

SUMMARY
The RIG-I like receptors (RLRs) signal innate immune defenses upon RNA virus infection but their roles in adaptive immunity have not been clearly defined. Here we showed that the RLR LGP2 was not essential for induction of innate immune defenses, but rather was required for controlling antigen-specific CD8+ T cell survival and fitness during peripheral T cell number expansion in response to virus infection. Adoptive transfer and biochemical studies demonstrated that T cell receptor signaling induced LGP2 expression wherein LGP2 operated to regulate death receptor signaling and imparted sensitivity to CD95-mediated cell death. Thus, LGP2 promotes an essential pro-survival signal in response to antigen stimulation to confer CD8+ T cell number expansion and effector functions against divergent RNA viruses, including West Nile virus and lymphocytic choriomeningitis virus.
INTRODUCTION
Pathogen recognition and signaling of cell intrinsic innate immunity is a crucial process for initiation of the immune response to virus infection. Early recognition of RNA viruses and induction of innate antiviral immunity are largely dependent on the RIG-I like receptors (RLRs) (Loo et al., 2008). The RLR family of cytosolic RNA helicases, which include RIG-I, MDA5, and LGP2, are expressed basally at low levels in most tissues and induced by type 1 interferon (IFN). RIG-I and MDA5 encode amino-terminal tandem caspase activation and recruitment domains (CARDs) that function in downstream signaling to induce the expression of IFN and other proinflammatory cytokines; in contrast, LGP2 (encoded by Dhx58) neither possesses CARDs nor has an analogous signaling effector domain (Yoneyama et al., 2005). Upon binding to RNA pathogen associated molecular patterns (PAMP), the RLRs undergo conformational changes and modifications that promote their signaling functions. In particular, RIG-I and MDA5 signal downstream through a direct interactions with the MAVS (also known as IPS-1) adaptor protein mediated by CARD-CARD interaction to initiate signaling of IRF-3 and NF-κB transcription factors activation. This process induces expression of immune effector genes, IFN, and interferon-stimulated genes (ISGs) that impart innate and adaptive immune defenses to limit virus replication and spread (Wilkins and Gale, 2010). In contrast LGP2 can function as a negative regulator of RLR signaling by binding viral dsRNA (Rothenfusser et al., 2005), inhibiting RIG-I multimerization (Saito et al., 2007), or competing for a common docking site with IKK-ε on MAVS to suppress innate immune signaling actions (Komuro and Horvath, 2006). More recent studies with Dhx58−/− mice have suggested that LGP2 can also function as a positive cofactor of RLR signaling of innate immune defenses (Satoh et al., 2010; Venkataraman et al., 2007), although the exact mechanism by which LGP2 contributes to RIG-I or MDA5 signaling actions remains unknown.
West Nile virus (WNV) is an emerging flavivirus of public health importance, and is now a major cause of epidemic encephalitis worldwide (Cdc, 2008). RLR signaling and adaptive immune responses are essential for immune protection against WNV infection. Within infected cells, WNV is recognized as a pathogen through the combined actions of RIG-I and MDA5 to temporally induce innate immune responses that are essential in controlling virus replication and modulating B and T cell responses (Fredericksen et al., 2008; Suthar et al., 2010). Whereas humoral immune responses are important for controlling systemic virus infection (Diamond et al., 2003a; Diamond et al., 2003b), cell-mediated responses, specifically CD8+ T cells (Brien et al.; Shrestha and Diamond, 2004; Shrestha et al., 2006; Szretter et al.) are critical in controlling virus replication and virus-induced pathology within the central nervous system (CNS). CD4+ T cells play a more prominent role in providing help in developing virus-specific antibody responses and clearance of WNV from the CNS at later times in infection (Sitati and Diamond, 2006). Regulation of the adaptive immune response is dependent on RLR signaling through MAVS (Suthar et al., 2010). RLR signaling also modulates the quality and balance of the adaptive immune response, including governance of T cell number expansion, inflammatory cell infiltration into the CNS, and generation of neutralizing antibodies (Suthar et al., 2010). While these observations indicate that RIG-I and MDA5 signaling through MAVS is important for an effective adaptive immune response against virus infection, the role of LGP2 in antiviral immunity and pathogenesis of WNV infection has remained poorly understood.
In this study, we generated a Dhx58−/− mouse line on a pure C57BL/6 background and evaluated the role of LGP2 in RNA virus infection and immunity. Our results revealed an essential role for LGP2 in promoting CD8+ T cell survival and fitness by regulating sensitivity to death receptor-mediated cell death. We also confirmed a role for LGP2 function as a positive regulator of RLR signaling of innate immune defenses in primary fibroblasts and myeloid-derived cells ex vivo, while demonstrating that LGP2 is not essential for induction of innate immunity in lymphoid organs and within the CNS in response to WNV challenge. Importantly, our study defines a cell-intrinsic role for LGP2 in regulation of T cell responses during RNA virus infection, and further demonstrates the importance of RLR signaling in regulating immunity and infection.
RESULTS
LGP2 is not required for innate defenses but serves to enhance RLR-dependent IFN induction
To define the role of LGP2 in directing immunity to virus infection, we generated a line of Dhx58−/− mice on a pure C57BL/6 background (Figure 1A), thus alleviating confounding variables due to mixed genetic background present in existing Dhx58−/− mouse lines (Satoh et al., 2010; Venkataraman et al., 2007). We engineered a gene-targeting vector that uniquely replaced Dhx58 exons 2 to 8, including the translation start codon, with a neomycin cassette. This construct was used to target Dhx58 in C57BL/6 embryonic stem cells, which were injected into a C57BL/6 donor mouse embryo. Southern blot analysis and qRT-PCR confirmed the deletion of LGP2 (Figure 1B and data not shown, respectively). Progeny mice from Dhx58+/− or Dhx58−/− intercrosses were born at a normal Mendelian ratio and showed no overt physical defects, in contrast to other Dhx58−/− mouse lines (Satoh et al., 2010). Expression of LGP2 was not detected in Dhx58−/− derived mouse embryo fibroblasts (MEF) cultured in the presence of IFN (Figure 1C). When infected with Sendai virus (SenV), Dengue virus type 2 (DENV2) or WNV (WNV-TX-02; representing the pathogenic and emerging strain of WNV (Keller et al., 2006)), Dhx58−/− MEFs produced slightly reduced amounts of IFN-β but retained temporal induction of IFIT2 and IFIT3 expression (Figure 1D). IFN-β induction by SenV, DENV2, and WNV is dependent on RLR signaling (Loo et al., 2008) and IFIT2 and IFIT3 are ISGs whose expression can be driven acutely by IRF-3 alone (Grandvaux et al., 2002). These results indicate that virus-induced RLR signaling to IRF-3 remains intact but IFN-β induction is moderately attenuated in MEFs in the absence of LGP2.
Figure 1. LGP2 is a positive regulator of RLR signaling in primary fibroblasts and myeloid-derived cells.

(A) LGP2 gene targeting strategy and predicted gene disruption. (B) Southern blot analysis of Dhx58+/+ and Dhx58+/− mice (WT allele = 5.7kb; Dhx58−/− allele = 3.7 kb). (C) Immunoblot analysis of mouse embryo fibroblasts (MEFs) from C57BL/6 (WT) and Dhx58−/− mice treated with type I IFN for 24 hours. (D) WT and Dhx58−/− MEFs were mock-infected (M) or infected with Sendai virus (200HA units/ml; left); DENV-2 (MOI 1.0; middle); and WNV-TX (MOI 1.0; right). IFN-β in the supernatant was measured by ELISA (upper) and interferon-stimulated gene (ISG) expression assessed by immunoblotting (lower) at the indicated hours post-infection. (E–I) Primary bone-marrow derived dendritic cells (DC) and macrophages (Mφ) recovered from WT and Dhx58−/− mice were mock-infected or infected with WNV-TX at an MOI of 1. Cells and culture media were harvested at the times indicated for determination of IFN-β production (DCs=E, Mφ=F), ISG expression (DCs=G, Mφ=H), and virus load (DCs=I, Mφ=J). Dotted line represents the limit of assay sensitivity. Graphs show the mean +/− standard deviation from triplicate samples from three independent experiments. Asterisks denote P < 0.05.
To determine the role of LGP2 in triggering innate immune defenses in myeloid cells, we evaluated the response of bone marrow-derived dendritic cells (DCs) and macrophages to WNV infection. Both cell types are targets of WNV infection in vivo (Samuel and Diamond, 2006). Dhx58−/− DCs (Figure 1E) and macrophages (Figure 1F) infected with WNV produced reduced amounts of IFN-β when compared to wild-type (WT) cells, suggesting a positive regulatory role for LGP2 in facilitating IFN-β production in myeloid cells. In contrast, whereas LGP2 was highly expressed in WT infected DCs (Figure 1G) and macrophages (Figure 1H), the absence of LGP2 neither altered the expression kinetics nor magnitude of IRF-3-target genes or ISGs, including those that encode RIG-I, MDA5, STAT-1, IFIT2, and IFIT3 during WNV infection. We observed significantly higher virus replication at 24 hours post-infection in WNV-infected Dhx58−/− DCs (Figure 1I) and macrophages (Figure 1J) when compared to WT infected cells. However, by later time points WT and Dhx58−/− infected cells produced similar amounts of infectious virus. Taken together, these results validate LGP2 as a non-essential but positive regulator to RLR signaling of innate immune defenses, and demonstrate that it can function to enhance IFN-β production during acute RNA virus infection.
LGP2 is essential for protection against WNV infection and virus control in the CNS
To examine the role of LGP2 in mediating protection against virus infection in vivo, we challenged WT and Dhx58−/− mice with WNV (WNV-TX) and assessed clinical phenotype, virologic as well as immunologic responses. Following a subcutaneous inoculation in the footpad, WNV replicates in the draining popliteal lymph node (pDLN), which results in viremia and spread to the spleen and central nervous system tissues (e.g., brain and spinal cord), thus recapitulating the pathogenesis of human infection (Samuel and Diamond, 2006). Dhx58−/− mice were found to be more susceptible to WNV infection (Figure 2A) and exhibited a significant increase in mortality (87.5% compared to 13% in WT mice; P<0.0001), as compared to WT infected mice. Thus, LGP2 is required for protection against WNV infection in vivo.
Figure 2. LGP2 is required for protection against WNV infection.

(A) Survival of WT (n=26) and Dhx58−/− (n=17) adult mice infected subcutaneously with 100 PFU of WNV-TX. Viral burden analysis from WT and Dhx58−/− infected mice in the (B) pDLN, (C) spleen, and (D) kidney (also analyzed Ips-1−/− mice) (E) IFN-β, (F) IFIT1, and (G) IFIT2 expression in the pDLN were determined by qRT-PCR. (H) Immunoblot analysis of spleens from WT and Dhx58−/− mock infected (M) and WNV-TX infected mice at the indicated days post-infection. (I) Serum type I IFN measured by an L929 bioassay. Dotted line represents the limit of assay sensitivity. Graphs show the mean +/− standard deviation of triplicate biological samples from three independent experiments.
To begin to define the basis for the increased mortality in Dhx58−/− mice, we evaluated the viral burden in different tissues over time. Analysis of virus levels within the pDLN (Figure 2B) and spleen (Figure 2C) revealed no differences in tissue viral load between WT and Dhx58−/− infected mice. In contrast to Mavs−/− mice (Suthar et al., 2010), Dhx58−/− infected mice exhibited normal tissue tropism, with no expansion to organs (e.g., kidney) that normally are resistant to infection in WT mice. (Figure 2D). Furthermore, comparable peripheral innate immune responses were observed in the absence of LGP2 as evidenced by similar amounts of IFN-β production (Figure 2E) and ISG expression in the pDLN (Figure 2F–G), ISG expression in the spleen (Figure 2H), and type I IFN in the serum (Figure 2I).
Dhx58−/− infected mice displayed similar kinetics of viral neuroinvasion as WT infected mice (see day 6; Figure 3A), although significantly higher viral loads were observed in the brain at late time points during infection, indicating that LGP2 is required for controlling virus replication and/or spread after entering the CNS. Accordingly, we examined WNV replication and innate immune defenses in primary cortical neurons isolated from WT or Dhx58−/− mice after ex vivo infection. We found that LGP2 was not essential for controlling virus replication in neurons, as WNV reached similar end-point viral loads in Dhx58−/− cortical neurons as compared to WT neurons (Figure 3B). Furthermore, LGP2 was not required for the induction or enhancement of innate immune defenses against infection in cortical neurons, as IFN-β induction (Figure 3C) and ISG expression (Figure 3D) were similar between WT and Dhx58−/− infected neurons. Although we detected an elevated basal level of STAT1 in Dhx58−/− cortical neurons, IFN treatment induced ISG expression similarly between WT and Dhx58−/− cells. IFN treatment also induced RLR expression, revealing that RLRs are inducible in neuronal cells. Overall, these results demonstrate that in neurons LGP2 does not contribute significantly to regulating innate immune responses or controlling virus replication.
Figure 3. LGP2 is required for regulating CNS inflammation and pathology.

(A) Viral burden in the brains of infected mice was determined at the indicated days post-infection. The dotted line represents the limit of assay sensitivity. (B–D) Primary cortical neurons (CN) were generated from WT and Dhx58−/− mice and infected at an MOI of 1 and cell supernatants were collected at the indicated hours post-infection. (B) Viral titers in the supernatants were determined by plaque assay. (C) Type I IFN in the supernatants was measured by a bioassay. (D) Immunoblot analysis from cell lysates of mock (M), WNV-TX infected, and type I IFN treated CNs. (E) H&E stained sagittal brain tissue sections. Arrows denote areas of inflammation and pathology. (F–J) Brain leukocytes were recovered from WT and Dhx58−/− mice eight days post-infection. (F) Total cells of brain lymphocytes were determined after cell counting. (G) Total CD4+ T cells, (H) Total CD8+ T cells, (I) representative flow analysis and gating schematic for identifying NS4b tetramer-specific CD8+ T cells, and (J) total NS4b tetramer-specific CD8+ T cells. M=Mock; W=WNV-TX. Data are representative of two or more independent experiments. Graphs show the mean +/− standard deviation (n=3). Asterisks denote P < 0.05.
CNS inflammation in Dhx58−/− mice during WNV infection
Because of the absence of a direct effect on viral burden in neurons, we hypothesized that LGP2 might protect against WNV infection in the brain by modulating cell-extrinsic immune control mechanisms. Therefore, we assessed the extent of inflammation and pathology in the brains of WT and Dhx58−/− mice after WNV infection. Histological analysis of brain sections recovered from WT mice 8 days after infection revealed moderate inflammation and sparse neuronal damage, consistent with previously published studies (Figure 3E; (Suthar et al., 2010). In addition, mononuclear cell infiltrates were present in the hippocampus and cerebral cortex regions, implying the onset of the inflammatory response that mediates protective immunity to infection. In contrast, brains from Dhx58−/− infected mice displayed extensive damage to neurons in the hippocampus and cortex regions, with pyknotic and vacuolated neurons. Surprisingly, we observed little to no inflammation within the hippocampus and cortex of brains from infected Dhx58−/− mice, despite the near 3-log increase in brain viral load compared to WT mice. Furthermore, the number of total lymphocytes isolated from the brain of infected Dhx58−/− mice was significantly reduced (70% reduction) as compared to WT mice (Figure 3F). Although the brains of infected Dhx58−/− mice did not exhibit a significant decrease in the numbers of CD4+ T cells (Figure 3G), there were significantly reduced numbers of total CD8+ T cells (55% reduction; Figure 3H) and antigen-specific CD8+ T cells (62% reduction; Figure 3I–J). These results indicate that LGP2 regulates CD8+ T cell recruitment to the CNS.
LGP2 regulates CD8+ T cell accumulation
To define whether the defects in inflammation were specific to the CNS or indicative of a more global inflammatory control mechanism by LGP2, we analyzed immune cell composition within the spleens of WNV-infected mice. Spleens recovered from Dhx58−/− mice between 4 and 6 days post-infection exhibited similar increases in total lymphocytes as compared to spleen from WT infected mice (Figure 4A). However, by day 7 post-infection spleens from Dhx58−/− mice contained significantly lower total lymphocyte numbers as compared to spleens from WT infected mice and this trend continued through day 10 post-infection. We observed similar trends in the number of total CD8+ T cells (Figure 4B) and antigen-specific CD8+ T cells (Figure 4C). Interestingly, and in contrast to mice deficient in RLR signaling (i.e. Mavs−/− mice; Suthar et al., 2010), we did not observe differences in either the quantity or the quality of the WNV-specific humoral response (Supplementary Table 1), suggesting that LGP2 does not directly govern B cell development or CD4+ T cell help during WNV infection (Sitati and Diamond, 2006). Thus LGP2 modulates CD8+ T cell responses but not humoral immune responses against WNV infection.
Figure 4. LGP2 regulates CD8+ T cell survival and fitness.

WT and Dhx58−/− mice were mock-infected (M) or infected with WNV-TX. Spleens were harvested and immune cells were isolated, counted, and characterized by flow cytometry. (A) Total cells (splenocytes), (B) total number of CD8+ T cells, (C) total number of NS4b tetramer-specific CD8+ T cells, (D) BrdU incorporation on cells gated on NS4b tetramer-specific CD8+ T cells from day 6 post-infection (left-representative histogram; right-bar graph), (E) Annexin V+ (left) and CD44 (right) staining on NS4b tetramer-specific CD8+ T cells (representative histogram from day 10 p.i), (F) bar graph of Annexin V+ cells, (G) flow cytometry histogram of TNF-α- and IFN-γ- secreting CD8+ T cells upon restimulation with media (left) or NS4b peptide (right) on day 10 post-infection, (H) frequency and total number of IFN-γ, (I) TNF-α, and (J) IFN-γ and TNF-α-secreting CD8+ T cells (top-frequency; bottom-total cells). Graphs show the mean +/− standard deviation from triplicate samples from two or more experiments. Asterisks denote P < 0.05
LGP2 is required for CD8+ T cell fitness and survival
To define the mechanisms by which LGP2 regulates CD8+ T cells, we evaluated T cell priming, activation and expansion in response to virus infection. Ex vivo assessment of bone-marrow derived dendritic cells and macrophages revealed that that the kinetics of maturation of these antigen-presenting cells, as measured by expression of key costimulatory molecules (CD80, CD86, CD40) and MHC class I and class II, were unaltered in the absence of LGP2 (data not shown). Furthermore, in vivo analysis of splenic dendritic cells and macrophages revealed no differences in expansion or maturation status of each during WNV infection (data not shown), demonstrating that LGP2 does not regulate dendritic cell or macrophage maturation. As RLR signaling has been shown to control regulatory T cell expansion during WNV infection (Suthar et al., 2010), we examined this population of cells in WT and Dhx58−/− infected mice. In the absence of LGP2, there was no change in regulatory T cell expansion as compared to WT infected mice (Supplementary Figure 1), suggesting that reduction in CD8+ T cells in Dhx58−/− infected mice was not due to aberrant expansion of regulatory T cells during WNV infection.
Because CD8+ T cells are essential for protection against acute WNV infection and the development of WNV encephalitis (Shrestha and Diamond, 2004), and given the robustness of CD8+ T cell responses during WNV infection (Brien et al., 2007, 2008), we assessed the mechanisms by which LGP2 governs CD8+ T cell function. To determine if LGP2 is required for CD8+ T cell proliferation, WT and Dhx58−/− WNV-infected mice were pulsed with bromodeoxyuridine (BrdU) on day 6 post-infection, a time point that directly precedes the dramatic decrease in CD8+ T cell numbers in Dhx58−/− infected mice (Figure 4D). Total CD8+ T cells (Supplementary Figure 2A) and antigen-specific CD8+ T cells from Dhx58−/− infected mice showed similar levels of BrdU incorporation as compared to WT infected mice, indicating that LGP2 does not regulate CD8+ T cell proliferation. We also assessed a possible role of LGP2 in supporting CD8+ T cell survival after antigen stimulation by analyzing the percent of CD8+ T cells undergoing apoptosis (as determined by Annexin V+ staining) in WT and Dhx58−/− infected mice. On day 6 post-infection, antigen-specific CD8+ T cells from WT and Dhx58−/− infected mice showed low but similar frequency of apoptotic cells (Figure 4E–F). By day 10 post-infection, antigen-specific (NS4b tetramer+) CD8+ T cells from Dhx58−/− infected mice displayed a significant increase (2.7 fold) in apoptotic cell frequency, suggesting that LGP2 regulates CD8+ T cell survival. Similarly, after restimulation with the WNV immunodominant NS4b peptide (Brien et al., 2007; Purtha et al., 2007), splenocytes from Dhx58−/− infected mice had reduced frequency and numbers of IFN-γ- (Figure 4H), TNF-α (Figure 4I), and TNF-α- and IFN-γ- secreting CD8+ T cells (Figure 4J) as compared to WT infected mice, with maximum differences observed on day 10 post-infection. Furthermore, IL-2 expression, an important cytokine for regulating T cell growth and function, was not observed in CD8+ T cells from WT and Dhx58−/− mice between days 6 and 10 post-infection (data not shown). To determine if the reduced numbers of effector T cells in Dhx58−/− mice were specific to WNV infection, we evaluated CD8+ T cell survival and fitness after challenge of WT and Dhx58−/− mice with another RNA virus, lymphocytic choriomeningitis virus (LCMV)-Armstrong. Similar to that seen after WNV infection, LCMV antigen-specific (GP33 tetramer+) CD8+ T cells from infected Dhx58−/− mice showed enhanced apoptosis and reduced effector functions as compared to cells from WT infected mice (Supplementary Figure 2B–C). Together, these results demonstrate that LGP2 participates in the regulation of CD8+ T cell survival and fitness during the immune response to RNA virus infection.
LGP2 is expressed in CD8+ T cells and regulates survival in a cell-intrinsic manner
We next measured RLR expression and abundance in purified CD8+ T cells recovered from WT mice. In general, CD8+ T cells expressed a low basal level of each RLR, including LGP2, and that RLR abundance was increased along with other ISGs when the cells were treated with IFN-β (Figure 5). Stimulation of CD8+ T cells with plate-bound anti-CD3 and anti-CD28 induced RLR expression, with LGP2 expression accumulating to the highest levels among the RLRs (Figure 5). These findings demonstrate that LGP2 expression in CD8+ T cells is induced by type I IFN and TCR signaling, suggesting a cell intrinsic role for LGP2 in regulating CD8+ T cell fitness during RNA virus infection.
Figure 5. Type I IFN and TCR conjugation stimulate expression of LGP2 and RIG-I in CD8+ T cells.
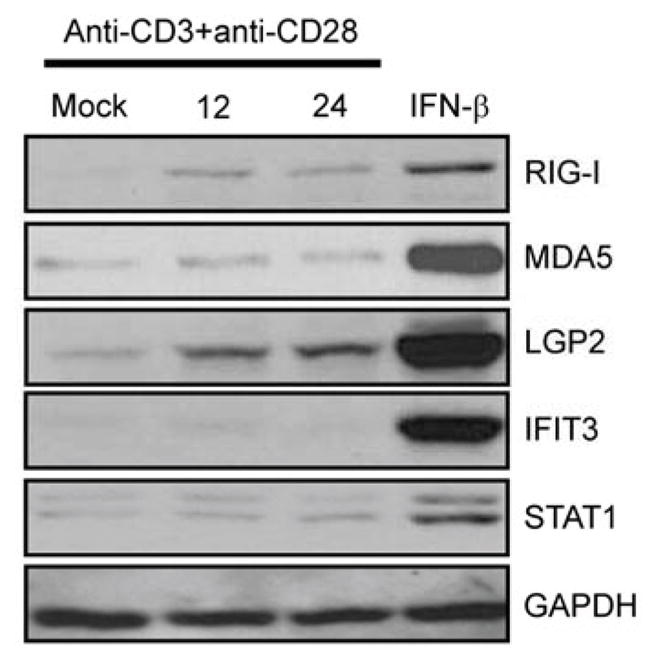
CD8+ T cells were purified from WT mice through negative selection, and CD8+ T cells were stimulated with plate-bound anti-CD3 and anti-CD28. Parallel cultures were treated with IFN-β (100 IU/ml) for 24 hours. Cell lysates were collected and immunoblot analysis performed using the indicated antibodies. Data are representative of three independent experiments.
To establish whether LGP2 functions in a cell-intrinsic or cell-extrinsic manner to promote CD8+ T cell survival, we isolated CD8+ T cells from CD45.1 congenic WT mice and Dhx58−/− (CD45.2) mice, and adoptively transferred them into Rag1−/− mice at a 1:1 cell ratio. One day after transfer, recipient mice were injected with diluent alone (uninfected control) or challenged with WNV (Figure 6A). Examination of donor cells in uninfected Rag1−/− recipient mice showed comparable division of CFSE labeled WT and Dhx58−/− CD8+ T cells (Figure 6B), with low Annexin V+ staining apparent in either WT or Dhx58−/− derived CD8+ T cells (data not shown). Thus, in the absence of infection, Dhx58−/− CD8+ T cells do not show overt defects in homeostatic proliferation or survival. However, the frequency and total numbers of antigen-specific Dhx58−/− CD8+ T cells were reduced by day 8 post WNV-infection (Figure 6C). To determine whether this reduction was due to turnover of cells or an inability of cells to effectively proliferate, we evaluated BrdU incorporation on day 8 post-infection. Antigen-specific Dhx58−/− CD8+ T cells displayed enhanced proliferation as compared to WT CD8+ T cells (Figure 6D), demonstrating that dysregulated cell proliferation does not account for the reduction in CD8+ T cells. Another possible explanation for the reduction in CD8+ T cells is dysregulated development of short-lived effector cell (SLEC) and memory-precursor effector cell (MPEC) populations in the absence of LGP2 (Joshi et al., 2007). Antigen-specific Dhx58−/− CD8+ T cells displayed comparable percentages of KLRG1 expressing cells, a marker for discriminating SLECs, and IL-7Rα (CD127) expressing cells, a marker for discriminating MPECs (Supplementary Figure 3). This suggests that LGP2 is not required for programming development of SLEC or MPEC populations during WNV infection. Consistent with our previous observations, antigen-specific Dhx58−/− CD8+ T cells displayed increased Annexin V+ staining compared to WT CD8+ T cells (Figure 6E). Furthermore, when restimulated with the immunodominant NS4b peptide ex vivo (Figure 6F), CD8+ T cells from Dhx58−/− mice exhibited compromised effector functions compared to WT cells, including a reduction in single-positive IFN-γ (41.5% reduction; P = 0.001), TNF-α (40.5% reduction; P = 0.004), and double-positive TNFα- and IFN-γ-secreting cells (43.9% reduction; P = 0.004).
Figure 6. LGP2 directly regulates CD8+ T cell survival and fitness.

(A) Schematic of CD8+ T cell adoptive transfer into Rag1−/− mice. (B) Homeostatic proliferation of CD8+ T cells from WT (CD45.1) and Dhx58−/− (CD45.2) measured by flow cytometry (left-representative flow cytometry analysis and gating schematic; right-bar graph displaying frequency of divided CD8+ T cells). (C) NS4b tetramer-specific CD8+ T cells from day 8 p.i (each connecting line represents cells isolated from the same Rag1−/− recipient host; top-frequency; right-relative ratio of NS4b tetramer+CD8+ T cells), (D) BrdU staining of NS4b tetramer-specific CD8+ T cells from WNV-TX infected mice on day 8 p.i. (representative flow analysis and bar graph displaying frequency of BrdU+ staining) (E) Annexin V+ staining of NS4b tetramer-specific CD8+ T cells (representative flow analysis and bar graph displaying frequency of Annexin V+ staining), (F) total number of WNV-specific CD8+ T cells secreting IFN-γ (left), TNF-α (middle), or both (right). Relative to WT (top panel) and actual (bottom panel) percentages of CD8+ T cells isolated from the same Rag1−/− recipient host. Graphs show the mean +/− standard deviation (n=3). Data are representative of two or more independent experiments. Asterisks denote P < 0.05.
LGP2 has been reported to bind to MAVS to regulate RLR signaling (Komuro and Horvath, 2006). Therefore, we assessed the role of MAVS in regulating CD8+ T cell survival using the adoptive transfer model. We observed no enhancement of cell death in Mavs−/− CD8+ T cells (data not shown), consistent with our previous studies (Suthar et al., 2010). Taken together, these results demonstrate that LGP2 functions in a cell-intrinsic manner to promote CD8+ cell survival and function after antigen stimulation independently of MAVS.
LGP2 imparts sensitivity to CD95-mediated cell death
Cell death in T cells can occur through activation of the intrinsic apoptosis pathway, involving Bcl-2, Bim, and Bcl-xL, or through the extrinsic pathway, initiated by death receptor (DR) signaling (Tourneur and Chiocchia, 2010). Using the adoptive transfer model, we evaluated key components in the intrinsic and extrinsic apoptosis signaling pathways to identify the mechanism by which LGP2 operates to control CD8+ T cell survival. In the absence of LGP2, anti-apoptotic factors, including Bcl-2 and Bcl-xL, as well as the proapoptotic factor Bim, displayed similar expression in antigen-specific CD8+ T cells as compared to WT CD8+ T cells, suggesting that LGP2 does not regulate the intrinsic apoptosis signaling pathway through altered expression of these factors (Supplementary Figure 4). In contrast, antigen-specific Dhx58−/− CD8+ T cells displayed enhanced caspase-8 (Figure 7A) and caspase-3 and -7 activity (Figure 7B), suggesting that LGP2 regulates the extrinsic apoptosis signaling pathway. Members of the tumor necrosis factor (TNF) superfamily are important in regulating the extrinsic apoptosis pathway in T cells. For example, ligation of TNFα, TNF-related apoptosis-inducing ligand (TRAIL), and CD95 ligand (CD95L) to their cognate receptors (TNF-RI and RII, TRAIL-R2, and CD95, respectively) leads to formation of the death-inducing signaling complex (DISC) followed by activation of caspase-8, caspase-3 and -7, and ultimately cell death (Aggarwal, 2003). In the absence of LGP2, antigen-specific CD8+ T cells displayed enhanced expression of TNF-RI (Figure 7C), TNF-RII (Figure 7D), TRAIL-R2 Figure 7E), and CD95 (Figure 7F) when compared to WT CD8+ T cells during virus infection. To determine whether the enhanced receptor expression correlated with increased sensitivity to cell death, splenocytes were treated exogenously with TNF–α, TRAIL, or CD95L and cell death was evaluated in antigen-specific CD8+ T cells. Consistent with ex vivo examination (Figure 6E), antigen-specific CD8+ T cells from Dhx58−/− mice displayed enhanced Annexin V+ staining as compared to WT cells in the absence of exogenous treatment, further demonstrating an overall increased sensitivity for cell death in the absence of LGP2 (Figure 7G–I). WT or Dhx58−/− antigen-specific CD8+ T cells displayed no enhancement of cell death upon treatment with TNF-α (Figure 7G) while treatment with TRAIL lead to similar increases in cell death between WT and Dhx58−/− antigen-specific CD8+ T cells (Figure 7H). In contrast, treatment with CD95L lead to an increase in cell death of antigen-specific CD8+ T cells from Dhx58−/− but not WT mice (Figure 7I). In fact, CD95L-treated Dhx58−/− CD8+ T cells displayed an increase in cell death from 21% to 32.5% upon ex vivo treatment as compared to WT CD8+ T cells (7.8% Annexin V+ cells in treated and untreated conditions). Although we did not observe a similar enhancement of sensitivity over WT CD8+ T cells upon TNF–α or TRAIL treatment, we cannot preclude the possibility that signaling through these pathways or other death receptors within the TNF superfamily contribute to the enhanced death of antigen-specific Dhx58−/− CD8+ T cells during virus infection. Taken together, these findings demonstrate a role for LGP2 in promoting CD8+ T cell survival by regulating sensitivity to death receptor-mediated signaling of cell death.
Figure 7. LGP2 controls sensitivity to CD95-mediated apoptosis signaling.

(A) Active caspase-8, (B) active caspase-3/7, (C) CD120a, (D) CD120b, (E) CD262, and (F) CD95 expression measured on NS4b tetramer-specific CD8+ T cells from WNV-TX infected mice on day 8 p.i. (representative flow analysis and bar graph displaying frequency of positive staining). Sensitivity to cell death upon ex vivo treatment with (G) TNF-α, (H) TRAIL or (I) CD95 ligand. Splenocytes from adoptively transferred Rag1−/− infected mice on were isolated and treated on day 8 p.i. Graphs show the mean +/− standard deviation (n=3). Data are representative of two independent experiments. Asterisks denote P < 0.05.
DISCUSSION
Our study defines an essential role for LGP2 in promoting immunity through cell intrinsic regulation of CD8+ T cell survival and fitness. We found that LGP2 promotes CD8+ T cell survival by controlling sensitivity to death receptor signaling. This finding is highly relevant to the outcome of WNV infection because CD8+ T cells are the critical immune cell that controls virus replication and spread within the CNS (Shrestha and Diamond, 2004). In the absence of LGP2, CD8+ T cells did not continue to expand to WT levels during viral infection but instead displayed increased cell death concomitant with decreased effector function. Furthermore, using an adoptive transfer system, we demonstrated that LGP2 acts in a T cell-intrinsic manner to mediate a pro-survival response. Although we found that LGP2 functions in activated CD8+ T cells independent of canonical MAVS signaling of RLRs, it remains possible that it may interact with either RIG-I or MDA5 to regulate pro-survival functions of T cells. Together, these findings reveal a role for LGP2 in CD8+ T cell mediated immunity to viral infection, and assigns an essential role for LGP2 in programming antigen-induced survival.
LGP2 has been described as both a positive and negative regulator of RLR signaling of innate immune defenses against RNA viruses in vitro (Komuro and Horvath, 2006; Rothenfusser et al., 2005; Saito et al., 2007) and it was proposed to enhance signaling induced by RIG-I or MDA5 in vivo (Satoh et al., 2010). Our analyses of Dhx58−/− primary cells ex vivo validates a positive albeit cell-specific role for LGP2 in RLR signaling of innate defenses, as LGP2 contributed to sustained RLR signaling of IFN-β expression. In the absence of LGP2, IFN-β production from myeloid cells but not MEFs was reduced in response to RNA virus infection, including WNV and DENV2, two flaviviruses which trigger innate immune defenses through a combination of RIG-I and MDA5 actions (Fredericksen et al., 2008; Loo et al., 2008). However, our studies also showed that LGP2 is not essential for RLR signaling of innate antiviral defenses. These findings contrast with a previous report that concluded that LGP2 was essential for RLR signaling of innate immunity (Satoh et al., 2010). These differences can likely be attributed to differences in the genetic background of the mutant mice and gene targeting strategies between each study. Importantly, our studies utilized a Dhx58−/− mouse that was generated directly on a pure C57BL/6 background and through the complete disruption of LGP2 transcription, differing from prior Dhx58−/− mouse lines which exist in mixed genetic backgrounds and with gene targeting approaches that retained the transcriptional start site to render possible mRNA expression from the targeted gene (Satoh et al., 2010; Venkataraman et al., 2007). While our data overall support a role for LGP2 as a positive cofactor of innate immune signaling, it remains possible that under certain conditions LGP2 also may function to suppress RLR signaling, as high LGP2 expression were shown to mediate RLR signaling suppression in several independent studies (Komuro and Horvath, 2006; Rothenfusser et al., 2005; Saito et al., 2007; Venkataraman et al., 2007; Yoneyama et al., 2005). Further studies under conditions of regulated LGP2 expression in vivo are required to fully ascertain its role in RLR signaling control.
During virus infection or antigen stimulation, CD8+ T cells receive multiple signals to undergo rapid proliferation and maintain survival. Both cell-intrinsic and extrinsic apoptotic signaling pathways are important in regulating peripheral deletion of activated CD8+ T cells (Michalek and Rathmell, 2010). During the expansion phase, expression of pro- and anti-apoptotic Bcl-2 family members is suppressed and remain low through the cell contraction or death phase wherein dysregulation of these processes can lead to premature T cell death (Grayson et al., 2000). In the absence of LGP2 however, we observed comparable expression of Bcl-2 family members during virus infection, suggesting that LGP2 does not impart regulation of the intrinsic apoptosis signaling pathway that is linked with altered expression of Bcl-2 family proteins. Among programs controlling effector T cell contraction, the cell-extrinsic apoptosis pathway is critical for peripheral deletion of activated CD8+ T cells through death receptor signaling (Zhang et al., 2005). In particular, CD95 signaling, and to a lesser degree TNF-α signaling, play important roles in maintaining CD8+ T cell homeostasis and shaping the peripheral T cell repertoire during infection. In contrast, ‘helpless’ CD8+ T cells undergo TRAIL-mediated cell death upon reactivation during a secondary challenge (Janssen et al., 2005). Under our experimental conditions, we observed enhanced expression of death receptors in antigen-specific Dhx58−/− CD8+ T cells, suggesting that LGP2 is important in controlling receptor expression and hence sensitivity to death receptor-mediated cell death. Using an ex vivo model, we determined that the absence of LGP2 lead to enhanced sensitivity to CD95-mediated cell death but not to TNF-α or TRAIL treatment. A lack of TNF-α or TRAIL enhancement of cell death in Dhx58−/− cells does not preclude the possibility that these death receptors contribute to the enhanced cell death that is observed in antigen-specific Dhx58−/− CD8+ T cells. Overall, our observations imply that LGP2 functions as a molecular switch to control (by preventing or limiting) death receptor signaling during the early stages of T cell activation. We propose three unique mechanisms by which cell death could be regulated by LGP2: (1) signaling initiated by protein or RNA interactions of LGP2 imparts LGP2 actions that regulate pro-survival or pro-apoptotic factor(s); (2) LGP2 signals the expression of pro-survival or apoptotic gene expression in response to binding of specific inducer RNA(s); (3) LGP2 regulates the synthesis of pro-survival microRNA species in antigen-stimulated T cells. Despite comparably low levels of RIG-I or MDA5 expression upon TCR conjugation, it is remains plausible that LGP2 could also work conjunction with the other RLRs to promote CD8+ T cell survival. Due to the embryonic lethality of RIG-I-deficient mice in a pure C57BL/6 background, we are unable to evaluate the role of RIG-I in CD8+ T cells at this time (Kato et al., 2006). However, we and others have evaluated WNV infection in MDA5-deficient mice and observed normal proliferation and survival of antigen-specific CD8+ T cells (data not shown), consistent with a lack of induction of MDA5 expression upon TCR conjugation. These observations suggest that LGP2 functions independently of MDA5 to govern CD8+ T cell survival during virus infection. In support of the third possibility, recent work has shown that the RNAi pathway protein Dicer, which regulates microRNA biogenesis, is required for mediating CD8+ T cell survival, proliferation and effector functions (Zhang and Bevan, 2010), and it has recently been demonstrated that LGP2 can directly interact with components of the RNAi biogenesis pathway, including Dicer and Argonaut2 (Li et al., 2011). In addition, evolutionary studies have suggested that the RLRs and Dicer have evolved through a common ancestor, suggesting commonalities in function (Sarkar et al., 2008; Zou et al., 2009). Thus, it is reasonable to speculate that LGP2 promotes T cell survival and fitness by regulating gene expression through governance of microRNA expression. These possibilities are currently under investigation.
The production of type I IFN and subsequent innate immune responses are a key linkage point between innate and adaptive immunity, specifically during T cell function (Havenar-Daughton et al., 2006; Kolumam et al., 2005; Thompson et al., 2006). Both TCR stimulation and type I IFN treatment induced RLR expression in T cells. Though there is little overlap in type I IFN and TCR signaling pathways, these findings strongly suggest that each may converge on a common transcriptional element that regulates the expression of LGP2 and the other RLRs.
In addition to LGP2, the Toll-like receptor adaptor molecule MyD88 functions in a cell-intrinsic manner to support CD8+ and CD4+ T cell survival (Gelman et al., 2004; Quigley et al., 2009; Rahman et al., 2008; Zhao et al., 2009). While we observed little requirement for LGP2 in the CD4+ T cell response after WNV or LCMV infection, it is possible that LGP2 or other RLRs may be required for CD4+ T cell response during infection by other viruses or intracellular pathogens. Consistent with this, MAVS provides an important signal that controls regulatory T cell expansion during viral infection (Suthar et al., 2010). Thus, RLRs and TLRs likely have a prominent role in coordinating signals that regulate priming and/or survival of T cell responses during viral infection.
Overall, our results reveal a role for LGP2 in survival of activated CD8+ T cells during virus infection. Cell-mediated responses are essential for protection and clearance of many virus infections, and are critical for successful vaccines against many pathogens (Pulendran and Ahmed, 2011). Thus, our findings have direct implications for vaccine and adjuvant design strategies aimed at eliciting sustained antiviral T cell-mediated responses. Drug designs that engage LGP2 or its downstream signaling pathways could enhance T cell survival and effector function for the control of virus infection, or alternatively promote cell death to limit immune-mediated pathology.
EXPERIMENTAL PROCEDURES
Cells and Viruses
BHK-21, L929, L929-ISRE reporter (kind gift from B. Beutler) cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), HEPES, L-glutamine, sodium pyruvate, antibiotic-antimycotic solution, and nonessential amino acids. WNV isolate TX 2002-HC (WNV-TX) was described previously (Keller et al., 2006) and titered by a standard plaque assay on BHK-21 cells. Working stocks of WNV-TX were generated by as previously described (Suthar et al., 2010). LCMV (Armstrong strain) was plaque purified and grown in BHK-21 cells and titered as previously described (Thompson et al., 2006). Dengue virus type 2 (DENV2) was a gift from Jay A. Nelson (Oregon Health and Sciences University). Sendai virus (SenV) strain Cantrell was obtained from Charles River Laboratory.
Generation of LGP2-deficient mice
A conventional mouse LGP2 gene (encoded by Dhx58) deletion procedure was performed by inGenious Targeting Laboratory, Inc. (Stonybrook, NY). The targeting vector was subcloned from a positively identified C57BL/6 Bacterial Artifical Chromosome (BAC) clone using a homologous recombination-based technique. The 13.9kb region that was subcloned contains exons 2–8 of LGP2. The long homology arm (LA) is located on the 5′ side of exon 2 and is 7.57kb long and the short homology arm (SA) extends 1.46kb 3′ to exon 8. A neomycin positive-selection cassette flanked by loxP sites replaces 4.8kb of the gene including exon 2–8 and the ATG start codon in exon 2. A NotI-linearized targeting construct was transfected into iTL IC1 C57BL/6 embryonic stem cells by electroporation. After selection in G418 antibiotic, surviving clones were expanded for PCR analysis to identify recombinant ES clones. The correctly targeted embryonic stem cell clones were microinjected into C57BL/6J blastocysts. The resulting chimeric mouse was mated with a wild type C57BL/6J mouse to establish germlime transmission. The heterozygous mice (F1 mice) were interbred to obtain homozygous Dhx58−/− mice. The genotypes of the mice were determined by Southern blot analysis (genomic DNA was cut with PvuII; 543 bp primer probe generated by primers: WPB1 5′-GAAGTCCCTTCTGGGTCCGGGTAC-3′; WPB2 5′-ATTCTCCCTAGCCTACTGAGTGAC-3′ and PCR (primers: F7: 5′–GGAACTTCGCTAGACTAGTACGCGTG-3′; A6: 5′-GACAGGACTTGGACATGGACACC-3′).
Mouse experiments
Sti−/− mice (C57BL/6 background; referred to in text as Mavs−/−) were generated in the Gale laboratory in a similar manner as the Dhx58−/− mice described earlier. The generation and characterization will be discussed in a future publication. C57BL/6 wild type inbred mice, CD45.1 mice and Rag1−/− mice were commercially obtained (Jackson Laboratories, Bar Harbor, ME). All mice were genotyped and bred in specific pathogen-free conditions in the animal facility at the University of Washington. Experiments were performed in accordance with the University of Washington Institutional Animal Care and Use Committee guidelines. Age-matched six to twelve week old mice were inoculated subcutaneously (s.c.) in the left rear footpad with 100 PFU of WNV-TX in a 10 μl inoculum diluted in Hanks balanced salt solution (HBSS) supplemented with 1% heat-inactivated FBS. Mice were monitored daily for morbidity and mortality. A total of 2 × 105 PFU of LCMV was injected via an intraperitoneal route.
Adoptive transfers
CD8+ T cells were isolated directly from CD45.1 and Dhx58−/− splenocytes using the CD8+ T cell negative selection Isolation kit (Miltenyi Biotec) according to the manufacturer’s instructions (greater than 90% purity). Purified CD8+ T cells (2×106 for each mouse strain) were resuspended in cold-PBS and mixed 1:1 in a total volume of 100 ul. Cells were transferred into sex-matched Rag1−/− mice by intravenous injection through the tail vein. The following day (24 hours post-transfer), mice were infected with 100 PFU WNV-TX through the s.c. route in the left rear footpad and mice monitored daily as described above.
Viral tissue burden and quantification
For in vivo studies, infected mice were euthanized, bled, and perfused with 20 ml of phosphate-buffered saline (PBS). Whole brain, kidney, and spleen were removed, weighed, and homogenized in 500ul of PBS containing 1% heat-inactivated FBS using a Precellys 24 at 1500 RPM for 20 seconds (Bertin Technologies, France). Sample homogenates were tittered by plaque assay on BHK cells. For analysis of viral load within the DLN, the popliteal DLN was harvested and total RNA extracted using the RNeasy kit (Qiagen), DNase treated (Ambion) and evaluated WNV RNA copy number was measured by RT-quantitative PCR (RT-qPCR) as previously described (Suthar et al., 2010).
Primary cell isolation and infection
Bone-marrow derived DC and macrophages were generated as described previously (Daffis et al., 2008). The DC or macrophages were infected with WNV-TX at an MOI of 1. At the indicated timepoints, supernatants were collected for evaluating viral titers and IFN-β by ELISA (described below). Cells were collected in parallel for immunoblot analysis. Cortical neurons were isolated from 15-day-old embryonic mice and cultured as described previously (Samuel et al., 2006). On day 6 of culture, neurons were infected with WNV-TX at an MOI of 1 and supernatants were collected for virus titration, and cells were collected for RNA analysis by RT-qPCR using primers described previously(Suthar et al., 2010).
Immunoblot analysis
Cells were lysed in radioimmunoprecipitation assay buffer containing a cocktail of protease and phosphatase inhibitors (Sigma). Protein extracts (25 μg) were analyzed by immunoblotting as described previously (Suthar et al., 2010). The following primary antibodies were used to probe blots: mouse anti-WNV from the Center for Disease Control; rabbit anti-ISG54 and rabbit anti-ISG49, kindly provided by Dr. G. Sen; rabbit anti-MDA5 from IBL; mouse anti-tubulin from Sigma; rabbit anti-GAPDH from Santa Cruz; and rabbit anti-STAT1 were from Cell Signaling; Rabbit anti-RIG-I was generated as previously described (Loo et al., 2008). Secondary antibodies (peroxidase-conjugated goat anti-rabbit, goat anti-mouse, donkey anti-rabbit, and donkey anti-mouse) were from Jackson Immunoresearch.
RNA extraction and analysis
For cultured cells, total RNA was extracted using the RNeasy kit (Qiagen), DNase treated (Ambion) and evaluated for ISG49, ISG54, and IFN-β RNA expression by SYBR Green RT-qPCR. Specific primer sets for ISG-49, ISG-54, and IFN-β have been described previously (Suthar et al., 2010).
Interferon bioassay and ELISA
IFN-α and -β were measured in sera using a biological assay as previously described (Suthar et al., 2010). IFN-β in cell culture supernatants was analyzed using mouse-specific ELISA kits from PBL Biomedical Laboratories according to the manufacturer’s protocol. One hundred microliters from mock infected and WNV-TX infected neuronal supernatant was used to treat L929-ISRE reporter cells, and luciferase units were measured 6 hours post-treatment.
WNV-specific antibody analysis
WNV-specific IgM and IgG levels were determined by an ELISA using purified recombinant E protein as previously described (Shrestha et al., 2006). The neutralization titer of serum antibody was determined by using a previously described plaque reduction neutralization assay (Diamond et al., 2003a). The dilution at which 50% of plaques were neutralized was determined after non-linear transformation of the data.
Histological analysis
Mock-infected or WNV-infected mice were perfused with PBS-4% paraformaldehyde, pH 7.3. Brains were embedded in paraffin and 10-μm sections were prepared and stained with hematoxylin and eosin (H&E) by the UW histology pathology laboratory. Sections were analyzed using a Nikon Eclipse E600 microscope (UW Keck microscope facility).
Flow cytometric analysis
Splenocytes were isolated, washed, and re-suspended in complete RPMI 1640 media containing 10% FBS; L-glutamine, and antibiotic-antimycotic solution (cRPMI) before in vitro stimulation. Cells were washed twice before flow cytometry staining. For isolation of CNS immune cells, mice were euthanized and perfused extensively with PBS to remove residual intravascular leukocytes. Brains were isolated and minced in RPMI media, triturated, and digested with Liberase (Roche) and type I DNase in serum-free RPMI media at 37°C for 45 min. Immune cells were isolated after g radient centrifugation from a 37/70% Percoll interface and washed twice with staining buffer. Immune cells were stained with directly-conjugated antibodies specific to CD3, CD8, CD4, CD95, CD127, KLRG1, CD44, CD262 (all reagents from eBiosciences), CD120a, and CD120b (from Biolegend). WNV-specific CD8+ T cells were identified using a Db-restricted NS4b peptide tetramer (produced at the immune monitoring core facility at the Fred Hutchinson Cancer Research Center; Seattle, WA) directly conjugated to either Phycoerythrin (PE) or allophycocyanin (APC). Annexin V+ staining was performed according to the manufacturer’s instruction (BD Biosciences). Pacific Blue-conjugated Annexin V+ (Biolegend) or FITC-conjugated Annexin V+ (BD Biosciences) was used for staining. Intracellular FoxP3 staining to identify regulatory T cells was performed as previously described (Lund et al., 2008; Suthar et al., 2010). Intracellular IFN-γ and TNF-α staining was performed on splenocytes as previous described (Suthar et al., 2010). Briefly, lymphocytes were stimulated with 1 μg/ml of the WNV NS4b peptide (SSVWNATTAI) or LCMV-GP33 (GP33-41) for 4 h at 37 °C. Cells were washed and stained f or cell surface markers followed by permeabilization-fixation using the Cytofix-Cytoperm Kit (BD-PharMingen) and stained with a Pacific Blue-conjugated IFN-γ and FITC-conjugated TNF-α antibody (eBiosciences) at 4 °C for 30 min, washed and analyzed by flow cytometry. Caspase activity was measured using the Vybrant FAM Caspase-3 and -7 or Caspase-8 assay kit (Invitrogen), according to the manufacturer’s instructions. For detection of intracellular Bcl-2, cells were fixed and treated with the FoxP3 intracellular staining kit (ebioscience) followed by staining with an APC-conjugated anti-Bcl-2 antibody (Biolegend). For detection of Bim and Bcl-xL, cells were fixed for 10 minutes with 4% paraformaldehyde at room temperature, permeabilized with 0.1% saponin for 30 minutes on ice, and stained with anti-Bim (Cell signaling), anti-Bcl-xL (Clone 54H6; Cell signaling), and isotype control (Rabbit IgG mAb clone DA1E) antibody for 30 minutes on ice. Alexaflour488 conjugated (Cell signaling) secondary antibody was used at a dilution of 1:1000 and cells were analyzed by flow cytometry. To measure CD8+ T cell proliferation in vivo, mice were injected with 1 mg of bromodeoxyuridine for 6 hours via intraperitoneal route and splenocytes were stained with CD8, PE-conjugated NS4b-tetramer, and APC-conjugated BrdU, according to the manufacturer’s instructions. Flow cytometry was performed on a BD LSRII machine using BD FACSDiva software. Cell analysis was performed on FlowJo (v.8.7.2) software.
Analysis of TNF-α, TRAIL, and CD95L mediated cell death
For TNF-α and TRAIL treatments, splenocytes from recipient Rag1−/− infected mice were resuspended in cRPMI and cultured at 37°C for 6 hours with either 2 ng/ml TN F-α or 20 ng/ml SuperKiller TRAIL (Enzo life sciences). For CD95L treatment, splenocytes were cultured at 37°C for 3 hours with 250 ng/ml FLAG-tagged recombinant CD95L (Alexis Biochemicals) cross-linked by 4 μg/ml M2 monoclonal anti-FLAG antibody (Sigma-Aldrich). Cells were stained and analyzed for cell death (Annexin V+) by flow cytometry.
Ex vivo TCR stimulation
CD8+ T cells were isolated directly from wild type and Dhx58−/− splenocytes using the CD8+ T cell negative selection Isolation kit (Miltenyi Biotec) according to the manufacturer’s instructions. For ex vivo TCR stimulation, 96-well plates were precoated overnight with 1 ug/ml anti-CD3 (Biolegend) and 5 ug/ml anti-CD28 (Biolegend). Isolated CD8+ T cells were plated (6 × 105 cells per well) in 200 ul cRPMI media supplemented with 500 IU/ml of murine IL-2.
Statistical Analysis
For in vitro studies and immune cell analysis an unpaired student T-test was used to determine statistical differences. For in vivo viral burden analysis, Mann-Whitney analysis was used to determine statistical differences. Kaplan-Meier survival curves were analyzed by the log-rank test. A P value ≤0.05 was considered statistically significant. All data were analyzed using Prism software (GraphPad Prism5).
Supplementary Material
HIGHLIGHTS.
LGP2 is nonessential for innate immune responses to WNV infection.
LGP2 regulates CD8+ T cell survival and fitness in a cell-intrinsic manner.
RLR expression in CD8+ T cells is driven by TCR and type I IFN signaling.
LGP2 imparts sensitivity to CD95-mediated cell death in CD8+ T cells.
Acknowledgments
We thank Drs. Sunil Thomas and Murali Krishna-Kaja for helpful discussion. We thank Brian P. Doehle and Stacy Horner for comments and manuscript revisions. Supported by NIH grants 1F32AI081490, R01AI074973, U19AI083019, U54AI081680.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- Brien JD, Daffis S, Lazear HM, Cho H, Suthar MS, Gale M, Jr, Diamond MS. Interferon regulatory factor-1 (IRF-1) shapes both innate and CD8 T cell immune responses against West Nile virus infection. PLoS Pathog. 7:e1002230. doi: 10.1371/journal.ppat.1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien JD, Uhrlaub JL, Nikolich-Zugich J. Protective capacity and epitope specificity of CD8(+) T cells responding to lethal West Nile virus infection. Eur J Immunol. 2007;37:1855–1863. doi: 10.1002/eji.200737196. [DOI] [PubMed] [Google Scholar]
- Brien JD, Uhrlaub JL, Nikolich-Zugich J. West Nile virus-specific CD4 T cells exhibit direct antiviral cytokine secretion and cytotoxicity and are sufficient for antiviral protection. J Immunol. 2008;181:8568–8575. doi: 10.4049/jimmunol.181.12.8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cdc. West Nile virus activity--United States, 2007. MMWR Morb Mortal Wkly Rep. 2008;57:720–723. [PubMed] [Google Scholar]
- Daffis S, Samuel MA, Suthar MS, Keller BC, Gale M, Jr, Diamond MS. Interferon regulatory factor IRF-7 induces the antiviral alpha interferon response and protects against lethal West Nile virus infection. Journal of virology. 2008;82:8465–8475. doi: 10.1128/JVI.00918-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Shrestha B, Marri A, Mahan D, Engle M. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J Virol. 2003a;77:2578–2586. doi: 10.1128/JVI.77.4.2578-2586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Sitati EM, Friend LD, Higgs S, Shrestha B, Engle M. A critical role for induced IgM in the protection against West Nile virus infection. The Journal of Experimental Medicine. 2003b;198:1853–1862. doi: 10.1084/jem.20031223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredericksen BL, Keller BC, Fornek J, Katze MG, Gale M., Jr Establishment and maintenance of the innate antiviral response to West Nile virus involves both RIG-I and MDA5 signaling through IPS-1. J Virol. 2008;82:609–616. doi: 10.1128/JVI.01305-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman AE, Zhang J, Choi Y, Turka LA. Toll-like receptor ligands directly promote activated CD4+ T cell survival. J Immunol. 2004;172:6065–6073. doi: 10.4049/jimmunol.172.10.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN, Lin R, Hiscott J. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. Journal of virology. 2002;76:5532–5539. doi: 10.1128/JVI.76.11.5532-5539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson JM, Zajac AJ, Altman JD, Ahmed R. Cutting edge: increased expression of Bcl-2 in antigen-specific memory CD8+ T cells. J Immunol. 2000;164:3950–3954. doi: 10.4049/jimmunol.164.8.3950. [DOI] [PubMed] [Google Scholar]
- Havenar-Daughton C, Kolumam GA, Murali-Krishna K. Cutting Edge: The direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J Immunol. 2006;176:3315–3319. doi: 10.4049/jimmunol.176.6.3315. [DOI] [PubMed] [Google Scholar]
- Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD, Griffith TS, Green DR, Schoenberger SP. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature. 2005;434:88–93. doi: 10.1038/nature03337. [DOI] [PubMed] [Google Scholar]
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Keller BC, Fredericksen BL, Samuel MA, Mock RE, Mason PW, Diamond MS, Gale M., Jr Resistance to alpha/beta interferon is a determinant of west nile virus replication fitness and virulence. J Virol. 2006;80:9424–9434. doi: 10.1128/JVI.00768-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. The Journal of Experimental Medicine. 2005;202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro A, Horvath CM. RNA and Virus-Independent Inhibition of Antiviral Signaling by RNA Helicase LGP2. J Virol. 2006 doi: 10.1128/JVI.01325-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Wang L, Berman M, Kong YY, Dorf ME. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity. 2011;35:426–440. doi: 10.1016/j.immuni.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, Garcia-Sastre A, Katze MG, Gale M., Jr. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. Journal of virology. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund JM, Hsing L, Pham TT, Rudensky AY. Coordination of early protective immunity to viral infection by regulatory T cells. Science. 2008;320:1220–1224. doi: 10.1126/science.1155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek RD, Rathmell JC. The metabolic life and times of a T-cell. Immunol Rev. 2010;236:190–202. doi: 10.1111/j.1600-065X.2010.00911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulendran B, Ahmed R. Immunological mechanisms of vaccination. Nat Immunol. 2011;12:509–517. doi: 10.1038/ni.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purtha WE, Myers N, Mitaksov V, Sitati E, Connolly J, Fremont DH, Hansen TH, Diamond MS. Antigen-specific cytotoxic T lymphocytes protect against lethal West Nile virus encephalitis. Eur J Immunol. 2007;37:1845–1854. doi: 10.1002/eji.200737192. [DOI] [PubMed] [Google Scholar]
- Quigley M, Martinez J, Huang X, Yang Y. A critical role for direct TLR2-MyD88 signaling in CD8 T-cell clonal expansion and memory formation following vaccinia viral infection. Blood. 2009;113:2256–2264. doi: 10.1182/blood-2008-03-148809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman AH, Cui W, Larosa DF, Taylor DK, Zhang J, Goldstein DR, Wherry EJ, Kaech SM, Turka LA. MyD88 plays a critical T cell-intrinsic role in supporting CD8 T cell expansion during acute lymphocytic choriomeningitis virus infection. J Immunol. 2008;181:3804–3810. doi: 10.4049/jimmunol.181.6.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, Fitzgerald KA. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol. 2005;175:5260–5268. doi: 10.4049/jimmunol.175.8.5260. [DOI] [PubMed] [Google Scholar]
- Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, Akira S, Fujita T, Gale M., Jr Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:582–587. doi: 10.1073/pnas.0606699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel MA, Diamond MS. Pathogensis of West NIle virus infection: a balance between virulence, innate and adaptive immunity, and viral evasion. Journal of virology. 2006;80:9349–9360. doi: 10.1128/JVI.01122-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel MA, Whitby K, Keller BC, Marri A, Barchet W, Williams BR, Silverman RH, Gale M, Jr, Diamond MS. PKR and RNase L contribute to protection against lethal West Nile Virus infection by controlling early viral spread in the periphery and replication in neurons. J Virol. 2006;80:7009–7019. doi: 10.1128/JVI.00489-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar D, Desalle R, Fisher PB. Evolution of MDA-5/RIG-I-dependent innate immunity: independent evolution by domain grafting. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:17040–17045. doi: 10.1073/pnas.0804956105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Kato H, Kumagai Y, Yoneyama M, Sato S, Matsushita K, Tsujimura T, Fujita T, Akira S, Takeuchi O. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1512–1517. doi: 10.1073/pnas.0912986107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha B, Diamond MS. Role of CD8+ T cells in control of West Nile virus infection. J Virol. 2004;78:8312–8321. doi: 10.1128/JVI.78.15.8312-8321.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha B, Samuel MA, Diamond MS. CD8+ T cells require perforin to clear West Nile virus from infected neurons. J Virol. 2006;80:119–129. doi: 10.1128/JVI.80.1.119-129.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitati EM, Diamond MS. CD4+ T-cell responses are required for clearance of West Nile virus from the central nervous system. Journal of virology. 2006;80:12060–12069. doi: 10.1128/JVI.01650-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthar MS, Ma DY, Thomas S, Lund JM, Zhang N, Daffis S, Rudensky AY, Bevan MJ, Clark EA, Kaja MK, et al. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog. 2010;6:e1000757. doi: 10.1371/journal.ppat.1000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szretter KJ, Daffis S, Patel J, Suthar MS, Klein RS, Gale M, Jr, Diamond MS. The innate immune adaptor molecule MyD88 restricts West Nile virus replication and spread in neurons of the central nervous system. Journal of virology. 2010;84:12125–12138. doi: 10.1128/JVI.01026-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LJ, Kolumam GA, Thomas S, Murali-Krishna K. Innate Inflammatory Signals Induced by Various Pathogens Differentially Dictate the IFN-I Dependence of CD8 T Cells for Clonal Expansion and Memory Formation. J Immunol. 2006;177:1746–1754. doi: 10.4049/jimmunol.177.3.1746. [DOI] [PubMed] [Google Scholar]
- Tourneur L, Chiocchia G. FADD: a regulator of life and death. Trends Immunol. 2010;31:260–269. doi: 10.1016/j.it.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Venkataraman T, Valdes M, Elsby R, Kakuta S, Caceres G, Saijo S, Iwakura Y, Barber GN. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J Immunol. 2007;178:6444–6455. doi: 10.4049/jimmunol.178.10.6444. [DOI] [PubMed] [Google Scholar]
- Wilkins C, Gale M., Jr Recognition of viruses by cytoplasmic sensors. Current opinion in immunology. 2010;22:41–47. doi: 10.1016/j.coi.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M, Jr, Akira S, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175:2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- Zhang N, Bevan MJ. Dicer controls CD8+ T-cell activation, migration, and survival. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21629–21634. doi: 10.1073/pnas.1016299107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Hartig H, Dzhagalov I, Draper D, He YW. The role of apoptosis in the development and function of T lymphocytes. Cell Res. 2005;15:749–769. doi: 10.1038/sj.cr.7290345. [DOI] [PubMed] [Google Scholar]
- Zhao Y, De Trez C, Flynn R, Ware CF, Croft M, Salek-Ardakani S. The adaptor molecule MyD88 directly promotes CD8 T cell responses to vaccinia virus. J Immunol. 2009;182:6278–6286. doi: 10.4049/jimmunol.0803682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Chang M, Nie P, Secombes CJ. Origin and evolution of the RIG-I like RNA helicase gene family. BMC Evol Biol. 2009;9:85. doi: 10.1186/1471-2148-9-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.