

Abstract
Significance: Hydrogen sulfide (H2S) is likely to join nitric oxide (NO) and carbon monoxide (CO) as the third gaseous transmitter, influencing an array of intracellular signaling cascades. Thus, H2S is implicated in numerous physiological processes and in the pathology of various diseases. Recent Advances: H2S-donating agents that liberate H2S slowly either alone or in combination with NO, the so-called NOSH compounds, are being synthesized, and these have been shown to have great potential against cancer. Critical Issues: An accurate determination of H2S levels is challenging. H2S and NO share many similar actions; do these similarities act to potentiate each other? Since many actions of H2S appear to be mediated through inhibition of inflammation and Nuclear factor kappa-light-chain-enhancer of activated B cells is a central player in this scenario, does S-nitrosylation of this transcription factor by NO affect its S-sulfhydration by H2S and vice versa? Future Directions: Deciphering the molecular targets of these novel hybrid agents and having genetically engineered animals should help us move toward targeted therapeutic applications. Human safety data with these new hybrids is essential. Antioxid. Redox Signal. 20, 831–846.
Introduction
Hydrogen sulfide, H2S, is a colorless gas with a strong odor, likened to rotten eggs, that until recently was only considered as being a toxic environmental pollutant with little or no physiological significance. However, the past few years have demonstrated its role in many biological systems, and it is becoming increasingly clear that H2S is likely to join nitric oxide (NO) and carbon monoxide (CO) as the third gaseous transmitter, influencing an array of intracellular signaling cascades. Thus, H2S is implicated in numerous physiological processes and in the pathology of various diseases.
In general, biosynthesis of H2S requires tissue-specific enzymes that generate l-cysteine, a metabolite of dietary methionine. Three such enzymes have been identified: cystathionine β-synthase (CBS, EC 4.2.1.22) and cystathionine γ-lyase (CSE, aka CGL, EC 4.4.1.1) that require pyridoxal 5′-phosphate as a cofactor and use l-cysteine as substrate (7, 17); and 3-mercaptopyruvate sulfurtransferase (EC 2.8.1.2) which produces H2S using 3-mercaptopyruvate, a metabolite of l-cysteine (56). H2S can also be generated endogenously through nonenzymatic reduction of elemental sulfur in the blood (67) using reducing equivalents supplied through the glycolytic pathway (55).
H2S is sparingly soluble in water up to ∼117 mmoles/L and when dissolved, it acts as a weak acid with the equilibrium: H2S ⇌ HS−+H+ ⇌ S2−+H+. Only the first reaction is physiology relevant, as the pKa1 is around 6.9, which is approximately equivalent to the intracellular pH in many tissues, and it is not far from the pH of mammalian blood (7.4) (23). In the presence of oxygen and metal catalysts such as iron (III), H2S is spontaneously oxidized, thus significantly reducing its concentration in a matter of hours (12). Mitochondrial oxidation via three consecutive reactions is how much of the H2S is metabolized (22). This oxidation requires one mole of oxygen for every mole of H2S that is oxidized along the electron transport chain.
Interest in the biological actions of H2S is growing, and, therefore, there has been a lot of focus on H2S-donating or H2S-releasing compounds and on the way in which these compounds deliver this volatile gas to target tissues. In this review, these two modes of H2S delivery are distinguished. H2S-releasing compounds are sulfide salts such as NaHS, Na2S, and CaS, which form HS− and H2S immediately on solvation in physiological buffers. Although extensively used in studying the biological actions of H2S, these compounds are of limited therapeutic value, as their effective residence time in tissues is relatively short. H2S is rapidly lost from solution by volatilization in laboratory conditions (12), or transferred across respiratory membranes (24).
H2S-donating compounds are either naturally occurring compounds (mainly in foods) or synthetic hybrids. There are a large number of “foods” that may release H2S when metabolized. For example, garlic and some of its metabolites (allicin, diallyl disulfide [DADS], and diallyl trisulfide) have been shown to release H2S in vivo (24, 26). Sulforaphane, the isothiocyanate compound from broccoli, and allyl isothiocyanate, derived from wasabi, mustard, and horseradish, have health benefits that are quite similar to those which are attributed to H2S or other H2S-donating compounds, although it has not yet been determined whether any of these are indeed metabolized to produce H2S.
There are a number of synthetic H2S-donating compounds that are in various stages of development as therapeutic agents, and the list keeps on growing. Of these, GYY4137 (morpholin-4-ium 4 methoxyphenyl(morpholino)), phosphinodithioate, Figure 1 is a water-soluble molecule from which H2S is slowly liberated (39). A good number of synthetic H2S-donating compounds use the dithiolethione moiety for H2S donation. These include sildenafil, L-DOPA, latanoprost, and nonsteroidal anti-inflammatory drugs (NSAIDs) such as aspirin (ASA), sulindac, indomethacin, diclofenac, naproxen, ibuprofen, and mesalamine. Other H2S-donating moieties such 3-(prop-2-en-1yldisulfanyl)propanoic acid (a derivative of DADS) have also been employed.
FIG. 1.

Structural components of hydrogen sulfide (H2S)-donating compounds. The structural components of HS-aspirin (HS-ASA) (salicylic acid derivative), HS-sulindac (indole derivative), and HS-ibuprofen, HS-naproxen (arylpropionic acid derivative), HS-diclofenac, HS-indomethacin, and HS-mesalamine are indicated; the traditional nonsteroidal anti-inflammatory drug (NSAID) is shown in the shaded box; this is covalently attached to a dithiolethione moiety [5-(4-hydroxyphenyl)-3H-1,2-dithiole-3-thione] that can release H2S. The chemical structure of GYY4137, a water-soluble H2S-donating compound, is also shown.
In the past few years, a number of articles have addressed the role of some of these H2S-donating compounds in the cardiovascular system (35), urology (57), neuroinflammation (34), and ophthalmology (51). The reader is also referred to an excellent review article by Sparatore et al., which highlights these general areas (59). Here, the role of H2S-donating compounds as agents that may have utility against cancer are focused on and have addressed some of the mechanisms of actions of these novel agents. Also discussed are the newly reported nitric oxide-and-hydrogen sulfide-donating dual hybrids of ASA (30).
Synthetic H2S-Donating Hybrids
GYY4137
This is a water-soluble molecule slowly liberating H2S when dissolved in acidic (pH 3.0) phosphate buffer; however, when dissolved in buffer at pH 7.4 or 8.5, relatively much smaller amounts of H2S are detected (39). When administered to rats either intrapertioneally (ip) or intravenously (iv), significant increases in plasma H2S were observed for approximately 3 h. However, the methodology used in determining H2S levels is questionable (28), casting doubt on actual in vivo H2S levels.
The anti-cancer effects of GYY4137 were evaluated by determining its cell growth inhibitory effects using seven different human cancer cell lines. These represented cervical carcinoma (HeLa), colorectal carcinoma (HCT-116), hepatocellular carcinoma (Hep G2), osteosarcoma (U2OS), breast adenocarcinoma (MCF-7), acute promyelocytic leukemia cells (HL-60), and myelomonocytic leukemia (MV4-11) (36). Five-day treatment with GYY4137 (400 μM) significantly reduced proliferation of MCF-7, MV4-11, and HL-60 cells. At 800 μM, GYY4137 killed greater than 90% of all these cancer cells (MCF-7, MV4-11, HL-60, HeLa, HCT-116, Hep-G2, and U2OS), but did not affect the survival of noncancer human diploid lung fibroblasts (IMR90 and WI-38). The apparent IC50 for GYY4137 in MCF-7, HL60, and MV4-11 cells was around 350 μM. GYY4137 promoted apoptosis as seen by the presence of cleaved poly(ADP-ribose)polymerase and cleaved caspase 9. In MCF-7 cells, it also caused cell-cycle arrest in the G2/M phase; however, it did not affect the noncancerous IMR90 cells. In a xenograft model using SCID mice, a daily i.p. injection of GYY4137 (at 100–300 mg/kg) also dose dependently decreased the growth of HL-60 and MV4-11 tumors (36). An inspection of the published reports in which GYY4137 has been used as the H2S-donating compound clearly indicates that the doses needed to see any biological/pharmacological effects are rather high; this limitation may damper enthusiasm for its further development as a pharmaceutical drug.
H2S-donating NSAIDs do not cause stomach ulcers
ADT-OH, (5-(4-hydroxyphenyl)-3H-1,2-dithiole-3-thione) the main metabolite of ADT (anethole dithiolethione (Fig. 1), has been extensively used as a H2S donor with many therapeutic agents, including a good number of NSAIDs, reviewed in (28). The rational for developing hybrid NSAIDs which release H2S was based on the observation that in animal models of chronic ulcer, an increase in expression of CSE and CBS enzymes as well as H2S levels were observed, suggesting a defensive response (65). Administration of structurally diverse H2S donors enhanced ulcer healing and since these agents did not affect gastric pH or inhibit acid secretion, KATP channel agonists or antagonists had no affect on ulcer healing; and since H2S is a vasodilator, it is possible that this may have contributed to the healing process observed in this study (65).
Long-term use of NSAIDs may lead to significant side effects, mainly gastrointestinal (GI) and renal. The GI side effects may range from dyspepsia to bleeding, obstruction, and perforation. Among patients using NSAIDs, it is estimated that about 16,500 deaths occur every year in the United States. This figure is similar to the number of deaths from the acquired immunodeficiency syndrome and considerably greater than the number of deaths from multiple myeloma, asthma, cervical cancer, or Hodgkin's disease (70). Unfortunately, many physicians and most patients are unaware of the magnitude of this problem. The gastric damage is a result of direct epithelial damage due to their acidic properties and also through the breakdown of mucosal defense mechanisms (leukocyte adherence, decreases in blood flow, bicarbonate and mucus secretions) due to a reduction of mucosal prostaglandin synthesis (62).
A number of H2S-donating NSAIDs have shown considerable protective effects in the stomach compared with their traditional counterparts. For example, H2S-diclofenac had significant GI sparing effects compared with diclofenac (37, 64, 65) and inhibited both cyclooxygenase (COX)-1 and COX-2 enzymatic activity to the same extent as its parent counterpart (64). It also reduced the plasma levels of the pro-inflammatory cytokines, IL-1β and Tumor-necrosis factor-alpha, while it elevated the anti-inflammatory, IL-10 (37). Similarly, H2S-indomethacin (61), H2S-mesalamine (64), and H2S-naproxen (63) were also shown to cause significantly less damage to the stomach compared with their respective parent compound. In a recent study, H2S-donating ASA (ACS14) was shown to spare the stomach of rats from the harmful side effects of ASA when given 30 min before ASA administration (41). ACS14 also abrogated ASA-induced up-regulation of COX-2 expression, but it had no effect on the reduced PGE2 levels; it also attenuated ASA-suppressed superoxide dismutase-1 expression and glutathione (GSH) activity (41). These data suggest that ACS14 enhances mucosal integrity by inhibiting oxidative stress in the gastric tissue.
H2S-donating NSAIDs are effective chemopreventive agents
The landmark study of Kune et al. (32) showing that subjects using NSAIDs for various indications had a significantly lower incidence of colon cancer ushered in a new era in chemoprevention. Many epidemiological studies, collectively describing results on >1 million subjects, have established NSAIDs in general and ASA in particular as the prototypical chemopreventive agents against cancer. Recently, Rothwell et al. (53) reported that daily ASA use, whether regular strength or low dose, resulted in reductions in cancer incidence and mortality. Moreover, the same group has also reported that ASA prevented distant metastasis (54). However, regular NSAID use may lead to serious side effects as described earlier. To overcome these limitations and to improve the safety profile of NSAIDs, numerous strategies have been employed. One such strategy mentioned earlier has been the use of H2S. Recently, a number of H2S-donating NSAIDs (HS-ASA, HS-sulindac, HS-ibuprofen, and HS-naproxen; these are the same as S-ASA, S-sulindac, S-ibuprofen, and S-naproxen) were shown to have utility as anti-cancer agents (8–10). Although human safety data on H2S-donating NSAIDs are not yet available, these compounds have shown great potential using in vitro and in vivo models of carcinogenesis.
Aspects of the mechanism of action of H2S-NSAIDs
Two approaches have been used to unravel the mechanisms underlying the properties of H2S-NSAIDs. First, the effect of H2S-NSAIDs on cell kinetics has been investigated, which, by controlling tissue mass, determines the development of cancer. Second, some of the molecular mechanisms that might mediate these effects have been evaluated. The effects of H2S-NSAIDs on at least five molecular targets, no doubt there are many others, that have been implicated in carcinogenesis have been studied, including the Wnt/β-catenin signaling pathway, Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), reactive oxygen species (ROS), thioredoxin/thioredoxin reductase-1 (Trx/TrxR), and modulations of xenobiotic-metabolizing enzymes.
Cell kinetics
H2S-NSAIDs have three effects on cultured cancer-cell kinetics. They inhibit cell proliferation, as demonstrated by a reduced expression of the proliferation marker proliferating cell nuclear antigen (PCNA); they enhance cell death by inducing apoptosis, and they block transitions through the cell cycle, predominantly that between G0/G1 (8, 9). The net result of these effects is a significant inhibition of cell growth, which, presumably, accounts for the ultimate inhibition of carcinogenesis that is observed in vivo.
HS-ASA, HS-Sulindac, HS-naproxen, and HS-ibuprofen inhibited growth of eleven different cancer cell lines of six different histological subtypes (9). The cell lines were that of colon (HT-29: COX-1 and COX-2 positive, HCT 15: COX null, and SW480: COX-1 positive, low levels of endogenous COX-2), breast (MCF7: [ER(+)], SKBR3: [ER(−) and MDA MB-231: [ER−, PR−, which does not overexpress Her2]); T-cell leukemia (Jurkat), pancreatic (BxPC3: both COX-1 and COX-2 positive, MIAPaCa-2: COX-null), prostate (LNCaP), and lung (A549).
All four HS-NSAIDs inhibited the growth of these cell lines (9) (Table 1). Interestingly, ASA, which was the least potent of the four NSAIDs in inhibiting the growth of these cell lines, its modified H2S-donating hybrid, became the most potent HS-NSAID. In a fold-comparison study of the IC50 values (Traditional NSAID/HS-NSAID), HS-ASA was at least 1100–3000-fold more potent than ASA. HS-SUL, HS-IBU, and HS-NAP were about 20–500-fold more potent than their corresponding traditional counterparts. Such fold increases imply that the H2S-related structural modification of the parent molecules imparts a differential enhancement in potency (9). Since colon HT-29 and pancreatic BxPC3 cells express both COX-1 and COX-2 whereas colon HCT15 and pancreatic MIA PaCa2 cells are COX null (21, 29), this suggests that the effects observed here are COX independent. The cell growth inhibitory effect of HS-NSAIDs is at least, in part, dependent on the release of H2S, as the preincubation of MDA MB-231 breast cancer cell with NaF, a carboxylesterase enzyme inhibitor, significantly reversed HS-ASA-mediated cell growth inhibition (8). It also appears that HS-NSAIDs preferentially inhibit the growth of cancer cells compared with normal cells. For example, treatment of normal human mammary epithelial cells (HMEpC) with 10 μM HS-ASA showed that 78% of the cells were viable after 24 h; however, under the same experimental conditions, only 28% of breast cancer cells (MDA MB-231) remained viable (8).
Table 1.
IC50 (μM) Values for Cell Growth Inhibition by Different HS-NSAIDs in Different Cancer Cell Lines
| Colon | Breast | Pancreas | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Agent | HT-29 | HCT15 | SW480 | MDA MB231 | SKBR3 | MCF7 | MIAPaCa2 | BxPC3 | Lung A549 | Prostate LNCAP | Leukemia Jurkat |
| ASA | >5000 | >5000 | >5000 | >5000 | >5000 | >5000 | >5000 | >5000 | >5000 | 4560±210 | >5000 |
| HS-ASA | 3.7±0.9 | 3.2±1.1 | 1.8±0.6 | 3.6±0.6 | 3.3±0.9 | 4.2±1.1 | 2.1±0.9 | 1.9±0.7 | 1.6±0.7 | 2.3±0.7 | 2.3±0.8 |
| Ratio | >1300 | >1500 | >2700 | >1300 | >1500 | >1100 | >2300 | >2600 | >3100 | 1983 | >2100 |
| NAP | 2730±121 | 3000±189 | 2980±151 | 3260±125 | 3100±129 | 2550±121 | 3300±150 | 2160±128 | 2770±133 | 3170±165 | 2650±161 |
| HS-NAP | 72±2 | 66±2 | 71±3 | 51±2 | 58±2 | 63±3 | 81±3 | 76±2 | 69±2 | 74±2 | 59±2 |
| Ratio | 38 | 45 | 42 | 64 | 54 | 40 | 41 | 28 | 40 | 43 | 45 |
| SUL | 740±32 | 863±43 | 678±46 | 987±37 | 865±41 | 1045±62 | 843±29 | 985±30 | 147±22 | 871±36 | 736±33 |
| HS-SUL | 6±1.6 | 7±0.9 | 8±2 | 10±2 | 9±1 | 10±1 | 7±1 | 5±1 | 7±2 | 7±1 | 5±0.9 |
| Ratio | 123 | 119 | 85 | 99 | 96 | 105 | 119 | 197 | 21 | 124 | 147 |
| IBU | 1090±77 | 1120±120 | 1135±121 | 789±47 | 763±53 | 843±61 | 567±40 | 621±51 | 968±40 | 1075±92 | 897±27 |
| HS-IBU | 2.8±0.9 | 3.5±0.7 | 3.1±0.8 | 5.4±0.7 | 6.2±0.9 | 4.1±0.9 | 2.9±0.8 | 3.7±0.9 | 1.9±0.5 | 2.8±0.9 | 4.3±0.7 |
| Ratio | 389 | 320 | 366 | 146 | 123 | 206 | 196 | 168 | 509 | 484 | 209 |
Colon, breast, pancreas, lung, prostate, and leukemia cancer cell lines were treated with various concentrations of HS-aspirin (HS-ASA), HS-naproxen (HS-NAP), HS-sulindac (HS-SUL), HS-ibuprofen (HS-IBU), and their traditional counterparts. Cell numbers were determined at 24 h from which IC50 values were calculated. The ratios of NSAID/HS-NSAID represent fold enhancement in potency of the HS-NSAID compared with the parent compound. Results are mean±SEM of at least four different experiments performed in triplicates. p<0.001 for all HS-NSAIDs compared with their respective NSAID in all cell lines. Reproduced from Chattopadhyay et al., 2012 (9) with permission from Elsevier.
HS-ASA, HS-NAP, HS-SUL, and HS-IBU reduced proliferation of HT-29 human colon cancer cells in a dose-dependent manner, as measured by the expression of PCNA (Fig. 2A), and also increased the apoptotic population of cells (Fig. 2B) (9). HS-NAP was the strongest contributor to apoptosis. Similarly, HS-ASA reduced PCNA expression of MDA MB-231 breast cancer cells in a dose-dependent manner as well as increased the proportion of cells in apoptosis (8).
FIG. 2.

Effect of HS-NSAIDs on HT-29 colon cancer cell kinetics. Cells were treated with the respective HS-NSAIDs at the concentration indicated for 24 h followed by determination of apoptosis (A) and proliferating cell nuclear antigen (B). Results are mean±SEM of three different experiments. *p<0.05; †p<0.01 compared with untreated cells. IC50s for HS-ASA, HS-NAP, HS-SUL, and HS-IBU were 3.7±0.9, 72±2, 6±1.6, and 2.8±0.9 μM, respectively. Reproduced from Chattopadhyay et al., 2012 (8) with permission from Elsevier.
Analysis of cell cycle phases by flow cytometry showed that treatment of HT-29 colon cancer cells by all four HS-NSAIDs was associated with a dose-dependent increase in the percentage of cells in G0/G1 phase, and this accumulation was accompanied by a corresponding reduction in the percentages of cells in S and G2/M phases (Fig. 3) (9). Similarly, HS-ASA also affected the cell cycle progression of MDA MB-231 breast cancer cells, causing a dose-dependent accumulation in G0/G1 phase (10). HS-ASA also dose-dependently inhibited the proliferation of Jurkat T-cell leukemia cells, increased apoptosis, and caused a G0/G1 block in the cell cycle (18). Thus, it appears that HS-NSAIDs inhibit the proliferation of HT-29 colon, MDA MB-231 breast, and Jurkat T-cell leukemia cancer cells by a combined induction of G0/G1 arrest and apoptosis.
FIG. 3.

Effect of HS-NSAIDs on cell cycle. Colon HT-29 cells were treated with various HS-NSAIDs and harvested at 24 h. The percentage of cells in each phase was determined by flow cytometry. Results are representative of two different experiments. This study was repeated twice, generating results within 10% of those presented here. Reproduced from Chattopadhyay et al., 2012 (8) with permission from Elsevier.
NF-κB signaling pathway
The transcription factor NF-κB has a central role in cancer biology. It has pleiotropic activities that relate to carcinogenesis, and it is an important regulator of the immune system; it is vital for the induction of cytokines, anti-apoptotic genes, and growth proliferation, collectively perpetuating the conversion of cells from normal to cancerous. NF-κB can suppress apoptosis, thus promoting tumorigenesis (25). Most chemopreventive agents (i) suppress the activation of the NF-κB through the inhibition of NF-κB signaling pathway, and (ii) they sensitize the tumors to chemotherapeutic agents through abrogation of NF-κB activation. As stated so eloquently, “NF-κB is an ideal target for chemoprevention and chemosensitization” (4). Since the field H2S-donating NSAIDs as chemotherapeutic agents is in its infancy, the number of reports describing the effects of this class of compounds on the NF-κB signaling pathway is rather limited.
One study showed that the H2S-donating agent, GYY4137, reduced lipopolysaccharide (LPS)-induced NF-κB activation in mouse macrophages (68). The same group has also reported that when rats were injected with LPS, GYY4137 decreased hepatic LPS-evoked rise in NF-κB activation (38). H2S-donating diclofanc was shown to reduce inflammation in rats after LPS administration, and this was attributed to inhibition of liver NF-κB activation (37). In these in vivo studies, nuclear proteins were extracted from rat livers and assayed for activity using TransAM NF-κB p65 kit.
There is one report detailing the effect of HS-ASA on the NF-κB signaling pathway using MDA MB-231 breast cancer cells. The rational for investigating this cell line was reported to be due to the fact that NF-κB is constitutively activated in estrogen receptor (ER)-negative breast cancer cell lines and primary tumors (5, 46, 52), and that this led to resistance to apoptosis (50, 66), and promotion of an invasive and metastatic phenotype (47).
In MDA-MB 231cells treated with HS-ASA for 6 h, there was a significant dose-dependent inhibition of DNA binding activity of NF-κB (p65) (Fig. 4A). HS-ASA at its IC50 for cell growth inhibition (3.6 μM) and 2xIC50 (7.2 μM) reduced the NF-κB-DNA binding activity to ∼71% and 50%, respectively. Levels of nuclear p65, which is the functionally active subunit of NF-κB, were also reduced by HS-ASA in a concentration-dependent manner (Fig. 4B). The decrease in DNA binding activity was associated with a decrease in NF-κB protein p65 levels in the nucleus. One of the most critical steps in NF-κB activation is its dissociation from IκB, which is mediated through phosphorylation and subsequent proteolytic degradation of this inhibitory subunit (6, 19). HS-ASA decreased phospho-IκBα levels and at 2xIC50, it completely inhibited IκBα phosphorylation, while total levels were unaltered (Fig. 4C and D). Thus, HS-ASA inhibits translocation of p65 to the nucleus through blockade of IκBα phosphorylation. The phosphorylation of the IκBs is known to be mediated by Ikappa kinases (IκKs), namely IκKα and IκKβ, which are activated by phosphorylation at specific serine residues in the activation loop of IκKβ (6). HS-ASA decreased the phosphorylation of both IκKα and IκKβ without affecting total IκK levels (Fig. 4C and D). Therefore, HS-ASA prevents activation of IκKs, inhibits phosphorylation-mediated degradation of IκBα, and prevents translocation of NF-κB-p65 into the nucleus (8).
FIG. 4.

HS-ASA inhibited NF-κB transcription factor activity in MDA-MB-231 cells. (A) Cells were treated with vehicle or HS-ASA for 24 h, and nuclear proteins were harvested. DNA binding activity of NF-κB was determined from nuclear extracts by ELISA for each sample relative to the vehicle control (100%). (B) Nuclear protein extracts was examined for levels of p65 subunit of NF-κB by immunoblotting. (C) Cytoplasmic fractions of HS-ASA-treated cells were examined for total IκBα, IKKα, and IKKβ levels, and (D) for phosphorylated IκBα,–IκKα, and–IκKβ by immunoblotting. Results are mean±SEM of three different sets of experiments. *p<0.05 compared with vehicle-treated controls. Reproduced from Chattopadhyay et al., 2012 (8) with permission from Elsevier.
Recently, using ER(−) breast cancer cells, it has been reported that dithiolethiones inhibit NF-κB transcriptional activity not through H2S release but rather via a covalent reaction with the NF-κB p50 and p65 subunits (60).
Reactive oxygen species
As a part of their mechanism of action, many anticancer agents induce ROS. High ROS levels can disrupt redox homeostasis, leading to cellular damage and apoptosis (15, 16). Using molecular probes that detect intracellular levels of peroxides and superoxide anions (O2•−), it was shown that HS-ASA dose-dependently induced ROS levels in MDA MB-231 breast cancer cells (8) (Fig. 5).
FIG. 5.

HS-ASA induces reactive oxygen species (ROS) levels. MDA-MB-231 cells were treated with HS-ASA for 1 h followed by staining with a general ROS probe 2′,7′-dichlorodidydrofluorescin diacetate (A, B) or dihydroethidium, which detects superoxide anions in cells (C, D). Representative histograms are shown. Values are the mean±SEM of three independent experiments. *p<0.05, †p<0.01 compared with untreated controls. The IC50 value for cell growth inhibition was 3.6±0.6 μM at 24 h. Reproduced from Chattopadhyay et al., 2012 (8) with permission from Elsevier. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
When the equilibrium between ROS formation and endogenous antioxidant defense mechanisms is disturbed, a state of oxidative stress is generated within the cell, which ultimately may lead to cell death. Thus, the observation that HS-ASA induced cell cycle changes and apoptosis in MDA-MB-231 cells, which was associated with parallel increases in ROS levels, appears to favor cell death.
Thioredoxin/thioredoxin reductase-1
Trx is a ubiquitous redox-active thiol protein associated with the enzyme TrxR that maintains it in the reduced state (45). Trx can suppress apoptosis (1), and increases in Trx and TrxR are found in several human primary cancers that are associated with aggressive tumor growth, including breast carcinoma (40). In addition, some relationships exist between TrxR inactivation and growth inhibition, cell cycle arrest, and apoptosis in colorectal cancer cell lines (71). In MDA-MB 231 breast cancer cells, it was shown that TrxR activity was strongly inhibited by HS-ASA (8). Moreover, it was also shown that HS-ASA caused a moderate inhibition of NF-κB-activity through inhibition of IKK activation (8). Thus, targeting the Trx/TrxR system, which is required for the maintenance of essential cysteine residues in the active thiol forms in certain proteins, including the NF-κB protein (43), are attractive targets for cancer drug development.
Wnt/β-catenin signaling pathway
The Wnt signaling cascade has a major role in human colon carcinogenesis (13), and its deregulation has also been implicated in T-cell acute lymphoblastic leukemia (ALL) as well as in B-cell chronic lymphocytic leukemia (14, 42). The β-catenin protein is intimately involved in the Wnt signaling pathway that regulates cell–cell adhesion and may promote leukemia cell proliferation (14). The stabilization and accumulation of β-catenin has a powerful regulatory role in proliferation and differentiation (44, 49). β-Catenin is expressed in T-ALL cells, tumor lines of hematopoietic origin, and primary leukemia cells but is undetectable in normal peripheral blood T cells. Among the leukemic cell lines, β-catenin is expressed in high levels in Jurkat T cells (14). Using this cell line, it has been shown that HS-ASA dose-dependently reduced β-catenin protein levels; whereas its parent counterpart, ASA, for approximately 5 mM had no effect on total β-catenin expression (18). Using synchronized cells, the effects of HS-ASA on the expression of β-catenin/T-cell factor transcriptional target, cyclin D1, were also investigated. HS-ASA dose-dependently reduced cyclin D1 protein expression (18). These results are in agreement with cell growth inhibition and G1 phase blockade. In some B- and T-cell leukemias, ASA and its NO donating derivatives are reported to activate caspases (3, 48). HS-ASA is also reported to increase caspases-3 enzyme activity and protein expression in a dose-dependent manner (18). The caspase inhibitor zVAD-fmk was able to completely abrogate HS-ASA-mediated caspase activation, confirming specificity. Preincubation of the cells with the caspase inhibitor also partially reversed HS-ASA-mediated cell growth inhibition, increasing its IC50 for cell growth inhibition by about five-fold, from 1.8±0.6 μM to 8.9±0.5 μM (18).
Modulation of xenobiotic-metabolizing enzymes
The xenobiotic-metabolizing enzymes are classified as phase-I (oxidation and reduction reactions) and phase-II (conjugation reactions) enzymes. The balance between the phase-I carcinogen-activating enzymes and the phase-II detoxifying enzymes is critical to determining an individual's risk for cancer [reviewed in (69)]. Phase-I enzymes primarily consist of the cytochrome P450 (CYP) superfamily. In humans, four CYP gene families (CYP1–CYP4) are considered the most important (31). Phase-II enzymes comprise many enzyme superfamilies, including the NAD(P)H:quinone oxireductase (NQO), glutathione S-transferases (GSTs), and UDP-glucuronyltransferases (UGTs). NQO protects cells against the toxicity of quinones and their metabolic precursors by promoting the obligatory two-electron reduction of quinones to semiquinones. The human GSTs consist of four main classes: α (A), μ (M), θ (T), and π (P), each of which is divided into one or more isoforms (20). GSTs detoxify a number of carcinogenic electrophiles by transforming them into their conjugates with reduced glutathione. Pharmacological strategies that modulate the levels of phase-I and phase-II enzymes can enhance the elimination of the carcinogenic reactive species. In particular, identification of compounds that preferentially activate phase-II compared with phase-I enzymes can be beneficial as chemopreventive agents.
There is a consensus enhancer element, known as antioxidant response element (ARE), in the regulatory domains of many phase-II genes, and the ARE-binding transcription factor Nrf2 has been implicated in the action of many chemopreventive agents (33). Nrf2 is a regulator of the inducible expression of the enzymes GST and NQO1, which are important in catalyzing the detoxification of reactive electrophiles and oxidants [reviewed in (58)].
Using human hepatoma HepG2 and human colonic adenocarcinoma LS180 cells, it has been shown that both H2S-donating diclofenac and H2S-donating sulindac inhibit the activity and expression of the carcinogen-activating enzymes CYP1A1, CYP1B1, and CYP1A2 (2). This inhibition was mediated by transcriptional regulation of the aryl hydrocarbon receptor (AhR). Both H2S-donating diclofenac and H2S-donating sulindac inhibited carcinogen-induced CYP enzyme activity through direct inhibition as well as through decreased transcriptional activation of the AhR. H2S-donating sulindac also induced expression of GST A2, a phase-II detoxifying enzymes (2).
Using mouse hepatoma Hepa 1c1c7 and human HT-29 colorectal cancer cells, it has also been reported that GST (Fig. 6A and B) and NQO1 (Fig. 6C and D) activities were dose-dependently induced by HS-ASA (10). The increase in enzyme activity was shown to be H2S dependent, as preincubation of the cells with NaF, a carboxylesterase enzyme inhibitor that dampens release of H2S from HS-ASA, reversed HS-ASA-mediated induction of NQO1 activity to near basal levels (Fig. 6E and F) Therefore, H2S release from HS-ASA appears to be required for induction of NQO1 activity. HS-ASA also caused a dose-dependent increase in NQO1, GST, and UGT1 protein levels as well as increased protein levels of CYP1A1 and CYP2E in these cell lines (Fig. 7). The transcription factor Nrf2, which is involved in induction of many phase-II enzymes, showed enhanced expression in the HS-ASA-treated cells (10) (Fig. 7).
FIG. 6.

The activity of the phase-II enzymes glutathione S-transferase (GST) and NAD(P)H:quinone oxireductase (NQO)1 is induced by HS-ASA. Hepa 1c1c7 or HT-29 cells were treated with the indicated concentrations of HS-ASA for 24 h before enzyme activity was measured. HS-ASA increased GST and NQO1 enzyme activities in a concentration-dependent manner. GST in Hepa 1c1c7 cells (A), in HT-29 cells (B); NQO1 in Hepa 1c1c7 (C) and in HT-29 (D). This increase in enzyme activity was dependent on H2S, as preincubation of the cells with NaF, a carboxylesterase inhibitor, abrogated the increase in enzyme activity back to control levels (E, F). Results are mean±SEM of three different experiments performed in duplicate. *p<0.05 compared with untreated cells, **p<0.02 compared with untreated cells, and †p<0.02 compared with HS-ASA at its IC50 in absence of NaF. Reproduced from Chattopadhyay et al., 2012 (9) with permission from Elsevier.
FIG. 7.

Effect of HS-ASA on phase-I and phase-II metabolizing enzymes. Hepa 1c1c7 (A) or HT-29 (B) cells were incubated with the indicated concentrations of HS-ASA for 24 h, after which cell extracts were prepared and subjected to immunoblotting. Representative blots (one of two determinations) are shown here. HS-ASA increased NQO1, GST, UGT, CYP1A1, and CYP2E1 protein levels. Nuclear proteins were also immunoblotted for Nrf2 and Lamin (bottom panel). Reproduced from Chattopadhyay et al., 2012 (9) with permission from Elsevier.
Treatment of rats with HS-ASA (100 mg/kg/day for 21 days) induced hepatic GST activity by 3.4-fold (Fig. 8A), GSH levels were increased 2-fold (Fig. 8B), and NQO1 enzyme activity was increased 1.5-fold (10) (Fig. 8C). Immunoblot analysis (Fig. 8D) demonstrated that HS-ASA moderately increased the liver cytosolic GST, NQO1, and substantially increased UGT1A1 protein levels (Fig. 8E). The levels of the phase-I enzymes, CYP1A1 and CYP2E1, did not differ between the untreated and treated groups (10) (Fig. 8E). This finding strongly suggests that in vivo HS-ASA is a monofunctional inducer of phase-II metabolizing enzymes, and it underscores the dangers of translating in vitro data to whole animals.
FIG. 8.

HS-ASA induces phase-II enzyme activity and glutathione (GSH) levels in vivo. Male Wistar rats (n=4 per group) were treated with HS-ASA (100 mg/kg body weight) for 3 weeks, after which liver microsomes and cytosol were prepared. HS-ASA treatment induced GST and NQO1 enzyme activities A and C, respectively. GSH levels were also increased in the livers of the HS-ASA-treated rats (B). Results are mean±SEM for four rats in each group. *p<0.05; †p<0.01 compared with vehicle-treated rats. HS-ASA is a mono-functional inducer of hepatic phase-II enzymes. (D) Liver microsomes and cytosol from HS-ASA-treated Wister rats were subjected to immunoblotting for phase-I (CYP1A1 and CYP2E1) and phase-II (GST, UGT1A1, and NQO1) enzymes. Actin was used as a housekeeping gene. The four lanes represent individual rats. (E) The expression of each protein (fold compared with control) for each group of animals (mean±SEM, n=4). *p<0.05, †p<0.01 compared with the corresponding control. Reproduced from Chattopadhyay et al., 2012 (9) with permission from Elsevier.
Animal studies
In a xenograft model of ER(−) breast cancer, HS-ASA significantly reduced tumor volume and tumor mass, through induction of apoptosis and inhibition of both proliferation and NF-κB (8). Athymic SCID male mice were injected subcutaneously with MDA-MB-231cells in the right flank, allowing for the development of subcutaneous tumors after 7 days (Fig. 9). After tumor formation, mice were treated with vehicle or 100 mg/kg HS-ASA for 23 days. HS-ASA-treated mice showed a significant reduction in tumor volume compared with untreated mice (Fig. 9), mean tumor volume was decreased by 44% (p<0.05), and average tumor mass was reduced by 47% (p<0.05). Immunohistochemical expression of HS-ASA-treated and untreated mouse tumor sections showed a strong expression for proliferation and NF-κB (Fig. 10A and C, control panel), whereas HS-ASA-treated tumors showed a diminished expression of NF-κB and proliferation (Fig. 10A and C, treated panel). HS-ASA treatment was also accompanied with increased number of cells undergoing apoptotic death (20%) compared with control (1.5%) (Fig. 10B).
FIG. 9.

HS-ASA inhibits tumor xenograft growth. Athymic SCID male mice were injected subcutaneously with MDA-MB-231 cells for the development of subcutaneous tumors. After tumor formation, the mice were randomly divided into two groups (n=6) and treated daily with vehicle or 100 mg/kg HS-ASA for 23 days according to the protocol in A. Representative tumor sizes for untreated and treated mice are shown in B. Average tumor volume as function of time and tumor mass at sacrifice are shown in C and D, respectively. HS-ASA significantly reduced tumor volume from day 20 post inoculation to sacrifice, p<0.05. Tumor mass was also significantly reduced by HS-ASA treatment, p<0.05. Reproduced from Chattopadhyay et al., 2012 (8) with permission from Elsevier. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 10.

HS-ASA inhibits proliferation (A), induces apoptosis (B), and decreases NF-κB p65 (C) in vivo. Tumors sections were probed and scored. Average mitotic index at sacrifice was determined by Ki-67 staining (p<0.05), TdT-mediated dUTP nick-end labeling staining (p<0.02), and p65 staining (p<0.02). Representative fields used for quantification of the staining are shown. The scale bar represents 200 μm. Reproduced from Chattopadhyay et al., 2012 (8) with permission from Elsevier. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
NOSH-Aspirin
Recently, a new class of anti-inflammatory agents that are capable of donating both NO and H2S (termed NOSH compounds) have been described (11, 30). Four NOSH agents have been reported; in all of these, ASA was used as a scaffold to which NO and H2S-releasing moieties were coupled with one of the 1, 2 positions. Nitrate (−ONO2) was used for NO release, and this was attached to the ASA through an aliphatic spacer; while one of the following H2S-releasing moieties, ADT-OH, or 4-hydroxy benzothiazamide, or lipoic acid, was directly coupled to ASA (30), Figure 11. NOSH-1 (NOSH-ASA, NBS-1120) was shown to have strong anti-inflammatory properties similar to its parent compound, ASA (30) (Fig. 12). Using the carrageenan rat's paw edema model, NOSH-1 showed a reduction in paw inflammation, which was dose dependent. A comparison of PGE2 content of paw exudates from control, ASA-treated, and NOSH-1-treated animals showed a clear and significant COX inhibition by ASA and NOSH-1. An evaluation of COX expression in paw exudates showed that COX-2, which produces inflammatory PGE2, was barely detectable in the controls, was significantly induced by carrageenan, and dose-dependently inhibited by NOSH-1, as shown in Figure 12.
FIG. 11.

The chemical structures of NOSH compounds. NOSH-1: (4-(3-thioxo-3H-1, 2-dithiol-5-yl) phenyl 2-((4-(nitrooxy)butanoyl)oxy) benzoate); NOSH-2: (4-(nitrooxy)butyl (2-((4-(3-thioxo-3H-1,2-dithiol-5-yl)phenoxy)carbonyl)phenyl)); NOSH-3: (4-carbamothioylphenyl 2-((4-(nitrooxy)butanoyl)oxy)benzoate); and NOSH-4: (4-(nitrooxy)butyl 2-(5-((R)-1,2-dithiolan-3-yl)pentanoyloxy)benzoate).
FIG. 12.

Anti-inflammatory properties of NOSH-1. Rat paw edema was induced by carrageenan injection. (A) ASA and NOSH-1 caused a significant reduction in paw volume at all time points. Results are mean±S.E.M. of four rats in each group, *p<0.05 versus vehicle-treated rats at all time points. (B) ASA and NOSH-1 caused a significant reduction in PGE2 levels in the paw exudate. Results are mean±S.E.M. for four rats in each group, *p<0.01 versus vehicle. (C) NOSH-1 inhibits induction of cyclooxygenase (COX)-1 and COX-2 by carrageenan. Results show one animal in controls, four in carrageenan injected, and two in NOSH-1 treated at two different doses. Reproduced with permission from Kodela et al., (30), Copyright 2012 American Chemical Society.
The cell growth inhibitory properties of the four NOSH-compounds were evaluated in 11 different human cancer cell lines of six different histological subtypes. The cell lines were those of colon (HT-29: COX-1 and COX-2 positive, HCT 15: COX null, and SW480: COX-1 positive, low levels of endogenous COX-2), breast (MCF7: [ER(+)], MDA MB-231 and SKBR3: [ER(−)]); T-cell leukemia (Jurkat), pancreatic (BxPC3: both COX-1 and COX-2 positive, MIAPaCa-2: COX-null), prostate (LNCaP), and lung (A549). All four NOSH compounds were extremely effective in inhibiting the growth of these cell lines; however, NOSH-1 was the most potent, with IC50s for cell growth inhibition ranging from 48–280 nM at 24 h (30).
In a fold comparison of the IC50 values (ASA/NOSH 1–4), NOSH-1 was at least 100,000-fold more potent than ASA in HT-29 colon cancer cells. The increase in potency for NOSH-2, -3, and -4 in the same cell line after 24 h of treatment was >60,000-fold, >600-fold, and >16,000-fold, respectively (30). In HT-29 colon cancer cells, the enhanced potency of NOSH-1 increased to ∼125,000-fold and to ∼250,000-fold after 48 h and 72 h of treatment (11).
NOSH-ASA (NBS-1120) treatment of athymic nude bearing a human colon cancer xenograft caused an 85% reduction in tumor volume after 18 days of treatment (Fig. 13) (11).
FIG. 13.
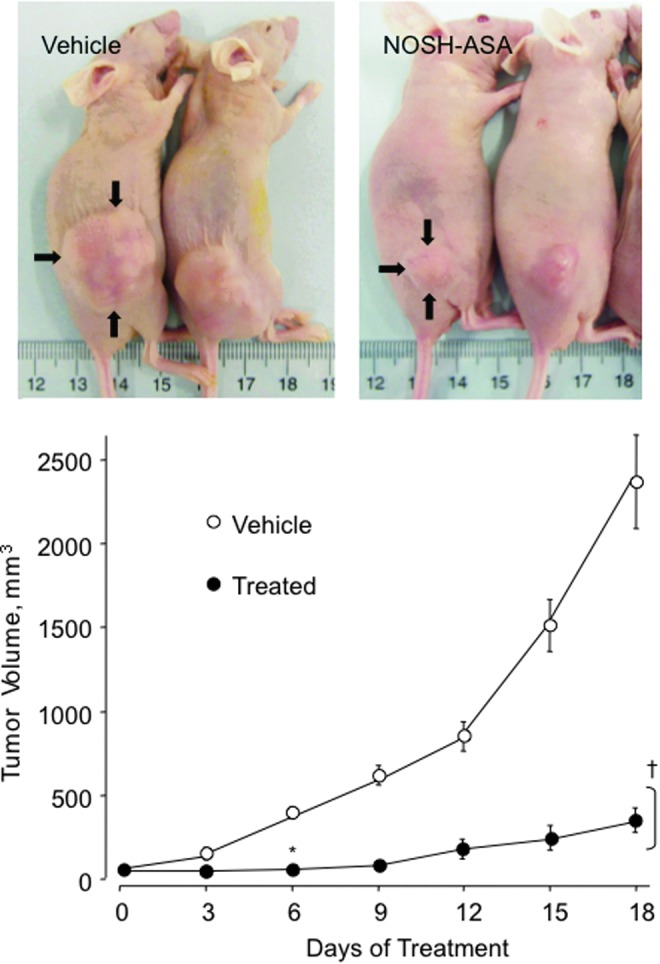
NOSH-ASA inhibits tumor xenograft growth. Athymic nude mice were injected subcutaneously with HT-29 cells for the development of subcutaneous tumors as described in Material and Methods. NOSH-ASA significantly reduced tumor volume 6 days after treatment to sacrifice, *p<0.05 at day 6, †p<0.01 days 9–18. Reproduced from Chattopadhyay et al., 2012 (10) with permission from Elsevier. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Conclusion
There appears to be no doubt in the utility of H2S in modulating an array of physiological and pathological conditions. H2S-donationg and NOSH-donating prodrugs are under development with great potential in influencing the treatment or prevention of a number of diseases. Similar to NO, H2S appears to be multifaceted, having opposing effects, which appear to be dose dependent. This is most evident in the context of inflammation, where both pro-inflammatory and anti-inflammatory effects have been reported [reviewed in (27)]. Various H2S-donationg prodrugs are actively being studied for the general treatment of conditions pertaining to arthritis, cardiovascular, urological, neuroinflammatory, and ophthalmological diseases, [reviewed in (54)]. H2S-donationg NSAIDs, apart from sparing the stomach from the serious side effects of the parent NSAID, also appear to have great potential as anti-cancer agents. The mechanisms and pathways affected by H2S-donationg NSAIDs in reducing NSAID-induced side effects, inflammation, and cancer are illustrated in Figure 14.
FIG. 14.

Mechanisms of action of H2S-releasing NSAIDs. When hydrolyzed, H2S-releasing NSAIDs produce the parent NSAID and the H2S-releasing moiety from which H2S is released. The NSAID component inhibits COX-1 and COX-2, resulting in compromised mucosal defense mechanisms, which may lead to ulcers. NSAIDs can reduce renal perfusion, which can lead to increases in blood pressure, leading to cardiovascular (CV) damage. The released H2S counteracts many of the detrimental effects of NSAIDs. These protective effects appear to be mediated through activation of KATP channels. H2S enhances the mucosal defense mechanisms; causes vasodilation, thus reducing BP and leading to cardioprotective effects. Both the NSAID and H2S have anti-inflammatory effects, the former through inhibition of COX and the latter through inhibition of nuclear transcription factor κB (NF-κB); collectively, these contribute to reduction in tumor mass. Modified and reproduced from Kashfi and Olson 2012 (27) with permission from Elsevier. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
An important issue that needs to be resolved is the question of how to accurately measure tissue H2S concentrations. Many methods reported in the literature are fraught with flaws; see (27) for details. To maximize the potential in developing long-lasting modalities for the treatment or prevention of any disease, we need to think about genetically modified animals which will allow us to delve into the mechanism(s) of cell signaling that involve H2S. With all that has been done in this field thus far, I believe that we are still at the dawn of an exciting new era.
Abbreviations Used
- AhR
aryl hydrocarbon receptor
- ALL
acute lymphoblastic leukemia
- ARE
antioxidant response element
- ASA
aspirin
- CBS
cystathionine β-synthase
- CO
carbon monoxide
- COX-1
cyclooxygenase-1
- COX-2
cyclooxygenase-2
- CSE
cystathionine γ-lyase
- CYP
cytochrome P450
- ER−
estrogen receptor negative
- ER+
estrogen receptor positive
- GST
glutathione S-transferase
- H2S
hydrogen sulfide
- HS-ASA
hydrogen sulfide-donating aspirin
- IκK
Ikappa kinases
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NO
nitric oxide
- NOSH
nitric oxide-hydrogen sulfide donating
- NQO
NAD(P)H:quinone oxireductase
- NSAIDs
nonsteroidal anti-inflammatory drugs
- PR-
progesterone receptor negative
- ROS
reactive oxygen species
- Trx/TrxR
thioredoxin/thioredoxin reductase-1
- UGT
UDP-glucuronyltransferase
Acknowledgment
This study was supported in part by National Institutes of Health grant R24 DA018055.
References
- 1.Arner ES. and Holmgren A. The thioredoxin system in cancer. Semin Cancer Biol 16: 420–426, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Bass SE, Sienkiewicz P, Macdonald CJ, Cheng RY, Sparatore A, Del Soldato P, Roberts DD, Moody TW, Wink DA, and Yeh GC. Novel dithiolethione-modified nonsteroidal anti-inflammatory drugs in human hepatoma HepG2 and colon LS180 cells. Clin Cancer Res 15: 1964–1972, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bellosillo B, Pique M, Barragan M, Castano E, Villamor N, Colomer D, Montserrat E, Pons G, and Gil J. Aspirin and salicylate induce apoptosis and activation of caspases in B-cell chronic lymphocytic leukemia cells. Blood 92: 1406–1414, 1998 [PubMed] [Google Scholar]
- 4.Bharti AC. and Aggarwal BB. Nuclear factor-kappa B and cancer: its role in prevention and therapy. Biochem Pharmacol 64: 883–888, 2002 [DOI] [PubMed] [Google Scholar]
- 5.Biswas DK, Cruz AP, Gansberger E, and Pardee AB. Epidermal growth factor-induced nuclear factor kappa B activation: a major pathway of cell-cycle progression in estrogen-receptor negative breast cancer cells. Proc Natl Acad Sci U S A 97: 8542–8547, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonizzi G. and Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol 25: 280–288, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Bukovska G, Kery V, and Kraus JP. Expression of human cystathionine beta-synthase in Escherichia coli: purification and characterization. Protein Expr Purif 5: 442–448, 1994 [DOI] [PubMed] [Google Scholar]
- 8.Chattopadhyay M, Kodela R, Nath N, Barsegian A, Boring D, and Kashfi K. Hydrogen sulfide-releasing aspirin suppresses NF-kappaB signaling in estrogen receptor negative breast cancer cells in vitro and in vivo. Biochem Pharmacol 83: 723–732, 2012 [DOI] [PubMed] [Google Scholar]
- 9.Chattopadhyay M, Kodela R, Nath N, Dastagirzada YM, Velazquez-Martinez CA, Boring D, and Kashfi K. Hydrogen sulfide-releasing NSAIDs inhibit the growth of human cancer cells: a general property and evidence of a tissue type-independent effect. Biochem Pharmacol 83: 715–722, 2012 [DOI] [PubMed] [Google Scholar]
- 10.Chattopadhyay M, Kodela R, Nath N, Street CR, Velazquez-Martinez CA, Boring D, and Kashfi K. Hydrogen sulfide-releasing aspirin modulates xenobiotic metabolizing enzymes in vitro and in vivo. Biochem Pharmacol 83: 733–740, 2012 [DOI] [PubMed] [Google Scholar]
- 11.Chattopadhyay M, Kodela R, Olson KR, and Kashfi K. NOSH-aspirin (NBS-1120), a novel nitric oxide- and hydrogen sulfide-releasing hybrid is a potent inhibitor of colon cancer cell growth in vitro and in a xenograft mouse model. Biochem Biophys Res Commun 419: 523–528, 2012 [DOI] [PubMed] [Google Scholar]
- 12.Chen KY. and Morris JC. Oxidation of sulfide by O2: catalysis and inhibition. J Sanit Eng Div 98: 215–227, 1972 [Google Scholar]
- 13.Chiang JM, Chou YH, Chen TC, Ng KF, and Lin JL. Nuclear beta-catenin expression is closely related to ulcerative growth of colorectal carcinoma.PG - 1124–9. Br J Cancer 86: 1124–1129, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung EJ, Hwang SG, Nguyen P, Lee S, Kim JS, Kim JW, Henkart PA, Bottaro DP, Soon L, Bonvini P, Lee SJ, Karp JE, Oh HJ, Rubin JS, and Trepel JB. Regulation of leukemic cell adhesion, proliferation, and survival by beta-catenin. Blood 100: 982–990, 2002 [DOI] [PubMed] [Google Scholar]
- 15.Circu ML. and Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med 48: 749–762, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dalle-Donne I, Giustarini D, Colombo R, Rossi R, and Milzani A. Protein carbonylation in human diseases. Trends Mol Med 9: 169–176, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Erickson PF, Maxwell IH, Su LJ, Baumann M, and Glode LM. Sequence of cDNA for rat cystathionine gamma-lyase and comparison of deduced amino acid sequence with related Escherichia coli enzymes. Biochem J 269: 335–340, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gan Z, Chattopadhyay M, Kodela R, Boring D, Crowell J, and Kashfi K. Hydrogen sulfide-releasing aspirin inhibits the growth of leukemic Jurkat cells and modulates β- catenin expression. American Association for Cancer Research, 102nd Annual Meeting, April2–6, Orlando FL2011 [Google Scholar]
- 19.Ghosh S. and Karin M. Missing pieces in the NF-kappaB puzzle. Cell 109Suppl: S81–S96, 2002 [DOI] [PubMed] [Google Scholar]
- 20.Grubben MJ, Nagengast FM, Katan MB, and Peters WH. The glutathione biotransformation system and colorectal cancer risk in humans. Scand J Gastroenterol Suppl 234: 68–76, 2001 [DOI] [PubMed] [Google Scholar]
- 21.Hanif R, Pittas A, Feng Y, Koutsos MI, Qiao L, Staiano-Coico L, Shiff SI, and Rigas B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem Pharmacol 52: 237–245, 1996 [DOI] [PubMed] [Google Scholar]
- 22.Hildebrandt TM. and Grieshaber MK. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J 275: 3352–3361, 2008 [DOI] [PubMed] [Google Scholar]
- 23.Hughes MN, Centelles MN, and Moore KP. Making and working with hydrogen sulfide: the chemistry and generation of hydrogen sulfide in vitro and its measurement in vivo: a review. Free Radic Biol Med 47: 1346–1353, 2009 [DOI] [PubMed] [Google Scholar]
- 24.Insko MA, Deckwerth TL, Hill P, Toombs CF, and Szabo C. Detection of exhaled hydrogen sulphide gas in rats exposed to intravenous sodium sulphide. Br J Pharmacol 157: 944–951, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karin M. NF-kappaB and cancer: mechanisms and targets. Mol Carcinog 45: 355–361, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Kaschula CH, Hunter R, Hassan HT, Stellenboom N, Cotton J, Zhai XQ, and Parker MI. Anti-proliferation activity of synthetic ajoene analogues on cancer cell-lines. Anticancer Agents Med Chem 11: 260–266, 2011 [DOI] [PubMed] [Google Scholar]
- 27.Kashfi K. Anti-inflammatory agents as cancer therapeutics. Adv Pharmacol 57: 31–89, 2009 [DOI] [PubMed] [Google Scholar]
- 28.Kashfi K. and Olson KR. Biology and therapeutic potential of hydrogen sulfide and hydrogen sulfide-releasing chimeras. Biochem Pharmacol 24: 019, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kashfi K, Ryan Y, Qiao LL, Williams JL, Chen J, Del Soldato P, Traganos F, and Rigas B. Nitric oxide-donating nonsteroidal anti-inflammatory drugs inhibit the growth of various cultured human cancer cells: evidence of a tissue type-independent effect. J Pharmacol Exp Ther 303: 1273–1282, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Kodela R, Chattopadhyay M, and Kashfi K. NOSH-aspirin: a novel nitric oxide- hydrogen sulfide-releasing hybrid: a new class of anti-inflammatory pharmaceuticals. ACS Med Chem Lett 3: 257–262, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kong AN, Owuor E, Yu R, Hebbar V, Chen C, Hu R, and Mandlekar S. Induction of xenobiotic enzymes by the MAP kinase pathway and the antioxidant or electrophile response element (ARE/EpRE). Drug Metab Rev 33: 255–271, 2001 [DOI] [PubMed] [Google Scholar]
- 32.Kune G, Kune S, and Watson L. Colorectal cancer risk, chronic illnesses, operations, and medications: case-control results from the Melbourne Colorectal Cancer Study. Cancer Res 48: 4399–4404, 1988 [PubMed] [Google Scholar]
- 33.Lee JS. and Surh YJ. Nrf2 as a novel molecular target for chemoprevention. Cancer Lett 224: 171–184, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Lee M, Sparatore A, Del Soldato P, McGeer E, and McGeer PL. Hydrogen sulfide- releasing NSAIDs attenuate neuroinflammation induced by microglial and astrocytic activation. Glia 58: 103–113, 2010 [DOI] [PubMed] [Google Scholar]
- 35.Lee M, Tazzari V, Giustarini D, Rossi R, Sparatore A, Del Soldato P, McGeer E, and McGeer PL. Effects of hydrogen sulfide-releasing L-DOPA derivatives on glial activation: potential for treating Parkinson disease. J Biol Chem 285: 17318–17328, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee ZW, Zhou J, Chen CS, Zhao Y, Tan CH, Li L, Moore PK, and Deng LW. The slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel anti-cancer effects in vitro and in vivo. PLoS One 6: e21077, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li L, Rossoni G, Sparatore A, Lee LC, Del Soldato P, and Moore PK. Anti-inflammatory and gastrointestinal effects of a novel diclofenac derivative. Free Radic Biol Med 42: 706–719, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Li L, Salto-Tellez M, Tan CH, Whiteman M, and Moore PK. GYY4137, a novel hydrogen sulfide-releasing molecule, protects against endotoxic shock in the rat. Free Radic Biol Med 47: 103–113, 2009 [DOI] [PubMed] [Google Scholar]
- 39.Li L, Whiteman M, Guan YY, Neo KL, Cheng Y, Lee SW, Zhao Y, Baskar R, Tan CH, and Moore PK. Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): new insights into the biology of hydrogen sulfide. Circulation 117: 2351–2360, 2008 [DOI] [PubMed] [Google Scholar]
- 40.Lincoln DT, Ali Emadi EM, Tonissen KF, and Clarke FM. The thioredoxin-thioredoxin reductase system: over-expression in human cancer. Anticancer Res 23: 2425–2433, 2003 [PubMed] [Google Scholar]
- 41.Liu L, Cui J, Song CJ, Bian JS, Sparatore A, Soldato PD, Wang XY, and Yan CD. H(2)S-releasing aspirin protects against aspirin-induced gastric injury via reducing oxidative stress. PLoS One 7: e46301, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu D, Zhao Y, Tawatao R, Cottam HB, Sen M, Leoni LM, Kipps TJ, Corr M, and Carson DA. Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 101: 3118–3123, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matthews JR, Wakasugi N, Virelizier JL, Yodoi J, and Hay RT. Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res 20: 3821–3830, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moon RT, Bowerman B, Boutros M, and Perrimon N. The promise and perils of Wnt signaling through beta-catenin. Science 296: 1644–1646, 2002 [DOI] [PubMed] [Google Scholar]
- 45.Mustacich D. and Powis G. Thioredoxin reductase. Biochem J 346Pt 1: 1–8, 2000 [PMC free article] [PubMed] [Google Scholar]
- 46.Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ, Jr., and Sledge GW., Jr.Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Mol Cell Biol 17: 3629–3639, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakshatri H. and Goulet RJ., Jr.NF-kappaB and breast cancer. Curr Probl Cancer 26: 282–309, 2002 [DOI] [PubMed] [Google Scholar]
- 48.Nath N, Labaze G, Rigas B, and Kashfi K. NO-donating aspirin inhibits the growth of leukemic Jurkat cells and modulates beta-catenin expression. Biochem Biophys Res Commun 326: 93–99, 2005 [DOI] [PubMed] [Google Scholar]
- 49.Nelson WJ. and Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303: 1483–1487, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patel NM, Nozaki S, Shortle NH, Bhat-Nakshatri P, Newton TR, Rice S, Gelfanov V, Boswell SH, Goulet RJ, Jr., Sledge GW, Jr., and Nakshatri H. Paclitaxel sensitivity of breast cancer cells with constitutively active NF-kappaB is enhanced by IkappaBalpha super-repressor and parthenolide. Oncogene 19: 4159–4169, 2000 [DOI] [PubMed] [Google Scholar]
- 51.Perrino E, Uliva C, Lanzi C, Soldato PD, Masini E, and Sparatore A. New prostaglandin derivative for glaucoma treatment. Bioorg Med Chem Lett 19: 1639–1642, 2009 [DOI] [PubMed] [Google Scholar]
- 52.Romieu-Mourez R, Landesman-Bollag E, Seldin DC, Traish AM, Mercurio F, and Sonenshein GE. Roles of IKK kinases and protein kinase CK2 in activation of nuclear factor-kappaB in breast cancer. Cancer Res 61: 3810–3818, 2001 [PubMed] [Google Scholar]
- 53.Rothwell PM, Price JF, Fowkes FG, Zanchetti A, Roncaglioni MC, Tognoni G, Lee R, Belch JF, Wilson M, Mehta Z, and Meade TW. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 379: 1602–1612, 2012 [DOI] [PubMed] [Google Scholar]
- 54.Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, and Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet 379: 1591–1601, 2012 [DOI] [PubMed] [Google Scholar]
- 55.Searcy DG. and Lee SH. Sulfur reduction by human erythrocytes. J Exp Zool 282: 310–322, 1998 [DOI] [PubMed] [Google Scholar]
- 56.Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, and Kimura H. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid Redox Signal 11: 703–714, 2009 [DOI] [PubMed] [Google Scholar]
- 57.Shukla N, Rossoni G, Hotston M, Sparatore A, Del Soldato P, Tazzari V, Persad R, Angelini GD, and Jeremy JY. Effect of hydrogen sulphide-donating sildenafil (ACS6) on erectile function and oxidative stress in rabbit isolated corpus cavernosum and in hypertensive rats. BJU Int 103: 1522–1529, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Slocum SL. and Kensler TW. Nrf2: control of sensitivity to carcinogens. Arch Toxicol 85: 273–284, 2011 [DOI] [PubMed] [Google Scholar]
- 59.Sparatore A, Santus G, Giustarini D, Rossi R, and Del Soldato P. Therapeutic potential of new hydrogen sulfide-releasing hybrids. Expert Rev Clin Pharmacol 4: 109–121, 2011 [DOI] [PubMed] [Google Scholar]
- 60.Switzer CH, Cheng RY, Ridnour LA, Murray MC, Tazzari V, Sparatore A, Del Soldato P, Hines HB, Glynn SA, Ambs S, and Wink DA. Dithiolethiones inhibit NF-kappaB activity via covalent modification in human estrogen receptor-negative breast cancer. Cancer Res 72: 2394–2404, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wallace JL. Hydrogen sulfide-releasing anti-inflammatory drugs. Trends Pharmacol Sci 28: 501–505, 2007 [DOI] [PubMed] [Google Scholar]
- 62.Wallace JL. Prostaglandins, NSAIDs, and gastric mucosal protection: why doesn't the stomach digest itself? Physiol Rev 88: 1547–1565, 2008 [DOI] [PubMed] [Google Scholar]
- 63.Wallace JL, Caliendo G, Santagada V, and Cirino G. Markedly reduced toxicity of a hydrogen sulphide-releasing derivative of naproxen (ATB-346). Br J Pharmacol 159: 1236–1246, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wallace JL, Caliendo G, Santagada V, Cirino G, and Fiorucci S. Gastrointestinal safety and anti-inflammatory effects of a hydrogen sulfide-releasing diclofenac derivative in the rat. Gastroenterology 132: 261–271, 2007 [DOI] [PubMed] [Google Scholar]
- 65.Wallace JL, Dicay M, McKnight W, and Martin GR. Hydrogen sulfide enhances ulcer healing in rats. FASEB J 21: 4070–4076, 2007 [DOI] [PubMed] [Google Scholar]
- 66.Weldon CB, Burow ME, Rolfe KW, Clayton JL, Jaffe BM, and Beckman BS. NF-kappa B-mediated chemoresistance in breast cancer cells. Surgery 130: 143–150, 2001 [DOI] [PubMed] [Google Scholar]
- 67.Westley AM. and Westley J. Biological sulfane sulfur. Anal Biochem 195: 63–67, 1991 [DOI] [PubMed] [Google Scholar]
- 68.Whiteman M, Li L, Rose P, Tan CH, Parkinson DB, and Moore PK. The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid Redox Signal 12: 1147–1154, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wilkinson Jt. and Clapper ML. Detoxication enzymes and chemoprevention. Proc Soc Exp Biol Med 216: 192–200, 1997 [DOI] [PubMed] [Google Scholar]
- 70.Wolfe MM, Lichtenstein DR, and Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N Engl J Med 340: 1888–1899, 1999 [DOI] [PubMed] [Google Scholar]
- 71.Zhang H, Zhi L, Moore PK, and Bhatia M. Role of hydrogen sulfide in cecal ligation and puncture induced sepsis in the mouse. Am J Physiol Lung Cell Mol Physiol 290: L1193–L1201, 2006 [DOI] [PubMed] [Google Scholar]