8, 55.0, 52.9, 49.7, 41.6 ppm. LRMS: mass calcd for C33H27N2O8S (M – H) 611.15, mass found (M – H) 611.0.
(6R,7R)-4-Methoxybenzyl 8-Oxo-3-(((2-oxo-2H-chromen-7-yl)oxy)methyl)-7-(2-phenylacetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate 5-Oxide (5)
To a suspension of compound 4 (122 mg, 0.2 mmol) in dry CH2Cl2 (5 mL) cooled to 0 °C was added mCPBA (45 mg, 0.2 mmol, 1 equiv). The reaction was stirred at 0 °C for 30 min followed by an additional 1 h at rt (a white precipitate was formed). The crude product was dry-loaded onto silica and purified by column chromatography (CH2Cl2/EtOAc, 8:2) to yield compound 5 as a cream white solid (60 mg, 48%). Rf = 0.80 (CH2Cl2/MeOH, 9:1). 1H NMR (400 MHz CDCl3): δ = 7.64 (d, J = 9.5 Hz, 1H), 7.27–7.40 (m, 8H), 6.91 (d, J = 8.5 Hz, 2H), 6.74–6.78 (m, 2H), 6.67 (d, J = 10.0 Hz, 1H), 6.30 (d, J = 9.5 Hz, 1H), 6.10 (dd, J = 10.0, 5.0 Hz, 1H), 5.22–5.35 (m, app mix of d and q, 3H), 4.80 (d, J = 13.5 Hz, 1H), 4.45 (dd, J = 5.0, 1.5 Hz, 1H), 3.99 (d, J = 19.0 Hz, 1H), 3.81 (s, 3H) 3.65 (m, app d, J = 5.5 Hz, 2H), 3.28 (d, J = 19.0 Hz, 1H) ppm. 13C NMR (101 MHz, DMSO-d6): δ = 171.1, 164.5, 160.7 (2C), 160.2, 159.4, 155.2, 144.3, 135.8, 130.4 (2C), 129.5, 129.1 (2C), 128.3 (2C), 126.8, 126.6, 125.0, 119.6, 113.7 (2C), 112.8, 101.6, 67.5, 67.3, 66.5, 58.4, 55.1, 45.4, 41.4 ppm. LRMS: mass calcd for C33H27N2O9S (M – H) 627.14, mass found (M – H) 627.0.
(6R,7R)-4-Methoxybenzyl 8-Oxo-3-(((2-oxo-2H-chromen-7-yl)oxy)methyl)-7-(2-phenylacetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate 5,5-Dioxide (6)
To a suspension of compound 4 (100 mg, 0.16 mmol) in dry CH2Cl2 (10 mL) cooled to 0 °C was added mCPBA (74 mg, 0.35 mmol, 2 equiv). The reaction was stirred at 0 °C for 30 min (formation of sulfoxide observed), after which the reaction was warmed to rt and stirred overnight. Upon completion of the reaction, the crude product was dry-loaded onto silica and purified by column chromatography (CH2Cl2/MeOH, 9:1) to yield the desired product as a white solid (35 mg, 34%). Rf = 0.85 (CH2Cl2/MeOH, 9:1). 1H NMR (400 MHz, DMSO-d6) (small amount of mCBA present): δ = 8.93 (d, J = 8.5 Hz, 1H), 8.01 (d, J = 9.5 Hz, 1H), 7.87–7.93 (m, 1H), 7.63 (d, J = 8.5 Hz, 1H), 7.18–7.34 (m, 7H), 6.94–6.88 (m, 2H, 2 signals overlapping), 6.81–6.86 (m, app d, 2H), 6.33 (d, J = 9.5 Hz, 1H), 6.01 (dd, J = 8.5, 5.0 Hz, 1H), 5.43 (d, J = 4.5 Hz, 1H), 5.22 (s, 2H), 4.89–4.84 (m, app d, 2H), 4.46 (d, J = 18.5 Hz, 1H) (part of AB system), 4.23 (d, J = 18.5 Hz, 1H) (part of AB system), 3.70 (s, 3H), 3.60 (d, J = 5.0 Hz, 2H) ppm. 13C NMR (101 MHz, DMSO-d6): δ = 170.9, 164.5, 160.6 (2C), 160.2, 159.4, 155.2, 144.3, 135.6, 130.4 (2C), 129.5, 129.2 (2C), 128.2 (2C), 126.6, 126.5, 124.6, 124.0, 113.7 (2C), 112.9, 112.8, 101.6, 67.8, 66.8, 66.2, 58.4, 55.1, 50.9, 41.2 ppm. LRMS: mass calcd for C33H27N2O10S (M – H) 643.14, mass found (M – H) 643.0.
(6R,7R)-8-Oxo-3-(((2-oxo-2H-chromen-7-yl)oxy)methyl)-7-(2-phenylacetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid 5-Oxide (FC4)
Compound 5 (50 mg, 81.6 μmol) was cooled to 0 °C prior to the addition of TFA/anisole (5:1, 3 mL). The resulting reaction mixture was stirred at 0 °C for 30 min with constant monitoring of the conversion by TLC analysis (CH2Cl2/EtOAC, 8:2). Upon completion of the reaction, cold Et2O (5 mL) was added, which resulted in the formation of a precipitate. The solid material was filtered off, washed with Et2O (2 × 5 mL), and subsequently dried under high vacuum to yield the desired product as an off-white solid (15 mg, 37%). Rf = 0.10 (CH2Cl2/MeOH +AcOH, 9:1). 1H NMR (400 MHz, DMSO-d6): δ = 8.45 (d, J = 8.5 Hz, 1H), 8.00 (d, J = 9.5 Hz, 1H), 7.65 (d, J = 8.5 Hz, 1H), 7.27–7.32 (m, 4H), 7.24 (td, J = 8.5, 4.0 Hz, 1H), 6.93–7.01 (m, 1H), 6.31 (d, J = 9.5 Hz, 1H), 5.81 (dd, J = 8.0, 4.5 Hz, 1H), 5.14 (d, J = 12.5 Hz, 1H), 4.87–4.93 (m, 1H), 3.98 (d, J = 18.0 Hz, 1H), 3.70 (d, J = 14.0 Hz, 1H) (part of AB system), 3.62 (d, J = 18.5 Hz, 1H), 3.54 (d, J = 14.0 Hz, 1H) (part of AB system) ppm. 13C NMR (101 MHz, DMSO-d6): δ = 171.1, 164.2, 162.2, 161.1, 155.3, 144.3, 135.8, 129.6, 129.1 (2C), 128.3 (2C), 126.6, 112.8 (2C), 101.6, 67.5, 66.3, 58.3, 45.3, 41.5 ppm. LRMS: mass calcd for C25H19N2O8S (M – H) 507.09, mass found (M – H) 507.0. Retention time: 7.15 min (purity 99%).
(6R,7R)-8-Oxo-3-(((2-oxo-2H-chromen-7-yl)oxy)methyl)-7-(2-phenylacetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic Acid 5,5-Dioxide (FC5)
Compound 6 (20 mg, 0.03 mmol) was cooled to 0 °C prior to the addition of TFA/anisole (5:1, 1.2 mL). The reaction was stirred at 0 °C for 30 min followed by 30 min at rt with constant monitoring of the conversion by TLC analysis (CH2Cl2/MeOH, 9:1). Upon completion of the reaction, cold Et2O (5 mL) was added, which resulted in the formation of a precipitate. The solid material was filtered off, washed with Et2O (2 × 3 mL), and subsequently dried under high vacuum to yield the desired product as an off-white solid (10 mg, 63%). Rf = 0.10 (CH2Cl2/MeOH +AcOH, 9:1). 1H NMR (400 MHz, DMSO-d6): δ = 8.92 (d, J = 9.0 Hz, 1H), 8.01 (d, J = 9.5 Hz, 1H), 7.66 (d, J = 8.5 Hz, 1H), 7.19–7.31 (m, 5H) 7.04 (d, J = 2.5 Hz, 1H), 6.99 (dd, J = 8.5, 2.5 Hz, 1H), 6.32 (d, J = 9.5 Hz, 1H), 5.97 (dd, J = 8.5, 4.5 Hz, 1H), 5.42 (d, J = 4.5 Hz, 1H), 4.90–5.00 (m, app q, 2H), 4.41 (d, J = 18.0 Hz, 1H), 4.19 (d, J = 18.0 Hz, 1H), 3.54–3.65 (m, app q, 2H) ppm. 13C NMR (101 MHz, DMSO-d6): δ = 170.9, 164.3, 162.1, 160.8, 160.2, 155.2, 144.3, 135.6, 129.6, 129.2 (2C), 128.2 (2C), 126.5, 125.2, 123.8, 112.9, 112.9, 101.7, 66.8, 66.3, 58.3, 50.8, 41.2 ppm. LRMS: mass calcd for C25H19N2O8S (M – H) 507.09, mass found (M – H) 507.0. Retention time: 8.98 min (purity 95%).
Acknowledgments
We thank the Medical Research Council (MRC)/Canadian Grant G1100135 and Biotechnology and Biological Sciences Research Council (BBSRC) for support of J.B., R.S., R.C., and A.V. Cancer Research UK (CRUK) is kindly acknowledged for the support of S.S.v.B. The Oxford Protein Production Facility UK (OPPF-UK) is supported by the MRC and BBSRC.
Glossary
Abbreviations Used
- MBL
metallo-β-lactamase
- SBL
serine-β-lactamase
- FC
fluorogenic cephalosporin
- DiPEA
N,N′-diisopropylethylamine
- DMF
dimethylformamide
- 7-HC
7-hydroxycoumarin
- MeCN
acetonitrile
- mCPBA
m-chloroperbenzoic acid
- MMC
7-mercapto-4-methylcoumarin
- PMB
p-methoxybenzyl
- AcOH
acetic acid
- TFA
trifluoroacetic acid
Supporting Information Available
Experimental procedures, characterization of intermediates and target compounds, description of protein production and purification, biological assays, determination of residual activity measurements and IC50 values, and in silico docking data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
# S.S.v.B. and J.B. contributed equally to this work.
The authors declare no competing financial interests.
Supplementary Material
References
- Spellberg B.; Guidos R.; Gilbert D.; Bradley J.; Boucher H. W.; Scheld W. M.; Bartlett J. G.; Edwards J. Jr. The epidemic of antibiotic-resistant infections: A call to action for the medical community from the Infectious Diseases Society of America. Clin. Infect. Dis. 2008, 46, 155–164. [DOI] [PubMed] [Google Scholar]
- World Health Organization. The evolving threat of antimicrobial resistance: Options for action, 2012. http://whqlibdoc.who.int/publications/2012/9789241503181_eng.pdf (accessed December 2012).
- Bush K.; Jacoby G. A. Updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A. R.; Kaye C.; Poupard J.; Pypstra R.; Woodnutt G.; Wynne B. J. Augmentin (amoxicillin/clavulanate) in the treatment of community-acquired respiratory tract infection: A review of the continuing development of an innovative antimicrobial agent. Antimicrob. Chemother. 2004, 53Suppl. S1i3–i20. [DOI] [PubMed] [Google Scholar]
- Drawz S. M.; Bonomo R. A. Three decades of β-lactamase inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oelschlaeger P.; Ai N.; DuPrez K. T.; Welsh W. J.; Toney J. H. Evolving carbapenemases: Can medicinal chemists advance one step ahead of the coming storm?. J. Med. Chem. 2010, 53, 3013–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp-Wallace K. M.; Endimiani A.; Taracila M. A.; Bonomo R. A Carbapenems: Past, present, and future. Antimicrob. Agents Chemother. 2011, 5, 4943–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehmann D. E.; Jahić H.; Ross P. L.; Gu R.-F.; Hu J.; Kern G.; Walkup G. K.; Fisher S. L. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 11663–11668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellar D. J.; Lundgren D. G. Fine structure of sporulation in Bacillus cereus grown in a chemically defined medium. J. Bacteriol. 1966, 92, 1748–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T. R. Emerging carbapenemases: A global perspective. Int. J. Antimicrob. Agents 2010, 36Suppl. 3S8–S14. [DOI] [PubMed] [Google Scholar]
- Cornaglia G.; Giamarellou H.; Maria Rossolini G. Metallo-β-lactamases: A last frontier for β-lactams?. Lancet Infect. Dis. 2011, 11, 381–393. [DOI] [PubMed] [Google Scholar]
- a Chen P.; Horton L. B.; Mikulski R. L.; Deng L.; Sundriyal S.; Palzkill T.; Song Y. 2-Substituted 4,5-dihydrothiazole-4-carboxylic acids are novel inhibitors of metallo-β-lactamases. Bioorg. Med. Chem. Lett. 2012, 22, 6229–6232. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Toney J. H.; Hammond G. G.; Fitzgerald P. M. D.; Sharma N.; Balkovec J. M.; Rouen G. P.; Olson S. H.; Hammond M. L.; Greenlee M. L.; Gao Y. D. Succinic acids as potent inhibitors of plasmid-borne IMP-1 metallo-β-lactamase. J. Biol. Chem. 2001, 276, 31913–31918. [DOI] [PubMed] [Google Scholar]
- a Toney J. H.; Fitzgerald P. M.; Grover-Sharma N.; Olson S. H.; May W. J.; Sundelof J. G.; Vanderwall D. E.; Cleary K. A.; Grant S. K.; Wu J. K.; Kozarich J. W.; Pompliano D. L.; Hammond G. G. Antibiotic sensitization using biphenyl tetrazoles as potent inhibitors of Bacteroides fragilis metallo-β-lactamase. Chem. Biol. 1998, 5, 185–196. [DOI] [PubMed] [Google Scholar]; b Weide T.; Saldanha S. A.; Minond D.; Spicer T. P.; Fotsing J. R.; Spaargaren M.; Frère J.-M.; Bebrone C.; Sharpless K. B.; Hodder P. S.; Fokin V. V. NH-1,2,3-Triazole-based inhibitors of the VIM-2 metallo-β-lactamase: Synthesis and structure–activity studies. ACS Med. Chem. Lett. 2010, 1, 150–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Liénard B. M. R.; Garau G.; Horsfall L.; Karsisiotis A. I.; Damblon C.; Lassaux P.; Papamicael C.; Roberts G. C. K.; Galleni M.; Dideberg O.; Frère J.-M.; Schofield C. J. Structural basis for the broad-spectrum inhibition of metallo-β-lactamases by thiols. Org. Biomol. Chem. 2008, 6, 2282–2294. [DOI] [PubMed] [Google Scholar]; b Liénard B. M., R.; Hüting R.; Lassaux P.; Galleni M.; Frère J.-M.; Schofield C. J. Dynamic combinatorial mass spectrometry leads to metallo-β-lactamase inhibitors. J. Med. Chem. 2008, 51, 684–688. [DOI] [PubMed] [Google Scholar]
- Walter M. W.; Felici A.; Galleni M.; Paul Soto R.; Adlington R. M.; Baldwin J. E.; Frère J.-M.; Gololobov M.; Schofield C. J. Trifluoromethyl alcohol and ketone inhibitors of metallo-β-lactamases. Bioorg. Med. Chem. Lett. 1996, 6, 2455–2458. [Google Scholar]
- a Fast W.; Sutton L. D. Metallo-β-lactamase: Inhibitors and reporter substrates. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 1648–1659. [DOI] [PubMed] [Google Scholar]; b Toney J. H.; Moloughney J. G. Metallo-β-lactamase inhibitors: Promise for the future?. Curr. Opin. Invest. Drugs 2004, 5, 823–826. [PubMed] [Google Scholar]; c Spencer J.; Walsh T. R.; New A. Approach to the inhibition of metallo-β-lactamases. Angew. Chem., Int. Ed. 2006, 45, 1022–1026. [DOI] [PubMed] [Google Scholar]
- a Moali C.; Anne C.; Lamotte-Brasseur J.; Groslambert S.; Devreese B.; Van Beeumen J.; Galleni M.; Frère J.-M. Analysis of the importance of the metallo-β-lactamase active site loop in substrate binding and catalysis. Chem. Biol. 2003, 10, 319–329. [DOI] [PubMed] [Google Scholar]; b Saradhi Borra P.; Samuelsen Ø.; Spencer J.; Walsh T. R.; Sjo Lorentzen M.; Leirose H.-K. S. Crystal structures of Pseudomonas aeruginosa GIM-1: Active-site plasticity in metallo-β-lactamases. Antimicrob. Agents Chemother. 2013, 57, 848–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Viswanatha T.; Marrone L.; Goodfellow V.; Dmitrienko G. I. Assays for β-lactamase activity and inhibition. Methods Mol. Med. 2008, 142, 239–260. [DOI] [PubMed] [Google Scholar]; b Kocaoglu O.; Calvo R. A.; Sham L.-T.; Cozy L. M.; Lanning B. R.; Francis S.; Winkler M. E.; Kearns D. B.; Carlson E. E. Selective penicillin-binding protein imaging probes reveal substructure in bacterial cell division. ACS Chem. Biol. 2012, 7, 1746–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zheng X.; Sallium U. W.; Verma S.; Athar H.; Evans C. L.; Hasan T. Exploiting a bacterial drug-resistance mechanism: A light-activated construct for the destruction of MRSA. Angew. Chem., Int. Ed. 2009, 48, 2148–2151. [DOI] [PubMed] [Google Scholar]
- Jones R. N.; Wilson H. W.; Novick W. J. Jr.; Barry A. L.; Thornberry C. In vitro evaluation of CENTA, a new β-lactamase-susceptible chromogenic cephalosporin reagent. J. Clin. Microbiol. 1982, 15, 954–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones R. N.; Wilson H. W.; Novick Jr. In vitro evaluation of pyridine-2-azo-p-dimethylaniline cephalosporin, a new diagnostic chromogenic reagent, and comparison with nitrocefin, cephacetrile, and other β-lactam compounds. J. Clin. Microbiol. 1982, 15, 677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon K.; Phillips I. β-Lactamase detection by three simple methods: Intralactam, nitrocefin and acidimetric. J. Antimicrob. Chemother. 1980, 6, 617–621. [DOI] [PubMed] [Google Scholar]
- a Gao W.; Xing B.; Tsien R. Y.; Rao J. Novel fluorogenic substrates for imaging β-lactamase gene expression. J. Am. Chem. Soc. 2003, 125, 11146–11147. [DOI] [PubMed] [Google Scholar]; b Rukavishnikov A.; Gee K. R.; Johnson I.; Corry S. Fluorogenic cephalosporin substrates for β-lactamase TEM-1. Anal. Biochem. 2011, 419, 9–16. [DOI] [PubMed] [Google Scholar]; c Zhang Y.-L.; Xiao J.-M.; Feng J.-L.; Yang K.-W.; Feng L.; Zhou L.-S.; Crowder M. W. A novel fluorogenic substrate for dinuclear Zn(II)-containing metallo-β-lactamases. Bioorg. Med. Chem. Lett. 2013, 23, 1676–1679. [DOI] [PubMed] [Google Scholar]
- Yao H.; So M.-K.; Rao J.; Bioluminogenic A. Substrate for in vivo imaging of β-lactamase activity. Angew. Chem., Int. Ed. 2007, 46, 7031–7034. [DOI] [PubMed] [Google Scholar]
- a Watanabe S.; Mizukami S.; Hori Y.; Kikuchi K. Multicolor protein labeling in living cells using mutant β-lactamase-tag technology. Bioconjugate Chem. 2010, 21, 2320–2326. [DOI] [PubMed] [Google Scholar]; b Mizukami S.; Watanabe S.; Akimoto Y.; Kikuchi K. No-wash protein labeling with designed fluorogenic probes and application to real-time pulse-chase analysis. J. Am. Chem. Soc. 2012, 134, 1623–1629. [DOI] [PubMed] [Google Scholar]; c Shao Q.; Xing B. Enzyme responsive luminescent ruthenium(II) cephalosporin probe for intracellular imaging and photoinactivation of antibiotics resistant bacteria. Chem. Commun. 2012, 48, 1739–1741. [DOI] [PubMed] [Google Scholar]
- References for plasmid production:; a Griffin D. H.; Richmond T. K.; Sanchez C.; Jon Moller A.; Breece R. M.; Tierney D. L.; Bennett B.; Crowder M. W. Structural and kinetic studies on metallo-β-lactamase IMP-1. Biochemistry 2011, 50, 9125–9134(IMP-1). [DOI] [PubMed] [Google Scholar]; b de Seny D.; Prosperi-Meys C.; Bebrone C.; Maria Rossolini G.; Page M. I.; Noel P.; Frère J.-M.; Galleni M. Mutational analysis of the two zinc-binding sites of the Bacillus cereus 569/H/9 metallo-β-lactamase. Biochem. J. 2002, 363, 687–696(Bc II). [DOI] [PMC free article] [PubMed] [Google Scholar]; c Green V. L.; Verma A.; Owens R. J.; Phillipsa S. E. V.; Carr S. B. Structure of New Delhi metallo-β-lactamase 1 (NDM-1). Acta Crystallogr. 2011, F67, 1160–1164(NDM-1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrow N. S.; Alderton D.; Sainsbury S.; Nettleship J.; Assenberg R.; Rahman N.; Stuart D. I.; Owens R. J. A versatile ligation-independent cloning method suitable for high-throughput expression screening applications. Nucleic Acids Res. 2007, 35, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebrone C.; Moali C.; Mahy F.; Rival S.; Docquier J.-D.; Maria Rossolini G.; Fastrez J.; Pratt R. F.; Frère J.-M.; Galleni M. CENTA as a chromogenic substrate for studying β-lactamases. Antimicrob. Agents Chemother. 2001, 45, 1868–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For experimental details see the Supporting Information.
- Typically mCPBA oxidation to give the (S)-sulfoxide; see:Kaiser G. V.; Cooper R . D. G.; Koehler R. E.; Murphy C. F.; Webber J. A.; Wright I. G.; van Heyningen E. M. Transformation of Δ2-cephem to Δ3-cephem by oxidation-reduction at sulfur. J. Org. Chem. 1970, 35, 2430–2433. [DOI] [PubMed] [Google Scholar]
- Goddard J.-P.; Reymond J.-L. Enzyme assays for high-throughput screening. Curr. Opin. Biotechnol. 2004, 15, 314–322. [DOI] [PubMed] [Google Scholar]
- Xie H.; Mire J.; Kong Y.; Chang M.; Hassounah H. A.; Thornton C. N.; Sacchettini J. C.; Cirillo J. D.; Rao J. Rapid point-of-care detection of the tuberculosis pathogen using a BlaC-specific fluorogenic probe. Nat. Chem. 2012, 4, 802–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The optimal absorption wavelength for umbelliferone in HEPES buffer was found to be 330 nm with a second absorption band at 380 nm. Fluorogenic substrates FC3–FC5 did not show this second absorption band at 380 nm, allowing the specific excitation of umbelliferone at this wavelength.
- Kim Y.; Tesar C.; Mire J.; Jedrzejczak R.; Binkowski A.; Babnigg G.; Sacchettini J.; Joachimiak A. Structure of apo- and monometalated forms of NDM-1—A highly potent carbapenem-hydrolyzing metallo-β-lactamase. PLoS One 2011, 6, e24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docquier J.-D.; Lamotte-Brasseur J.; Galleni M.; Amicosante G.; Frère J.-M.; Maria Rossolini G. On functional and structural heterogeneity of VIM-type metallo-β-lactamases. J. Antimicrob. Chemother. 2003, 51, 257–266. [DOI] [PubMed] [Google Scholar]
- Laraki N.; Franceschini N.; Rossolini G. M.; Santucci P.; Meunier C.; de Pauw E.; Amicosante G.; Frère J.-M.; Galleni M. Biochemical characterization of the Pseudomonas aeruginosa 101/1477 metallo-β-lactamase IMP-1 produced by Escherichia coli. Antimicrob. Agents Chemother. 1999, 43, 902–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy T. A.; Simm A. M.; Toleman M. A.; Jones R. N.; Walsh T. R. Biochemical characterization of the acquired metallo-β-lactamase SPM-1 from Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2003, 47, 582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul-Soto R.; Hernadez-Valladares M.; Fonzé E.; Goussard S.; Courvalin P.; Frère J.-M. Mono- and binuclear Zn-β-lactamase from Bacteroides fragilis: Catalytic and structural roles of the zinc ions. FEBS Lett. 1998, 438, 137–140. [DOI] [PubMed] [Google Scholar]
- Felici A.; Amicosante G. Kinetic Analysis of extension of substrate specificity with Xanthomonas maltophilia, Aeromonas hydrophila, and Bacillus cereus metallo-β-lactamases. Antimicrob. Agents Chemother. 1995, 39, 192–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young D.; Toleman M. A.; Giske G. C.; Cho C. H.; Sundman K.; Lee K.; Walsh T. R. Characterization of a new metallo-β-lactamase gene, blaNDM-1, and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob. Agents Chemother. 2009, 53, 5046–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badarau A.; Llinás A.; Laws A. P.; Damblon C.; Page M. I. Inhibitors of metallo-β-lactamase generated from β-lactam antibiotics. Biochemistry 2005, 44, 8578–8589. [DOI] [PubMed] [Google Scholar]
- Astrid Zervosen A.; Hernandez Valladares M.; Devreese B.; Prosperi-Meys C.; Adolph H.-W.; Sandra Mercuri P.; Vanhove M.; Amicosante G.; van Beeumen J.; Frère J.-M.; Galleni M. Inactivation of Aeromonas hydrophila metallo-β-lactamase by cephamycins and moxalactam. Eur. J. Biochem. 2001, 268, 3840–3850. [DOI] [PubMed] [Google Scholar]
- Fonseca F.; Arthur C. J.; Bromley E. H. C.; Samyn B.; Moerman P.; Saavedra M. J.; Correia A.; Spencer J. Biochemical characterization of Sfh-I, a subclass B2 metallo-β-lactamase from Serratia fonticola UTAD54. Antimicrob. Agents Chemother. 2011, 55, 5392–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low KM values can result in a low signal readout; in particular, when chromogenic substrates are used, this can decrease the sensitivity of the method (i.e., lead to high interexperimental error during kinetic measurements). Moreover, by using a high-affinity substrate (low KM), it is generally not possible to detect potential slow-binding inhibitors as, for example, in the case of NDM-1/nitrocefin, where the substrate presented a low micromolar range KM, with the enzyme concentration being in the same micromolar range (also see ref (44)).
- Siemann S.; Clarke A. J.; Viswanatha T.; Dmitrienko G. I. Thiols as classical and slow-binding inhibitors of IMP-1 and other binuclear metallo-β-lactamases. Biochemistry 2003, 42, 1673–1683. [DOI] [PubMed] [Google Scholar]
- a Badarau A.; Llinas A.; Laws A. P.; Damblon C.; Page M. I. Inhibitors of metallo-β-lactamase generated from β-lactam antibiotics. Biochemistry 2005, 44, 8578–8589. [DOI] [PubMed] [Google Scholar]; b Murphy T. A; Catto L. E.; Halford S. E.; Hadfield A. T.; Minor W.; Walsh T. R.; Spencer J. Crystal structure of Pseudomonas aeruginosa SPM-1 provides insights into variable zinc affinity of metallo-β-lactamases. J. Mol. Biol. 2006, 357, 890–903. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Hao Q. Crystal structure of NDM-1 reveals a common β-lactam hydrolysis mechanism. FASEB J. 2011, 25, 2574–2582. [DOI] [PubMed] [Google Scholar]
- Garcia-Saez I.; Docquier J. D.; Rossolini G. M.; Dideberg O. The three-dimensional structure of VIM-2, a Zn-β-lactamase from Pseudomonas aeruginosa in its reduced and oxidised form. J. Mol. Biol. 2008, 375, 604–611. [DOI] [PubMed] [Google Scholar]
- a Goodsell D. S.; Morris G. M.; Olson A. J. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. [DOI] [PubMed] [Google Scholar]; b Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gillet V.; Johnson A. P.; Mata P.; Sike S.; Williams P. SPROUT: A program for structure generation. J. Comput.-Aided Mol. Des. 1993, 7, 127–153. [DOI] [PubMed] [Google Scholar]; b Gillet V. J.; Newell W.; Mata P.; Myatt G.; Sike S.; Zsoldos Z.; Johnson A. P. SPROUT: Recent developments in the de novo design of molecules. J. Chem. Inf. Comput. Sci. 1994, 34, 207–217. [DOI] [PubMed] [Google Scholar]
- Cadag E.; Vitalis E.; Lennox K. P.; Zhou C. L. E.; Zemla A. T. Computational analysis of pathogen-borne metallo β-lactamases reveals discriminating structural features between B1 types. BMC Res. Notes 2012, 5, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Stubbs C. J.; Loenarz C.; Mecinović J.; Kheng Yeoh K.; Hindley N.; Liénard B. M.; Sobott F.; Schofield C. J.; Flashman E. Application of a proteolysis/mass spectrometry method for investigating the effects of inhibitors on hydroxylase structure. J. Med. Chem. 2009, 52, 2799–2805. [DOI] [PubMed] [Google Scholar]; b Tian Y.-M.; Yeoh K. K.; Lee M. K.; Eriksson T.; Kessler B. M.; Kramer H. B.; Edelmann M. J.; Willam C.; Pugh C. W.; Schofield C. J.; Ratcliffe P. J. Differential sensitivity of hypoxia inducible factor hydroxylation sites to hypoxia and hydroxylase inhibitors. J. Biol. Chem. 2011, 286, 13041–13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Heinz U.; Bauer R.; Wommer S.; Meyer-Klaucke W.; Papamichaels C.; Bateson J.; Adolph H.-W. Coordination geometries of metal ions in D- or L-captopril-inhibited metallo-β-lactamases. J. Biol. Chem. 2003, 278, 20659–20666. [DOI] [PubMed] [Google Scholar]; b King D. T.; Worrall L. J.; Gruninger R.; Strynadka N. C. New Delhi metallo-β-lactamase: Structural insights into β-lactam recognition and inhibition. J. Am. Chem. Soc. 2012, 134, 11362–11365. [DOI] [PubMed] [Google Scholar]
- Conversion of IC50 to Ki was performed according to Cer et al.:Cer R. Z.; Mudunuri U.; Stephens R.; Lebeda F. J. IC50-to-Ki: A Web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res. 2009, 37, W441–W445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.; Wang J.; Niu G.; Shui W.; Sun Y.; Zhou H.; Zhang Y.; Yang C.; Lou Z.; Rao Z. A structural view of the antibiotic degradation enzyme NDM-1 from a superbug. Protein Cell 2011, 2, 384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.