

Abstract
Hypervirulent BI/NAP1/027 strains of Clostridium difficile have been associated with increased mortality of C. difficile infection (CDI). The emergence of highly fluoroquinolone (FLQ)-resistant BI/NAP1/027 strains suggests that FLQ exposure may be a risk factor for CDI development. However, the mechanism for this is not clear. We compared the effects of subinhibitory concentrations of ciprofloxacin on Toxin A and B gene expression and protein production in recent (strain 039) and historical (strain 5325) BI/NAP1/027 clinical isolates with high- and low-level ciprofloxacin resistance, respectively. In the highly ciprofloxacin-resistant isolate (strain 039), ciprofloxacin significantly and dose-dependently increased Toxin A gene expression and shifted its expression to earlier in its growth cycle; TcdB gene expression also increased but was less sensitive to low-dose ciprofloxacin. Maximal Toxin A/B production (4 ng ml−1) was increased twofold and occurred significantly earlier than in the untreated control. In strain 5325, ciprofloxacin at 0.25×MIC markedly increased both tcdA and tcdB expression but their temporal dynamics were unchanged. Maximal toxin production (250 ng ml−1) was reduced approximately threefold compared with that of the untreated control. These results demonstrate significant differences in ciprofloxacin-induced toxin gene expression and protein production among BI/NAP1/027 isolates, and offer a new paradigm for FLQ-associated CDI caused by recent, highly antibiotic-resistant strains.
Introduction
Clostridium difficile is the leading cause of nosocomial diarrhoea in hospitals and long-term healthcare facilities worldwide. The incidence, severity and mortality associated with C. difficile infection (CDI) have increased drastically over the past decade due to the emergence of the hypervirulent B1/NAP1/027 (NAP1) strain. C. difficile NAP1 strains are characterized by increased cytotoxin [Toxin A (TcdA) and Toxin B (TcdB)] production and by increased resistance to several classes of antibiotics (Carter et al., 2011; Hookman & Barkin, 2009; O’Connor et al., 2009; Bartlett, 1992; Saxton et al., 2009).
CDI is generally associated with recent or current administration of antibiotics, with clindamycin, the penicillins and cephalosporins carrying the greatest risk factors for the disease (O’Connor et al., 2009). Fluoroquinolone (FLQ) antibiotics are among the most commonly prescribed antimicrobials, and several NAP1 outbreaks in hospitals in the United States, Canada and Europe have been attributed to FLQ usage (O’Connor et al., 2009; Saxton et al., 2009; Deshpande et al., 2008). While FLQs are now considered by some to be a major risk factor for CDI because of hypervirulent, FLQ-resistant strains, several case–control studies have demonstrated no significant relationship between FLQ usage and CDI development (Dhalla et al., 2006; McFarland et al., 2007; Novell & Morreale, 2010).
β-Lactam antibiotic-induced toxin production has been clearly demonstrated for Staphylococcus aureus (Stevens et al., 2007) and other Gram-positive pathogens. Vancomycin, metronidazole, linezolid, amoxicillin and clindamycin have also been reported to induce toxin gene expression and increase toxin production by historical strains of C. difficile (Pultz & Donskey, 2005; Drummond et al., 2003; Gerber et al., 2008). Although FLQs have been shown to stimulate germination and growth of NAP1 strains (Saxton et al., 2009; Adams et al., 2007), the direct effects of FLQs on toxin gene expression and production by hypervirulent NAP1 strains have not been previously reported. Here, we describe the effects of subinhibitory doses of the FLQ antibiotic ciprofloxacin on tcdA and tcdB gene expression and soluble TcdA and TcdB production by two strains of C. difficile NAP1: the historical 5325 strain and a recent clinical NAP1 isolate, strain 039. These strains exhibit distinct differences in (i) exotoxin production in vitro, (ii) resistance to ciprofloxacin and (iii) their responses to antibiotic-induced toxin production following ciprofloxacin exposure. Our results demonstrate that recent, highly FLQ-resistant C. difficile isolates increase extracellular TcdA/B toxin production in response to ciprofloxacin. These findings offer a new mechanism to explain FLQ-associated CDI caused by NAP1 C. difficile.
Methods
C. difficile strains.
Two strains of C. difficile were studied. Strain 5325 is a historical NAP1 clinical isolate collected in 1993 (a kind gift from Dr Stewart Johnson, VA Chicago Health Care System, Chicago, IL, USA). C. difficile strain 039 was collected in 2005 from a critically ill patient with CDI at Johns Hopkins Medical Center, Baltimore, MD, USA (provided by Dr Karen Carroll). Both organisms were determined as toxinotype III strains by PFGE (data not shown).
Determination of MICs.
Ciprofloxacin was purchased from Sigma-Aldrich. The MIC of this antibiotic was determined for C. difficile NAP1 strains by using the microbroth dilution assay according to the National Committee for Clinical Laboratory Standards (NCCLS) guidelines for such testing in anaerobes (CLSI, 2004). In brief, 200 µl stationary overnight culture was used to inoculate 20 ml pre-reduced brain heart infusion (BHI) broth. Cultures were grown anaerobically for approximately 2 h at 37 °C until a turbidity equal to the 0.5 McFarland standard (OD630 of 0.08–0.1; ~1×106 c.f.u. ml−1) was achieved. The prepared C. difficile (50 µl) was then added to duplicate wells of a 96-well plate containing 50 µl of twofold serially diluted (0.5–1024 µg ml−1) ciprofloxacin. Plates were incubated anaerobically at 37 °C for 48 h and growth (turbidity) was assessed by a microplate reader (OD630). MICs were defined as the lowest antibiotic concentration that inhibited measurable bacterial growth (i.e. OD630 equal to the negative control). MICs were determined five times in triplicate.
Growth curves and RNA isolation.
NAP1 isolates were cultured anaerobically overnight in BHI, whereupon bacteria were collected by centrifugation and washed once with fresh BHI. The pellet was resuspended to the original volume (10 ml) in fresh BHI and an aliquot of washed bacteria (4–6 ml) was added to 100 ml fresh, pre-equilibrated BHI to an OD630 of 0.08–0.1. The culture was then added to 900 ml fresh, pre-equilibrated BHI and grown anaerobically to an OD630 of 0.08–0.1. Aliquots (199 ml) of this stock culture were divided among five individual sterile 500 ml flasks. Ciprofloxacin was prepared in sterile water as a ×200 stock solution with reference to the highest concentration required. Twofold serial dilutions were made from this stock in sterile water and 1 ml of each appropriate stock was added to 199 ml of C. difficile culture to give final concentrations of 0.25×, 0.125× and 0.0625×MIC, respectively. Sterile water (1 ml) served as a negative treatment control. At times −3 h (stock culture split), 0 h (antibiotics added) and 6, 12, 24 and 48 h after antibiotic addition, 10 ml samples were removed from each C. difficile culture and a small aliquot (20 µl) was used to determine viable c.f.u. The remaining organisms were collected by centrifugation (13 000 g) and used to prepare total RNA as described below for PCR analysis of toxin gene expression. Resultant supernatants were filter-sterilized and frozen at −70 °C for soluble TcdA/B production by ELISA.
Analysis of toxin gene expression and production.
RNA was isolated from collected bacterial pellets using the RiboPure-Bacteria kit (Ambion) according to the manufacturer’s recommendations. Contaminating genomic DNA was removed by two rounds of DNase treatment (DNA-free kit; Ambion), and the final RNA yield and quality were assessed by UV absorbance and agarose gel electrophoresis, respectively. Changes in tcdA and tcdB gene expression were assessed by real-time RT-PCR. For cDNA synthesis, 1 µg total RNA was converted to cDNA using M-MuLV reverse transcriptase (10 U ml−1; New England BioLabs), 1 mM random hexamer primers (Invitrogen) and 0.5 mM dNTPs (Invitrogen); all final concentrations. Reverse transcriptase reactions were performed at 37 °C for 2 h and were terminated by heating to 95 °C for 5 min. cDNA samples were diluted 1 : 4 and 1 : 40 for strains 039 and 5325, respectively, in sterile water and used for subsequent PCR. In brief, real-time RT-PCR was performed using a 7500 Fast Real-time PCR System (Applied Biosystems) by using the RT2 real-time SYBR green/Rox PCR master mix (SuperArray) and the following cycles: 10 min at 95 °C and then 40 cycles each at 95 °C for 15 s and 55 °C for 60 s. Primer sequences used for RT-PCR are listed in Table 1. Relative gene expression was determined using the 2–ΔΔCt method (Livak & Schmittgen, 2001). Sample mean Ct of 16S rRNA (internal control gene) was subtracted from the sample mean Ct of the tcdA and tcdB genes (ΔCT). Delta-Ct (ΔCt) of the no-treatment control for each time point was subtracted from the mean ΔCt of each experimental sample (ΔΔCt). This 2–ΔΔCt method yields fold change in gene expression of the gene of interest normalized to the expression of the 16S rRNA internal control and relative to the no-treatment control.
Table 1. Primer sequences for amplification of C. difficile tcdA and tcdB gene sequences.
Soluble TcdA and TcdB levels were measured (in combination) in collected culture supernatant samples using the Wampole Tox A/B II kit (TechLabs) as described by Merrigan et al. (2010). TcdB purified from a NAP1 isolate (a kind gift from Dr Jimmy Ballard, University of Oklahoma Health Sciences Center; stock concentration 300 µg ml−1) was used to construct a standard curve. Samples were diluted when necessary to obtain readings within the linear range of the standard curve. All samples were tested in triplicate.
Results
MICs of historical and recent NAP1 strains
The MIC of ciprofloxacin was determined for both the recent 039 and historical 5325 C. difficile NAP1 strains. According to the CLSI and NCCLS interpretative categories for resistance, sensitive organisms have an MIC <8.0 µg ml−1 (CLSI, 2004). In the recent 039 NAP1 strain, a strong resistance to ciprofloxacin was observed (MIC = 256 µg ml−1), while the historical 5325 strain displayed a lower resistance to the antibiotic (MIC = 8 µg ml−1).
Growth of recent and historical NAP1 strains in the presence of ciprofloxacin
The addition of subinhibitory concentrations of ciprofloxacin during the early exponential phase had little effect on growth of either strain 039 (Fig. 1) or strain 5325 (Fig. 2), although the absolute amounts of drug used differed by more than tenfold between strains. Some minor differences in growth dynamics were noted between strains, irrespective of the presence of antibiotic. Growth of both strains peaked at ~6 h; however, the recent 039 strain gradually declined thereafter, whereas the viability of the historical 5325 strain dropped by more than 1 log between 6 and 10 h and then maintained or slightly increased this level thereafter.
Fig. 1.

Effects of ciprofloxacin on the growth, toxin gene expression and soluble toxin production by the C. difficile NAP1 039 strain. (a) Antibiotic-free cultures, (b) 0.0625×MIC (16 µg ml−1), (c) 0.125×MIC (32 µg ml−1) and (d) 0.25×MIC (64 µg ml−1). Ciprofloxacin was added, at the final concentrations indicated, during early exponential phase growth (designated time 0). Samples were collected in duplicate over 48 h for quantification of viable C. difficile, and for measurement of toxin gene expression by real-time RT-PCR, and TcdA/B production by ELISA. Fold change in mRNA expression for each strain is made relative to that in its respective ‘No treatment’ control at 6 h (which was virtually zero in both strains). Filled line, tcdA mRNA; dotted line, tcdB mRNA.
Fig. 2.

Effects of ciprofloxacin on the growth, toxin gene expression and soluble toxin production by the C. difficile NAP1 5325 strain. (a) Antibiotic-free cultures, (b) 0.0625×MIC (0.5 µg ml−1), (c) 0.125×MIC (1 µg ml−1) and (d) 0.25×MIC (2 µg ml−1). Ciprofloxacin was added, at the final concentrations indicated, during early exponential phase growth (designated time 0). Samples were collected in duplicate over 48 h for quantification of viable C. difficile, and for measurement of toxin gene expression by real-time RT-PCR, and TcdA/B production by ELISA. Fold change in mRNA expression for each strain is made relative to that in its respective ‘No treatment’ control at 6 h (which was virtually zero in both strains). Filled line, tcdA mRNA; dotted line, tcdB mRNA.
Ciprofloxacin induces tcdA and tcdB gene expression and toxin production
Ciprofloxacin upregulated expression of both tcdA and tcdB in the recent NAP1 039 strain, although the dynamics of expression were subtly different between the two genes (Fig. 1). Specifically, ciprofloxacin significantly and dose-dependently induced tcdA expression by 6 h (Fig. 1, solid line), which returned to baseline by 10 h. Ciprofloxacin-induced tcdB expression demonstrated a biphasic increase, peaking first at 6 h and then again at 24 h (Fig. 1; broken line). For both genes, ciprofloxacin-induced upregulation of tcdA and tcdB was maximal in the 0.25×MIC (64 µg ml−1)-treated culture, being 40-fold and fourfold greater, respectively, than antibiotic-free controls (Fig. 1a, d).
The early and significant ciprofloxacin-induced increase in toxin gene expression in strain 039 resulted in an approximately twofold increase in TcdA/B production and shifted the time to peak toxin level from 48 h in untreated control cultures (Fig. 1a; peak 2.3 ng ml−1) to 24 h in ciprofloxacin-treated cultures (Fig. 1d; peak 4.0 ng ml−1). At 48 h, TcdA/B levels declined slightly but remained higher than untreated cultures (Fig. 3).
Fig. 3.
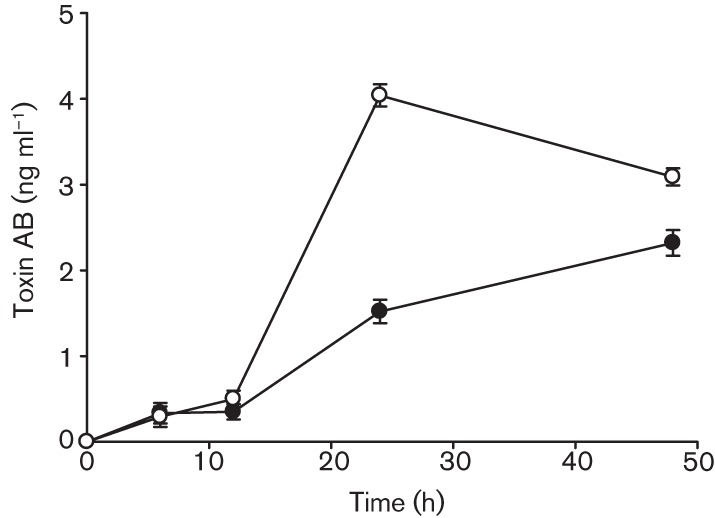
Effects of ciprofloxacin on Toxin A and B production by the C. difficile NAP1 039 strain. Soluble Toxin A and B measured by ELISA in the 64 µg ml−1-treated (0.25×MIC) culture. Ciprofloxacin was added at the final concentration during early exponential phase growth, designated time 0. Culture supernatant samples were collected over 48 h for detection of TcdA/TcdB by ELISA. Data shown are the means±sd of three replicates. •, No treatment; ○, 64 mg ml−1.
In contrast to the highly ciprofloxacin-resistant recent 039 NAP1 isolate, subinhibitory concentrations of ciprofloxacin minimally stimulated tcdA and tcdB expression by the historical 5325 NAP1 strain (Fig. 2), and did so during the stationary phase of growth. Specifically, 1.7- and 2.7-fold increases in tcdA and tcdB transcripts, respectively, were observed in cultures containing ciprofloxacin at 0.25×MIC (2 µg ml−1) at 24 h (Fig. 2d). Toxin expression levels were comparable with those of antibiotic-free cultures for all other ciprofloxacin treatments tested between 6 and 48 h. Consistent with the findings described for toxin gene expression, extracellular TcdA/B production by strain 5325 NAP1 was not increased by ciprofloxacin. In fact, 0.5–2.0 µg ciprofloxacin ml−1 (0.0625–0.25×MIC) dose-dependently decreased soluble TcdA/B levels in stationary phase supernatants when compared with no-treatment control cultures (Fig. 4).
Fig. 4.
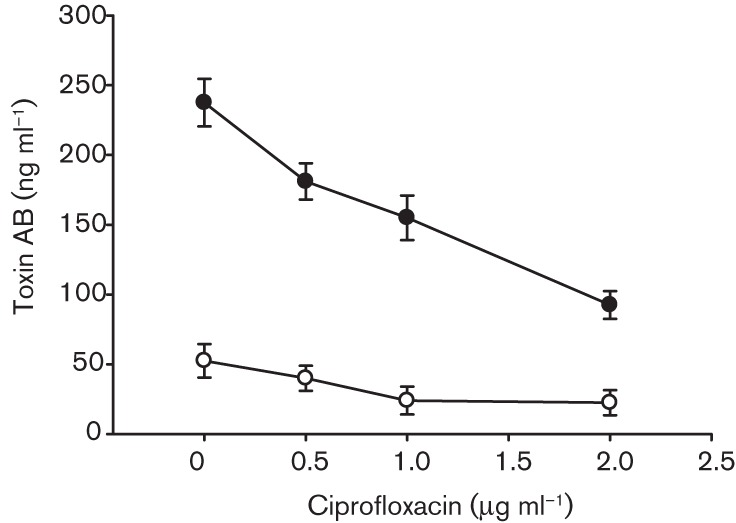
Dose-dependent decreases in Toxin A and B production in C. difficile NAP1 5325 strain following ciprofloxacin exposure. Culture supernatant samples were collected at 24 and 48 h following the addition of ciprofloxacin at final concentrations of 0, 0.5, 1.0 and 2.0 µg ml−1 (antibiotic free, 0.0625, 0.125 and 0.25×MIC, respectively) for detection of TcdA/TcdB by ELISA. Data shown are the means±sd of three replicates. ○, 24 h; •, 48 h.
Discussion
The incidence, severity and mortality associated with C. difficile-associated disease (CDI) have worsened during the last decade, largely due to the emergence of hypervirluent NAP1 strains with their increased toxin production and broad antibiotic resistance profile. FLQ resistance in NAP1 strains ranges from mild (8 µg ml−1) to strong (>512 µg ml−1), and more recent isolates are trending towards the high end of the resistance spectrum (Saxton et al., 2009). As a result, FLQ use has now become a major risk factor for CDI.
The traditional paradigm for the pathogenesis of antibiotic-associated CDI emphasizes antibiotic-induced alterations of the normal gut micro-organisms such that colonization and proliferation of toxinogenic C. difficile are favoured. However, a newer concept is emerging that antibiotic exposure induces CDI by not only changing colonic micro-organsisms but also by affecting the virulence properties of the organism. Honda et al. (1983) and Onderdonk et al. (1979) each demonstrated enhanced C. difficile enterotoxin production in response to sublethal amounts of antibiotics having different mechanisms of action. Similarly, Gerber et al. (2008) and Drummond et al. (2003) each showed that sub-MICs of drugs that either promote CDI (i.e. amoxicillin, clindamycin, cephalosporins) or those typically used to treat the infection (i.e. metronidazole, vancomycin, linezolid) upregulated toxin production by traditional (non-NAP1) strains of C. difficile. Both studies concluded that antibiotic-induced increases in C. difficile toxin production were part of a stress-induced response. Also, Gerber et al. (2008) have shown that antibiotic-induced toxin production in both laboratory strains and in traditional C. difficile clinical isolates is associated with a shift in toxin production to earlier stages of growth and that this is correlated with upregulation of tcdA and tcdB gene expression. Our findings extend these observations to include the toxigenic effects of FLQs on the more recent, hypervirulent NAP1 strains.
We further demonstrate that different NAP1 isolates, even within the same toxinotype, respond differently to subinhibitory antibiotic pressure based on their level of FLQ resistance, with increased toxin production being greatest in the highly FLQ-resistant strain. It is worth noting that the historical NAP1 5325 strain naturally produces nearly 250 times greater amounts of tcdA and tcdB transcripts and 100 times greater amounts of TcdA and TcdB than the recent NAP1 039 strain. Thus, the high background levels of toxin production in the no-treatment control may have masked any detectable antibiotic-induced increase in exotoxin production in this strain.
In our studies, we observed an ~18 h delay between the peaks of toxin gene expression and soluble toxin production in the recent 039 C. difficile isolate. Two mechanisms could explain this finding. First, Stevens et al. (2007) demonstrated that subinhibitory concentrations of nafcillin induced and prolonged mRNA transcripts for several toxins in S. aureus. Thus, it is plausible that sublethal doses of ciprofloxacin trigger a similar mechanism in C. difficile, inducing early tcdA and tcdB transcript production and increasing their half-lives until their translation in the stationary phase. Second, it is also possible that, following translation, TcdA and TcdB proteins remain intracellular during exponential phase growth and are released once organisms have entered the stationary phase. This latter scenario seems likely, as the holin-like protein, TcdE, is principally expressed in the stationary phase of growth where it is thought to facilitate exotoxin (TcdA and TcdB) release through permeabilization of the C. difficile cell wall (Tan et al., 2001). In addition, normal cell death and lysis of C. difficile organisms during the stationary phase probably contribute to TcdA and TcdB release into the external milieu. Both of these potential mechanisms are currently being investigated in our laboratory.
Marked ciprofloxacin-induced upregulation of toxin expression and production by the recent NAP1 039 strain, which is 32 times more resistant to ciprofloxacin than the historical 5325 isolate, suggests that the mechanisms governing high-level antibiotic resistance are linked to pathways controlling virulence factor production. It is not known whether this phenomenon can be generalized to all hyper-drug-resistant, toxin-producing human pathogens, such as Pseudomonas aeruginosa or meticillin-resistant S. aureus. Such a phenomenon occurring in vivo could severely complicate management and adversely affect outcomes for patients with life-threatening infections due to toxin-producing organisms. It would also suggest that a conserved mechanism exists which could be exploited as a therapeutic target for treatment of a broad spectrum of infections caused by hyper-resistant ‘superbugs’.
As the NAP1 039 and 5325 strains displayed diverse responses following challenge with ciprofloxacin, it is possible that these heterogeneous effects reflect strain-dependent responses to ciprofloxacin, given that only one FLQ antibiotic and two strains of C. difficile were examined. While our observations indicated that FLQ stimulated toxin production in some NAP1 strains, specific mechanisms of FLQ resistance have been identified in C. difficile that also contribute to FLQ-related complications of CDI (Ruiz, 2003).
In conclusion, exposure to several classes of antibiotics triggers CDI. Historically, most believed this was due to a suppression of normal bowel micro-organisms followed by colonization and proliferation of toxinogenic strains of C. difficile. However, our studies show that ciprofloxacin upregulates toxin gene expression and protein production in a hypervirulent NAP1 strain of C. difficile that is highly resistant to this antibiotic. Thus, our findings have important clinical implications when highly FLQ-resistant strains are prevalent in the community or hospital environments. In these instances, FLQ use could increase the incidence and severity of C. difficile colitis when patients are colonized with highly resistant organisms by increasing toxin production. Unfortunately, most clinical laboratories do not routinely culture C. difficile or determine susceptibility patterns of isolates. Thus, with the recent emergence of highly drug-resistant NAP1 strains, performance of routine antibiograms may gain renewed importance in the clinical microbiology laboratory and antibiotic stewardship committees may consider recommending the limited use of certain antibiotics when hyper-resistant strains are present.
Acknowledgements
This material is based upon work supported in part by the US Department of Veterans Affairs, Office of Research and Development Biomedical Laboratory Research Program (M. J. A., A. E. B., D. L. S.) and by the National Institutes of Health (grant P20 RR016454/GM103408 from the Idaho INBRE Program of the National Center for Research Resources).
Abbreviations:
- CDI
Clostridium difficile infection
- FLQ
fluoroquinolone
- TcdA
C. difficile Toxin A
- TcdB
C. difficile Toxin B
References
- Adams D. A., Riggs M. M., Donskey C. J. (2007). Effect of fluoroquinolone treatment on growth of and toxin production by epidemic and nonepidemic Clostridium difficile strains in the cecal contents of mice. Antimicrob Agents Chemother 51, 2674–2678 10.1128/AAC.01582-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett J. G. (1992). Antibiotic-associated diarrhea. Clin Infect Dis 15, 573–581 10.1093/clind/15.4.573 [DOI] [PubMed] [Google Scholar]
- Carter G. P., Douce G. R., Govind R., Howarth P. M., Mackin K. E., Spencer J., Buckley A. M., Antunes A., Kotsanas D. & other authors (2011). The anti-sigma factor TcdC modulates hypervirulence in an epidemic BI/NAP1/027 clinical isolate of Clostridium difficile. PLoS Pathog 7, e1002317 10.1371/journal.ppat.1002317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLSI (2004). Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria; Approved Standard, 6th ed NCCLS document M11-A6. Wayne, PA: NCCLS [Google Scholar]
- Deshpande A., Pant C., Jain A., Fraser T. G., Rolston D. D. (2008). Do fluoroquinolones predispose patients to Clostridium difficile associated disease? A review of the evidence. Curr Med Res Opin 24, 329–333 10.1185/030079908X253735 [DOI] [PubMed] [Google Scholar]
- Dhalla I. A., Mamdani M. M., Simor A. E., Kopp A., Rochon P. A., Juurlink D. N. (2006). Are broad-spectrum fluoroquinolones more likely to cause Clostridium difficile-associated disease? Antimicrob Agents Chemother 50, 3216–3219 10.1128/AAC.00592-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond L. J., Smith D. G., Poxton I. R. (2003). Effects of sub-MIC concentrations of antibiotics on growth of and toxin production by Clostridium difficile. J Med Microbiol 52, 1033–1038 10.1099/jmm.0.05387-0 [DOI] [PubMed] [Google Scholar]
- Gerber M., Walch C., Loffler R., Tischendorf K., Reischl U., Ackermann G. (2008). Effect of sub-MIC concentrations of metronidazole, vancomycin, clindamycin and linzolid on toxin gene transcription and production in Clostridium difficile. J Med Microbiol 57, 776–783 10.1099/jmm.0.47739-0 [DOI] [PubMed] [Google Scholar]
- Honda T., Hernadez I., Katoh T., Miwatani T. (1983). Stimulation of enterotoxin production of Clostridium difficile by antibiotics. Lancet 321, 655 10.1016/S0140-6736(83)91832-9 [DOI] [PubMed] [Google Scholar]
- Hookman P., Barkin J. S. (2009). Clostridium difficile associated infection, diarrhea and colitis. World J Gastroenterol 15, 1554–1580 10.3748/wjg.15.1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods 25, 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- McFarland L. V., Clarridge J. E., Beneda H. W., Raugi G. J. (2007). Fluoroquinolone use and risk factors for Clostridium difficile-associated disease within a Veterans Administration health care system. Clin Infect Dis 45, 1141–1151 10.1086/522187 [DOI] [PubMed] [Google Scholar]
- Merrigan M., Venugopal A., Mallozzi M., Roxas B., Viswanathan V. K., Johnson S., Gerding D. N., Vedantam G. (2010). Human hypervirulent Clostridium difficile strains exhibit increased sporulation as well as robust toxin production. J Bacteriol 192, 4904–4911 10.1128/JB.00445-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novell M. J., Morreale C. A. (2010). The relationship between inpatient fluoroquinolone use and Clostridium difficile-associated diarrhea. Ann Pharmacother 44, 826–831 10.1345/aph.1M696 [DOI] [PubMed] [Google Scholar]
- O’Connor J. R., Johnson S., Gerding D. N. (2009). Clostridium difficile infection caused by the epidemic B1/NAP1/027 strain. Gastroenterology 136, 1913–1924 10.1053/j.gastro.2009.02.073 [DOI] [PubMed] [Google Scholar]
- Onderdonk A. B., Lowe B. R., Bartlett J. G. (1979). Effect of environmental stress on Clostridium difficile toxin levels during continuous cultivation. Appl Environ Microbiol 38, 637–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pultz N. J., Donskey C. J. (2005). Effect of antibiotic treatment on growth of and toxin production by Clostridium difficile in the cecal contents of mice. Antimicrob Agents Chemother 49, 3529–3532 10.1128/AAC.49.8.3529-3532.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz J. (2003). Mechanisms of resistance to quinolones: target alterations, decreased accumulation and DNA gyrase protection. J Antimicrob Chemother 51, 1109–1117 10.1093/jac/dkg222 [DOI] [PubMed] [Google Scholar]
- Saxton K., Baines S. D., Freeman J., O’Connor R., Wilcox M. H. (2009). Effects of exposure of Clostridium difficile PCR ribotypes 027 and 001 to fluoroquinolones in a human gut model. Antimicrob Agents Chemother 53, 412–420 10.1128/AAC.00306-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens D. L., Ma Y., Salmi D. B., McIndoo E., Wallace R. J., Bryant A. E. (2007). Impact of antibiotics on expression of virulence-associated exotoxin genes in methicillin-sensitive and methicillin-resistant Staphylococcus aureus. J Infect Dis 195, 202–211 10.1086/510396 [DOI] [PubMed] [Google Scholar]
- Tan K. S., Wee B. Y., Song K. P. (2001). Evidence for holin function of tcdE gene in the pathogenicity of Clostridium difficile. J Med Microbiol 50, 613–619 [DOI] [PubMed] [Google Scholar]