

Abstract
To examine the role of TRF2 in epithelial tumorigenesis, we characterized conditional loss of TRF2 expression in the basal layer of mouse epidermis. These mice exhibit some characteristics of dyskeratosis congenita, a human stem cell depletion syndrome caused by telomere dysfunction. The epidermis in conditional TRF2 null mice exhibited DNA damage response and apoptosis which correlated with stem cell depletion. The stem cell population in conditional TRF2 null epidermis exhibited shorter telomeres than those in control mice. Squamous cell carcinomas induced in conditional TRF2 null mice developed with increased latency and slower growth due to reduced numbers of proliferating cells as the result of increased apoptosis. TRF2 null epidermal stem cells were found in both primary and metastatic tumors. Despite the low grade phenotype of the conditional TRF2 null primary tumors, the number of metastatic lesions was similar to control cancers. Basal cells from TRF2 null tumors demonstrated extreme telomere shortening and dramatically increased numbers of telomeric signals by fluorescence in situ hybridization due to increased genomic instability and aneuploidy in these cancers. DNA damage response signals were detected at telomeres in TRF2 null tumor cells from these mice. The increased genomic instability in these tumors correlated with 8 fold expansion of the transformed stem cell population compared to that in control cancers. We concluded that genomic instability resulting from loss of TRF2 expression provides biological advantages to the cancer stem cell population.
Keywords: telomere, DNA damage, apoptosis, genomic instability
INTRODUCTION
The telomeric shelterin complex contains the double stranded DNA binding proteins TRF2 and TRF1 (1). Dominant negative TRF2 induced end to end chromosomal fusions of telomeric DNA (2). These fusions have lost the single strand G tail, and cells expressing the dominant negative TRF2 protein undergo senescence or apoptosis. Programmed cell death was mediated by ATM kinase and p53 consistent with DNA damage checkpoint activation (3–5). Nonhomologous end joining of telomeres was dependent on DNA ligase IV (6). Telomeres lacking from cells lacking TRF2, p53, and DNA ligase IV did not undergo degradation but were recognized as DNA damage sites. Nonhomologous end joining in TRF2 deficient cells was dependent on 53BP1 which binds to damaged telomeres (7). The MRE11-RAD50-NBS1 (MRN) complex interacts with ATM to detect double strand breaks. MRN deficient cells do not activate ATM when TRF2 is removed from telomeres (8). Chromosome end to end fusions are reduced in these cells with persistence of the telomeric overhangs. Formation of DNA damage foci and telomere fusions was reduced in cells doubly deficient for NBS1 and TRF2 (9,10). Dominant negative TRF2 also induced catastrophic deletions of telomeric DNA dependent on XRCC3 and created t loop sized telomeric circles (11). TRF2 mediated end capping that occurred after telomere replication was limited to those produced by leading strand DNA synthesis (12). TRF2 overexpression accelerates the rate of telomere shortening without increasing senescence (13,14). These studies demonstrate the importance of TRF2 in regulating DNA damage response at telomeres.
Alterations in telomere length regulation have profound effects on stem cell populations in many tissues (15,16). Stem cells in many tissues have longer telomeres than the proliferating populations found in these anatomic locations (17). In mammalian epidermis, an important stem cell population resides in the adult hair follicle bulge (18,19). These slowly cycling CD34+/K15+ cells respond to external stimuli such as chemical carcinogens and wounding by increased cell division and migration, and are capable of regenerating components of the epidermis (20, 21). Telomere shortening due to loss of telomerase activity inhibited proliferation and mobilization of stem cells and impaired hair growth (22), while telomerase overexpression caused rapid transition to anagen phase with robust hair growth (23,24). TRF2 overexpression in epidermis (K5-TRF2 mice) results in short telomeres in the presence of telomerase activity leading to premature aging and increased cancer (25). Loss of telomerase activity in K5-TRF2 mice accelerates epithelial carcinogenesis by increased DNA damage and chromosomal instability (26). Epidermal skin cell dysfunction in these mice is rescued by p53 deletion but skin carcinogenesis is accelerated due to diminished p21 induction (27).
TRF2 expression is dysregulated in many types of cancer (28,29). Human cancer studies have suggested that increased TRF2 expression during multistage lung carcinogenesis stabilizes telomeres in advanced tumors (30). To provide a new perspective on the role of TRF2 in epithelial tumorigenesis, we characterized conditional loss of TRF2 expression in the basal layer of mouse epidermis. These mice exhibit some characteristics of dyskeratosis congenita, a rare human stem cell depletion syndrome caused by telomere dysfunction. However, these stem cell depleted mice exhibit significant stem cell expansion and metastasis during carcinogenesis due to severe telomere dysfunction, genomic instability, and aneuploidy.
RESULTS
To determine how loss of TRF2 expression affects epidermal development, homeostasis, and tumorigenesis we created TRF2 conditional null mutant mice in epidermis by crossing K14-Cre with TRF2f/f mice. These mice lack TRF2 expression in stratified epithelia such as epidermis. TRF2 genotyping and mRNA expression in sorted epidermal cells from these mice are shown in Fig. 1D,E. K14-Cre;TRF2f/f mice were born at the expected mendelian frequencies and were grossly normal with respect to appearance, weight, and behavior. By 8 weeks of age, some K14-Cre;TRF2f/f mice developed multiple distinct bends in the tail resembling the crinkled mouse phenotype which exhibits stem cell depletion (32; Fig. 1A,B). Older K14-Cre;TRF2f/f mice developed nail dystrophy which has been described in mouse models of dyskeratosis congenita exhibiting stem cell depletion resulting from short telomeres (33,34; Fig. 1C). These phenotypes were not observed in K14-Cre;TRF2+/+ mice. To determine if this phenotype correlated with apoptosis and telomeric DNA damage response in epidermal stem cells, we examined programmed cell death and localization of 53BP1 protein at telomeres in sorted stem cells. Apoptosis was increased in sorted TRF2 deficient stem cells compared to those expressing the gene product (0.03% vs. 4.5%; p<0.005; Fig. 2A,B and 2D,E). TRF2 deficient stem cells exhibited robust induction of 53BP1 DNA damage foci which localized to telomeres (Fig. 2C,F). These results indicate that TRF2 deficiency induces telomeric DNA damage response and apoptosis of epidermal stem cells resulting in stem cell depletion phenotypes in conditional null mutant mice.
Fig. 1.

TRF2 null mice exhibit some stem cell depletion phenotypes consistent with mouse models of dyskeratosis congenita (DKC). (A) Mouse tail from K14-Cre;TRF2+/+ mouse. (B) Crinkled tail from K14-Cre;TRF2f/f mouse. (C) Nail dystrophy in K14-Cre;TRF2f/f mouse. (D) Genotyping of sorted CD34+/K15+ and CD34−/K15− cells from epidermis of K14-Cre;TRF2f/f and K14-Cre;TRF2+/+ mice was performed by PCR using The Jackson Laboratory protocol (upper panel). TRF2 null, TRF2 wild type (wt), and Cre PCR products are shown. TRF2 mRNA expression in sorted CD34+/K15+ and CD34−/K15− cells from epidermis of K14-Cre;TRF2f/f and K14-Cre;TRF2+/+ mice by was performed by RT-PCR (lower panel). β-actin expression is shown as the internal control. Representative gels are shown. (E) Quantitative PCR of genotyping and TRF2 expression shown in (D). Error bars represent SEM.
Fig. 2.

TRF2 deficiency results in telomeric DNA damage response and apoptosis of CD34+/K15+ stem cells. TUNEL analysis of sorted CD34+/K15+ cells from K14-Cre;TRF2+/+ epidermis. DAPI (A) and FITC (B) fluorescence is shown. (C) Combined 53BP1 immunofluorescence (FITC) and telomere FISH (Cy3) in sorted CD34+/K15+ cells from K14-Cre;TRF2+/+ epidermis. Cells were counterstained with DAPI. TUNEL analysis of sorted CD34+/K15+ cells from K14-Cre;TRF2f/f epidermis. DAPI (D) and FITC (E) fluorescence is shown. (F) Combined 53BP1 immunofluorescence (FITC) and telomere FISH (Cy3) in sorted CD34+/K15+ cells from K14-Cre;TRF2f/f epidermis. Arrows indicate 53BP1 localization at telomeres (yellow foci). Cells were counterstained with DAPI.
We examined DNA damage response activation in K14-Cre;TRF2f/f skin by western blot. As shown in Fig. 3A, activated ATM (phospho-ATM) expression was induced by up to 10 fold in skin from K14-Cre;TRF2f/f mice. Expression of phospho-Chk2 expression was increased in the skin of K14-Cre;TRF2f/f mice by up to 20 fold. p53 expression was strongly induced in the skin of K14-Cre;TRF2f/f mice (10 fold). These results indicate that loss of TRF2 expression induces a robust DNA damage response in mouse epidermis.
Fig. 3.

TRF2 deficiency in basal layer of mouse epidermis induces DNA damage response, apoptosis, and stem cell depletion. (A) Western blot analysis demonstrating DNA damage response in K14-Cre;TRF2+/+ (wt) and K14-Cre;TRF2f/f (TRF) mice. Blots were incubated with antibodies indicated at left using independent protein samples. Epidermis from K14-Cre;TRF2+/+ (B) and K14-Cre;TRF2f/f mice (G). Stem cells (arrow), hair follicles (f), and sebaceous glands (s) are shown. Scale bar = 10 μm. TRF2 expression in skin from K14-Cre;TRF2+/+ (C) and K14-Cre;TRF2f/f mice (H) is shown by immunohistochemistry. Apoptosis in epidermis as determined by TUNEL analysis in K14-Cre;TRF2+/+ (D, DAPI; E, FITC) and K14-Cre;TRF2f/f (I, DAPI; J, FITC) mice. Cell cycle analysis of dissociated epidermal keratinocytes in K14-Cre;TRF2+/+ (F) and K14-Cre;TRF2f/f (K) mice. FACS of dissociated mouse epidermal keratinocytes incubated with control IgG (L), epidermal keratinocytes from K14-Cre;TRF2+/+ (M) or K14-Cre;TRF2f/f (N) mice incubated with phycoerythrin conjugated CD34 antibody. Hematocrit (Hct; O) and white blood cell (WBC) counts (P) in K14-Cre;TRF2+/+ and K14-Cre;TRF2f/f mice.
Histopathologic analysis by hematoxylin and eosin staining of skin in K14-Cre;TRF2f/f mice up to 18 months old revealed no differences in interfollicular epidermis compared to K14-Cre;TRF2+/+ animals (Fig. 3B,G). However follicular bulge cells in serial sections were decreased by 50% (p<0.001) in K14-Cre;TRF2f/f skin, and demonstrated nuclear condensation characteristic of early stages of apoptosis. Hair follicle diameter was decreased by 23% due to decreased numbers of root sheath epithelial cells in K14-Cre;TRF2f/f skin (p<0.03). We examined TRF2 protein expression in skin from K14-Cre;TRF2+/+ and K14-Cre;TRF2f/f mice (Fig. 3C,H). TRF2 protein expression was not detected in skin from K14-Cre;TRF2f/f mice but was readily detected in epidermis and hair follicles from K14-Cre;TRF2+/+ animals. To determine if the robust DNA damage response in epidermis from K14-Cre;TRF2f/f correlated with increased apoptosis in this cells, we examined programmed cell death by TUNEL analysis. K14-Cre;TRF2f/f mice (Fig. 3I,J) showed a significant increase in apoptotic cells in the proliferating basal layer of epidermis compared to K14-Cre;TRF2+/+ (Fig. 3D,E) animals (0.01% vs. 3.5%; p<0.0001). Cell cycle analysis of epidermal keratinocytes from K14-Cre;TRF2f/f mice showed a significant increase in the G2/M phase fraction compared to cells from K14-Cre;TRF2+/+ animals (15% vs. 28%; p<0.05; Fig. 3F,K). These results indicate that epidermal stem and basal layer cells from K14-Cre;TRF2f/f mice exhibit elevated DNA damage response and apoptosis. However increased proliferation of epidermal keratinocytes in these mice compensates for programmed cell death.
To determine if increased apoptosis in epidermal cells of K14-Cre;TRF2f/f mice resulted in stem cell depletion, we sorted the stem cell population from dissociated epidermal keratinocytes using the well characterized CD34 and K15 markers (35). Flow cytometric sorting of epidermal keratinocytes using control IgG is shown in Fig. 3L. Stem cells comprised 1.9% of dissociated epidermal keratinocytes isolated from K14-Cre;TRF2+/+ mice (Fig. 3M). However, stem cells comprised only 0.8% of epidermal keratinocytes isolated from K14-Cre;TRF2f/f mice (p<0.002; Fig. 3N). No differences in hematocrit or white blood cell count were detected in K14-Cre;TRF2f/f compared to K14-Cre;TRF2+/+ mice (Fig. 3 O,P), indicating normal function of the hematopoietic stem cell compartment in conditional TRF2 null animals. These results indicated the epidermal stem cell compartment was depleted in K14-Cre;TRF2f/f mice.
To determine the effects of conditional TRF2 deletion on epidermal carcinogenesis, we treated K14-Cre;TRF2f/f and K14-Cre;TRF2+/+ mice with DMBA using a protocol which consistently results in squamous cell carcinoma with metastasis to regional lymph nodes (36). As shown in Fig. 4A, tumor latency was significantly increased in K14-Cre;TRF2f/f mice. Tumor latency was 22 weeks in K14-Cre;TRF2f/f mice compared to 18 weeks in K14-Cre;TRF2+/+ animals (p<0.03). Tumor growth was significantly inhibited in K14-Cre;TRF2f/f mice compared to K14-Cre;TRF2+/+ animals (tumor volume 277 mm3 vs. 80 mm3; p<0.01; Fig. 4B). There were no significant differences in number of tumors between K14-Cre;TRF2+/+ and K14-Cre;TRF2f/f mice (13 vs. 14 tumors respectively). Tumor histopathology was similar in both K14-Cre;TRF2+/+ and K14-Cre;TRF2f/f mice, representing squamous cell carcinoma (Fig. 4C,D). TRF2 protein expression was detected in tumor cells from K14-Cre;TRF2+/+ mice by immunohistochemistry (Fig. 4E), but not in tumors from K14-Cre;TRF2f/f animals (Fig. 4F). Metastatic tumor cells were detected in regional lymph nodes in both K14-Cre;TRF2+/+ and K14-Cre;TRF2f/f mice (Fig. 4G,H). We performed immunofluorescent examination of primary tumors and lymph nodes using anti-CD34 and K15 antibodies (37,38). Epidermal stem cells were detected both in primary and metastatic tumors from mice of both genotypes (Fig. 4I,J). TRF2 expression also was decreased in human SCC lines (Fig. 4K). These results indicate that epidermal stem cells are present in primary and metastatic tumors from K14-Cre;TRF2f/f mice and that these tumors are characterized by increased latency and slow growth.
Fig. 4.

Increased tumor latency and reduced proliferation in chemically induced squamous cell carcinomas from K14-Cre;TRF2f/f mice. (A) Percent of K14-Cre;TRF2+/+ and K14-Cre;TRF2f/f mice with tumors is shown. (B) Tumor volume after ten weeks tumor induction in K14-Cre;TRF2+/+ and K14-Cre;TRF2f/f mice. Error bars represent SEM. H&E stained sections of primary SCC in K14-Cre;TRF2+/+ (C) and K14-Cre;TRF2f/f (D) mice. Immunohistochemical analysis of TRF2 protein expression in SCC from K14-Cre;TRF2+/+ (E) and K14-Cre;TRF2f/f mice (F). H&E stained sections of metastatic tumors from K14Cre;TRF2+/+ (G) and K14-Cre;TRF2f/f (H) mice. CD34 and K15 immunofluorescent localization of keratinocyte stem cells in primary (I) and metastatic (J) SCC. Scale bar = 5 μm. (K) TRF2 expression in normal human epidermal keratinocytes (NHEK) and SCC lines is shown by qRT-PCR. These experiments were performed three times with independent samples. Error bars indicate SEM.
To determine if slow growth of squamous cell carcinomas in K14-Cre;TRF2f/f mice was due to decreased tumor cell proliferation, we performed anti-PCNA immunohistochemistry on tumors from animals of both genotypes. Representative sections showing PCNA expression in tumors from K14-Cre;TRF2+/+ and K14-Cre;TRF2f/f mice are shown in Fig. 5A,B. The number of PCNA positive cells was reduced by 80% in K14-Cre;TRF2f/f tumors (p<0.002; Fig. 5C). To determine if the decrease in proliferative cells in K14-Cre;TRF2f/f tumors was due to increased apoptosis, we performed TUNEL analysis on tumor tissue of both genotypes. Representative sections of TUNEL positive cells is shown in Fig. 6A,B (K14-Cre;TRF2+/+) and Fig. 6C,D (K14-Cre;TRF2f/f). TUNEL positive cells were increased by 14 fold in tumors from K14-Cre;TRF2f/f mice (p<0.001; Fig. 6E). TUNEL positive cells were confined to the proliferative basal layer cells in K14-Cre;TRF2f/f tumors as shown in Fig. 6C,D. To determine if increased programmed cell death correlated with DNA damage response in K14-Cre;TRF2f/f tumors, we performed anti-p53 and 53BP1 immunofluorescent analysis. Representative sections of p53 positive cells are shown in K14-Cre;TRF2+/+ (Fig. 7A) and K14-Cre;TRF2f/f tumors (Fig. 7B). The number of p53 positive cells was increased by 6 fold in K14-Cre;TRF2f/f tumors (p<0.04; Fig. 7C). Representative sections of 53BP1 positive cells are shown in Fig. 7D, E. The number of 53BP1 positive cells was increased 25 fold in K14-Cre;TRF2f/f tumors (p<0.001; Fig. 7F). Nude mouse subcutaneous injection of 104 CD34+ tumor cells from K14-Cre;TRF2+/+ mice consistently formed tumors within 6 months. However, injection of 105 CD34+ tumor cells from K14-Cre;TRF2f/f mice failed to form tumors by 12 months (data not shown). We compared in vitro colony forming ability of isolated cells from K14-Cre;TRF2+/+ and K14-Cre;TRF2f/f epidermis and primary SCC. The percentage of colony forming cells in K14-Cre;TRF2+/+ epidermis, K14-Cre;TRF2+/+ SCC, and K14-Cre;TRF2f/f SCC was 0.1, 1.7, and 0.3% respectively. We were unable to establish K14-Cre;TRF2f/f epidermal cells in culture. These results indicate that K14-Cre;TRF2f/f tumors are characterized by increased DNA damage response, elevated apoptosis, decreased proliferating cells, and impaired colony and tumor formation.
Fig. 5.
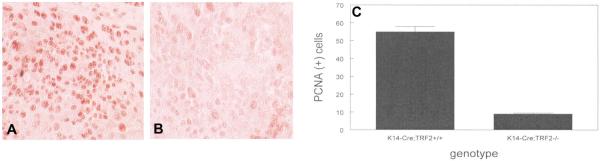
Decreased proliferating cells in SCC from K14-Cre;TRF2f/f mice. Immunohistochemical analysis of PCNA positive cells from SCC in K14Cre;TRF2+/+ (A) and K14-Cre;TRF2f/f (B) mice. Scale bar = 50 μm. (C) Quantification of PCNA positive cells. Error bars indicate SEM.
Fig. 6.
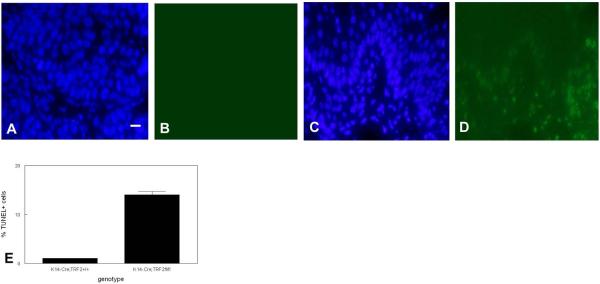
Increased apoptotic cells in basal cells of SCC from K14-Cre;TRF2f/f mice. TUNEL localization of apoptotic cells in SCC from K14-Cre;TRF2+/+ (A, DAPI; B, FITC) and K14-Cre;TRF2f/f (C, DAPI; D, FITC). Scale bar = 10 μm. (E) Quantification of TUNEL positive cells. Error bars indicate SEM.
Fig. 7.

Increased DNA damage signaling in SCC from K14-Cre;TRF2f/f mice. Localization of p53 positive cells in SCC from K14-Cre;TRF2+/+ (A) and K14-Cre;TRF2f/f mice (B). Scale bar = 10 μm. (C) Quantification of p53 cells. Localization of 53BP1 positive cells in SCC from K14-Cre;TRF2+/+ and K14-Cre;TRF2f/f mice (E). Quantification of 53BP1 positive cells (F). bars indicate SEM.
To determine if the DNA damage response in K14-Cre;TRF2f/f epidermis and squamous cell carcinomas correlated with telomere dysfunction, we examined telomere length in CD34 positive and negative cells from epidermis and squamous cell carcinomas in K14-Cre;TRF2+/+ and K14-Cre;TRF2f/f mice. Average telomere length ratios (ATLR) were determined using extracted DNA from sorted cell populations using an established quantitative PCR method for limiting amounts of genomic DNA (31). As shown in Fig. 8A, in K14-Cre;TRF2+/+ epidermis ATLR was significantly decreased in CD34 negative proliferative basal cells compared to the CD34 positive stem cell population (1.7 vs. 2.2; p<0.04) consistent with previous reports (17). ATLR in CD34 positive stem cells from K14-Cre;TRF2+/+ tumors was significantly decreased compared to the same population in K14-Cre;TRF2+/+ epidermis (0.5 vs. 2.2; p<0.0001). ATLR was significantly decreased in CD34 negative proliferative basal cells from tumors compared to CD34 negative cells in K14-Cre;TRF2+/+ epidermis (0.4 vs. 1.7; p<0.001). In K14-Cre;TRF2f/f epidermis, ATLR was significantly decreased in CD34 negative basal cells compared to CD34 positive stem cells (0.3 vs. 1.5; p<0.0002). In K14-Cre;TRF2f/f tumors, ATLR was significantly decreased in CD34 negative tumor cells compared to the CD34 positive population (0.2 vs. 0.5; p<0.01). ATLR was significantly decreased in CD34 positive cells from K14-Cre;TRF2f/f tumors compared to the same cell population from epidermis (0.4 vs. 1.5; p<0.003). ATLR in CD34 positive cells from K14-Cre;TRF2f/f epidermis was significantly decreased compared to K14-Cre;TRF2+/+ skin (1.5 vs. 2.2; p<0.04). ATLR in the CD34 negative proliferating basal cells was significantly decreased in K14-Cre;TRF2f/f epidermis compared to K14-Cre;TRF2+/+ skin (0.3 vs. 1.7; p<0.001). These results indicate telomeres in both epidermal stem and non-stem populations are significantly shorter in TRF2 null cells.
Fig. 8.

Telomere dysfunction in stem cells from epidermis leads to aneuploidy, cancer stem cell expansion, and metastasis in SCC from K14-Cre;TRF2f/f mice. (A) CD34+/K15+ stem cells were sorted from K14-Cre;TRF2+/+ and K14-Cre;TRF2f/f epidermis and SCC. Average telomere length ratios were determined in CD34+/K15+ and CD34−/K15− cells from epidermis and SCC in K14-Cre;TRF2+/+ and K14-Cre;TRF2−/− mice. Error bars indicate SEM. The number of telomere ends in tumor sections from K14-Cre;TRF2+/+ (B) and K14-Cre;TRF2f/f (C) mice was determined by FISH. Representative sections are shown with DAPI counterstain. Scale bar = 10 μm. Telomere signals (Cy3) and 53BP1 foci (FITC) in tumor cells from K14-Cre;TRF2+/+ (D) and K14-Cre;TRF2f/f (E) mice are shown with DAPI counterstain. Colocalized telomere and 53BP1 signals (yellow) in K14-Cre;TRF2f/f cells are shown at arrowheads. Scale bar = 2 μm. Telomere signals (Cy3) are shown by FISH in DAPI stained metaphase chromosomes from K14-Cre;TRF2+/+ (F) and K14-Cre;TRF2f/f (G) tumor cells. Chromosomal fusions are shown by arrowheads in K14-Cre;TRF2f/f metaphase spreads. Scale bar = 5 μm. (H) Percentage of histopathologically confirmed metastatic lymph nodes in K14-Cre;TRF2+/+ and K14-Cre;TRF2f/f mice. Error bars indicate SEM. FACS of dissociated SCC cells from K14-Cre;TRF2+/+ (I) or K14-Cre;TRF2f/f (J) mice incubated with phycoerythrin conjugated CD34 antibody. (K) Relative CXCR3 expression in K14-Cre;TRF2+/+ and K14-Cre;TRF2f/f SCC is shown by qRT-PCR. Error bars indicate SEM.
We also characterized the number of telomeric signals observed by fluorescence in situ hybridization in K14-Cre;TRF2+/+ and K14-Cre;TRF2f/f tumors. The number of telomeric FISH signals was significantly increased (10 fold; p<0.004) in tumors from K14-Cre;TRF2f/f mice (Fig. 8C) compared to tumors from K14-Cre;TRF2+/+ mice (Fig. 8B). Cultured tumor cells from K14-Cre;TRF2f/f mice showed increased telomere localization of 53BP1 foci compared to tumor cells from K14-Cre;TRF2+/+ animals by combined immunofluorescence/in situ hybridization (Fig. 8D,E; p<0.02). Metaphase spreads from wild type and K14-Cre;TRF2f/f tumor cells (Fig. 8F,G) showed significantly increased signal free ends (2.5% vs. 18.8% respectively; p<0.01), aneuploidy (modal chromosome number 51 vs. 98 respectively; p<0.002), and chromosomal fusions (1.0 vs. 4.6 per metaphase respectively; p<0.04). These results indicate that telomere shortening in the absence of TRF2 expression is associated with DNA damage response, genomic instability, and aneuploidy in K14-Cre;TRF2f/f tumors.
Despite the increased tumor latency and slow growth of K14-Cre;TRF2f/f tumors, we observed similar numbers of metastatic lymph nodes in these mice (48/72 nodes; 67%) compared to tumor bearing K14-Cre;TRF2+/+ animals (50/72 nodes; 69%; Fig. 8H). To determine if the unexpected increase in metastatic tumors in K14-Cre;TRF2f/f mice could result from expansion of the transformed stem cell population, we sorted these cells from K14-Cre;TRF2+/+ and K14-Cre;TRF2f/f tumors. K14-Cre;TRF2f/f tumors showed greater than 10 fold expansion of the CD34+/K15+ population compared to control tumors (0.8% vs. 8.2%; p<0.003; Fig. 8I,J). Expression of the metastasis gene CXCR3 was 7.7 fold higher in K14-Cre;TRF2f/f SCCs (Fig. 8K). These results indicate that the transformed stem cell population is significantly expanded in K14-Cre;TRF2f/f tumors.
DISCUSSION
Conditional deletion of TRF2 expression in stratified epithelium induced a dramatic DNA damage response in the epidermis characterized by ATM and Chk2 activation, and induction of p53 expression. These results correlated with significantly increased numbers of apoptotic cells in bulge cells and basal layer of epidermis, suggesting that loss of TRF2 results in telomeric DNA damage response and cell death. The finding that apoptotic cells were detected in the rapidly dividing basal layer but rarely in suprabasal cells suggests that TRF2 is important for telomere maintenance in this population. The sensitivity of basal layer cells to TRF2 depletion also may be due in part to telomere shortening as the result of increased cell proliferation rather than recruitment of XPF (25). Basal layer cells express low telomerase activity which is insufficient to halt telomere shortening with time (17). The increased percentage of G2/M phase cells observed in TRF2 null epidermis suggests that this tissue can maintain integrity by compensating for the loss of dividing cells with increased proliferation. In agreement with these results, acute ablation of follicular stem cells resulted in loss of hair follicles but survival of the epidermis (39). Late generation Terc deficient mice show follicular stem cell depletion and impaired hair growth without significant epidermal hypoplasia (22). Similarly, the stem cell depletion syndrome dyskeratosis congenita is characterized by telomere dysfunction but not epidermal hypoplasia (40), nor is the Pot1b null mouse model of this disease (33). Both Terc and Pot1b deficient mice demonstrate reduced stem cell populations in multiple tissue compartments but do not develop significant hair loss (22,33). These results indicate that epidermis and its appendages can adapt to reduction of the follicular stem cell population.
Conditional TRF2 null mutant mice exhibit stem cell depletion in epidermis resulting in nail dystrophy observed in dyskeratosis congenita (33,34). Conditional TRF2 null mutant mice also exhibit distinct bending of the tail which is reminiscent of the crinkled mouse phenotype. Stem cells of the hair follicle bulge were required for optimal lateral expansion of epidermis during growth of the tail (32), suggesting that depletion of this population may inhibit epidermal proliferation resulting in tail crinkling. Given that the keratin 14 promoter targets the follicular stem cell population (38), telomere dysfunction and resultant apoptosis in these cells may inhibit epidermal proliferation during rapid growth of the tail in neonatal development.
Epidermal tumorigenesis in TRF2 conditional null mutant mice was significantly delayed compared to control animals. These results complement previous studies indicating that TRF2 overexpression increases epithelial carcinogenesis (26). These cancers also grew substantially slower than those in K14-Cre;TRF2+/+ mice as indicated by tumor volume measurements and significantly decreased numbers of PCNA positive cells. These phenotypic results correlated with increased p53 expression and TUNEL positive cells in TRF2 null tumors. The percentage of apoptotic cells in DMBA treated TRF2 null tumors was significantly higher than in control cancers, indicating that carcinogen treatment exacerbated TRF2 mediated programmed cell death found in conditional null epidermis resulting in slow tumor onset and growth. This may be due to additional telomere shortening resulting from carcinogen damage. The ability of TRF2 null tumor cells to form colonies in vitro and tumors in immunocompromised mice also was substantially impaired. A previous study showed that epidermal tumorigenesis initiated by DMBA and promoted by TPA was delayed in CD34 null mutant mice (41). Depletion of proliferating cells including the CD34+ population inhibited mouse skin tumorigenesis (35). In contrast to the stem cell depletion observed in wild type tumors treated with DMBA alone, the TPA promoted mouse skin carcinogenesis protocol resulted in expansion of the CD34+ cell population (35). This result is likely due to increased mitosis promoted by TPA, compared to persistent DNA damage and apoptosis in tumors treated with DMBA alone.
Telomere length was shorter in follicular stem cells from TRF2 null epidermis, and this shortening was dramatically enhanced in non-stem basal layer cells. These results suggest that loss of TRF2 expression regulates telomere shortening in vivo. Previous studies have demonstrated that homologous recombination results in loss of telomeric DNA in cultured cells with diminished TRF2 function (11), and follicular stem cells exhibit short telomeres in Terc deficient mice (24). The effects of telomere dysfunction in rapidly proliferating epidermis contrasts with those of quiescent epithelia such as hepatocytes, in which TRF2 deficiency induced telomere fusions and aneuploidy in the absence of cell division (42). Telomeres were significantly shorter in tumors from both wild type and conditional TRF2 null mice, indicating that carcinogen induced DNA damage and increased cell proliferation are important for regulating telomere shortening in these cancers. DNA damage response at telomeres was enhanced in tumor cells from TRF2 conditional null mice which correlated with increased signal free ends, aneuploidy, and chromosomal fusions in these cancers. Inhibition of TRF2 function resulted in chromosomal fusions and anaphase bridges in cultured cells (2). In contrast, we did not observe tandem chromosomal fusions at telomeres in TRF2 null tumor cells which have been demonstrated by acute loss of TRF2 function in vitro (5). It is likely that these cells undergo programmed cell death in vivo which precludes their establishment in monolayer culture. Telomere fusions in human cancer cell lines have been shown to possess extremely short telomeric sequences or lack telomere repeats altogether (42).
Despite the increased latency and slow growth of TRF2 null tumors, the dramatically increased genomic instability in these cancers correlated with significant expansion of CD34+/K15+ cells. However unlike wild type CD34+/K15+ cancer cells, TRF2 deficient CD34+/K15+ tumor cells are highly niche dependent given their poor colony forming efficiency in vitro and tumor formation in immunocompromised mice. It is interesting to speculate that other cells in the tumor may support proliferation and metastasis of the CD34+/K15+ population, possibly through paracrine or cell adhesion signaling. Shortened telomeres have been shown to cause genomic instability in diverse model systems (42–44). While most genetically unstable clones are unable to expand, those with specific mutations and rearrangements can acquire proliferative and invasive capabilities. With regard to cancer stem cells, aneuploidy has been demonstrated in label retaining cells from human cancer cell lines, which acquire the ability to proliferate (45). DNA damage in side population cells can induce proliferation, and these cells are enriched in recurrent tumors (46). These results indicate that genomic instability can induce stem cell proliferation and promote development of metastatic clones.
TRF1 conditional null mutant mice exhibit severe epidermal defects that can be rescued by p53 deficiency (47). In contrast, the selective depletion of basal keratinocytes in TRF2 conditional null mutant mice suggests the involvement of additional telomere maintenance mechanisms such as telomerase which is normally expressed by these cells (22,23). It is possible that stochastic variations in telomere length within the epidermis make subpopulations of cells more susceptible to TRF2 mediated telomere dysfunction which may be revealed by inhibition of telomerase in this genetic background. Finally multiple distinct stem cell populations have been proposed in epidermis (48–50) which may have different sensitivities to telomere dysfunction and carcinogenesis.
MATERIALS AND METHODS
Transgenic Mouse Procedures
The Tg(KRT14-Cre)1Amc/J (K14-Cre) and B6;129P2-Terf2tm1Tdl/J (TRF2f/f; 6) mutant mouse strains were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice were backcrossed to create K14-Cre;TRF2f/f animals (n=12 each group). Genotyping was performed on extracted genomic DNA from sorted epidermal cells. One month old K14-Cre;TRF2+/+ and K14-Cre;TRF2f/f mice were dosed twice weekly with 25 μg dimethylbenzanthracene, which induces metastatic squamous cell carcinoma. The latency, number, and volume of tumors were recorded for each animal. Tail skin and tumors from 9 month old mice were frozen or fixed in 4% buffered formaldehyde.
RT-PCR
Human cell lines were obtained from the American Type Culture Collection. RNA was extracted from sorted cells and reverse transcribed according to manufacturer's instructions (Invitrogen, Carlsbad, CA). cDNA was amplified using mTRF2 primers 5'- ACTAGCTTACGGAGTCTGC -3' and 5'-AAGGGGGAGTTTCAGGAGAG -3. Human TRF2 primers were 5'-CTTCTGATGCAAATGCAAAGG -3' and 5'- AGACAGCAAGCACAACAC -3'. Mouse CXCR3 primers were 5'- ACAGCTCAGCATATATCCAGG -3' and 5'-GATACTGAGATGGGCACATTC -3'. β-actin was amplified using primers 5'-AAAAGCCACCCCCACTCCTAAG-3' and 5'- TCAAGTCAGTGTACAGGCCAGC-3' at 94° C for 25 seconds, 55° C for 1 minute, and 72° C for 1 minute. Quantitative PCR was performed using iCycler (Bio-Rad) and products separated by agarose gel electrophoresis.
Western Blot
Protein was extracted from mouse epidermis in 1x Laemmli buffer. 75 μg total cellular protein was separated by SDS-PAGE. Proteins were electroblotted to PVDF membranes. Blots were incubated with antibodies to phospho-ATM, total ATM, Chk2, p53, or β-actin for 16 hours at 4° C. After washing, blots were incubated for 30 minutes at room temperature with anti-IgG secondary antibody conjugated to horseradish peroxidase. Bands were visualized by the enhanced chemiluminescence method and quantitated by laser densitometry.
Immunofluorescence and Immunohistochemistry
Mouse skin and tumor tissue sections were deparaffinized and stained with hematoxylin and eosin. Sections were incubated with anti-CD34, p53, or 53BP1 antibody overnight at room temperature. Sections were incubated with secondary antibody conjugated to phycoerythrin or fluorescein. Sections were washed in PBS and visualized by fluorescence microscopy. For immunohistochemical analysis, sections were blocked with 10% normal serum followed by incubation with anti-TRF2 or PCNA antibodies. After three washes in PBS, sections were incubated with secondary antibody conjugated to biotin followed by streptavidin conjugated peroxidase. Antigen-antibody complexes were detected by incubation with substrate solution. Data were analyzed by Student t test.
Cell Death Analysis
Epidermal cells and tumor tissue were fixed, washed in PBS, and incubated with terminal deoxynucleotidyl transferase and dUTP-fluorescein for 1 hour at 37° C. Apoptotic cells were visualized by fluorescence microscopy. The percentage of fluorescent cells in 10 random high power fields was determined. Data were analyzed by Student t test.
Fluorescence Activated Cell Sorting
Epidermal keratinocytes and SCCs were dissociated by trypsinization, washed in PBS, and incubated with phycoerythrin conjugated CD34 and K15 antibodies. For cell cycle analysis, cells were incubated with Vybrant Dye Cycle Violet reagent (Invitrogen, Carlsbad, CA). Samples were washed in PBS followed by FACS.
Telomere Length Analysis of Sorted Cells
We used a quantitative PCR method to measure average telomere length ratios (31). Telomeric primers were 5'-CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTT-3' and 5'-GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT-3'. Primers for the mouse acidic ribosomal phosphoprotein PO (36B4) gene were 5'-ACTGGTCTAGGACCCGAGAAG-3' and 5'- TCAATGGTGCCTCTGGAGATT-3'. Each reaction for the telomere portion of the assay included 12.5 μl Syber Green PCR master mix (Applied Biosystems, Foster City, CA), 300 nM each primer, and 20 ng genomic DNA. Samples were amplified in triplicate with reaction conditions of 95° C for 10 minutes followed by 30 cycles at 95° C for 15 seconds and 56° C for 1 minute. For the 36B4 assay, reaction conditions were 95° C for 10 minutes followed by 35 cycles at 95° C for 15 seconds, 52° C annealing for 20 seconds, and extension at 72° C for 30 seconds. The relative input amount of telomere PCR was divided by the relative input amount of the 36B4 PCR. PCR was performed 3 times for each sample and the average of these ratios was reported as the average telomere length ratio.
Metaphase Spreads and Fluorescence In Situ Hybridzation
Tumor cell cultures were treated with 0.1 μg/ml colcemid for 3 hours at 37° C. Cell pellets were suspended in 60 mM KCl and incubated at room temperature for 30 minutes. After centrifugation, cell pellets were fixed 3 times in 3:1 methanol:acetic acid and spotted onto microscope slides. Cy3 labeled telomeric probe was hybridized to the metaphase spreads (Dako, Carpinteria, CA). After washing, slides were coverslipped and photographed using fluorescence microscopy.
ACKNOWLEDGEMENTS
This research was supported by National Institutes of Health grant DE14283.
Footnotes
CONFLICT OF INTEREST The authors have no conflicts of interest.
REFERENCES
- 1.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 2.van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end to end fusions. Cell. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- 3.Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. p53 and ATM dependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283:1321–1324. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- 4.Smogorzewska A, de Lange T. Different telomere damage signaling pathways in human and mouse cells. EMBO J. 2002;21:4338–4348. doi: 10.1093/emboj/cdf433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068–1071. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- 6.Celli GB, de Lange T. DNA processing is not required for ATM mediated telomere damage response after TRF2 deletion. Nature Cell Biol. 2005;7:712–718. doi: 10.1038/ncb1275. [DOI] [PubMed] [Google Scholar]
- 7.Dimitrova N, Chen YCM, Spector DL, de Lange T. 53BP1 promotes non homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng Y, Guo X, Ferguson DO, Chang S. Multiple roles for MRE11 at uncapped telomeres. Nature. 2009;460:914–918. doi: 10.1038/nature08196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Attwooll CL, Akpinar M, Petrini JHJ. The Mre11 complex and the response to dysfunctional telomeres. Mol Cell Biol. 2009;29:5540–5551. doi: 10.1128/MCB.00479-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimitrova N, de Lange T. Cell cycle dependent role of MRN at dysfunctional telomeres: ATM signaling dependent induction of nonhomologous end joining (NHEJ) in G1 and resection mediated inhibition of NHEJ in G2. Mol Cell Biol. 2009;29:5552–5563. doi: 10.1128/MCB.00476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang RC, Smogorzewska A, de Lange T. Homologous recombination generates t loop sized deletions at human telomeres. Cell. 2004;119:355–368. doi: 10.1016/j.cell.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 12.Bailey SM, Cornforth MN, Kurimasa A, Chen DJ, Goodwin EH. Strand specific postreplicative processing of mammalian telomeres. Science. 2001;293:2462–2465. doi: 10.1126/science.1062560. [DOI] [PubMed] [Google Scholar]
- 13.Ancelin K, Brunori M, Bauwens S, Koering CE, Brun C, Ricoul M, et al. Targeting assay to study the cis functions of human telomeric proteins: evidence for inhibition of telomerase by TRF1 and for activation of telomere degradation by TRF2. Mol Cell Biol. 2002;22:3474–3487. doi: 10.1128/MCB.22.10.3474-3487.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karlseder J, Smogorzewska A, de Lange T. Senescence induced by altered telomere state, not telomere loss. Science. 2002;295:2446–2449. doi: 10.1126/science.1069523. [DOI] [PubMed] [Google Scholar]
- 15.Rossi DJ, Jamieson CHM, Weissman IL. Stem cells and the pathways to aging and cancer. Cell. 2008;132:681–696. doi: 10.1016/j.cell.2008.01.036. [DOI] [PubMed] [Google Scholar]
- 16.Fuchs E. The tortoise and the hair: slow cycling cells in the stem cell race. Cell. 2009;137:811–819. doi: 10.1016/j.cell.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flores I, Canela A, Vera E, Tejera A, Cotsarelis G, Blasco MA. The longest telomeres: a general signature of adult stem cell compartments. Genes Dev. 2008;22:654–67. doi: 10.1101/gad.451008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morris RJ, Liu Y, Marles L, Yang Z, Trempus C, Li S, et al. Capturing and profiling adult hair follicle stem cells. Nature Biotechnol. 2004;22:411–417. doi: 10.1038/nbt950. [DOI] [PubMed] [Google Scholar]
- 19.Fuchs E. Scratching the surface of skin development. Nature. 2007;445:834–842. doi: 10.1038/nature05659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cotsarelis G. Epithelial stem cells: a folliculocentric view. J Invest Derm. 2006;126:1459–1468. doi: 10.1038/sj.jid.5700376. [DOI] [PubMed] [Google Scholar]
- 21.Fuchs E. Skin stem cells: rising to the surface. J Cell Biol. 2008;180:273–284. doi: 10.1083/jcb.200708185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flores I, Cayuela ML, Blasco MA. Effects of telomerase and telomere length on epidermal stem cell behavior. Science. 2005;309:1253–1256. doi: 10.1126/science.1115025. [DOI] [PubMed] [Google Scholar]
- 23.Sarin KY, Cheung P, Gilison D, Lee E, Tennen RI, Wang E, et al. Conditional telomerase induction causes proliferation of hair follicle stem cells. Nature. 2005;436:1048–1052. doi: 10.1038/nature03836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siegl-Cachedenier I, Flores I, Klatt P, Blasco MA. Telomerase reverses epidermal hair follicle stem cell defects and loss of long term survival associated with critically short telomeres. J Cell Biol. 2007;179:277–290. doi: 10.1083/jcb.200704141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munoz P, Blanco R, Blasco MA. Role of TRF2 telomeric protein in cancer and ageing. Cell Cycle. 2006;5:718–721. doi: 10.4161/cc.5.7.2636. [DOI] [PubMed] [Google Scholar]
- 26.Blanco R, Munoz P, Flores JM, Klatt P, Blasco MA. Telomerase abrogation dramatically accelerates TRF2 induced epithelial carcinogenesis. Genes Dev. 2007;21:206–220. doi: 10.1101/gad.406207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stout GJ, Blasco MA. Genetic dissection of the mechanisms underlying telomere associated diseases: impact of the TRF2 telomeric protein on mouse epidermal stem cells. Disease Models Mech. 2009;2:139–156. doi: 10.1242/dmm.002121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsutani N, Yokozaki H, Tahara E, Tahara H, Kuniyasu H, Haruma K, et al. Expression of telomeric repeat binding factor 1 and 2 and TRF1 interacting nuclear protein 2 in human gastric carcinoma. Int J Oncol. 2001;19:507–512. [PubMed] [Google Scholar]
- 29.Oh BK, Kim YJ, Park C, Park YN. Upregulation of telomere binding proteins TRF1, TRF2, and TIN2 is related to telomere shortening during human multistep hepatocarcinogenesis. Am J Pathol. 2005;166:73–80. doi: 10.1016/S0002-9440(10)62233-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lantuejoul S, Raynaud C, Salameire D, Gazzeri S, Moro-Sibilot D, Soria JC, et al. Telomere maintenance and DNA damage responses during lung carcinogenesis. Clin Cancer Res. 2010;16:2979–2988. doi: 10.1158/1078-0432.CCR-10-0142. [DOI] [PubMed] [Google Scholar]
- 31.Callicot RJ, Womack JE. Real time PCR assay for measurement of mouse telomeres. Comparative Med. 2006;56:17–22. [PubMed] [Google Scholar]
- 32.Heath J, Langton AK, Hammond NL, Overbeek PA, Dixon MJ, Headon DJ. Hair follicles are required for optimal growth during lateral skin expansion. J Invest Derm. 2009;129:2358–2364. doi: 10.1038/jid.2009.102. [DOI] [PubMed] [Google Scholar]
- 33.Hockemeyer D, Palm W, Wang RC, Couto SS, de Lange T. Engineered telomere degradation models dyskeratosis congenita. Genes Dev. 2008;22:1773–1785. doi: 10.1101/gad.1679208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He H, Wang Y, Guo X, Ramchandani S, Ma J, Shen MF, et al. Pot1b deletion and telomerase haploinsufficiency in mice initiate an ATR dependent DNA damage response and elicit phenotypes resembling dyskeratosis congenita. Mol Cell Biol. 2009;29:229–240. doi: 10.1128/MCB.01400-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malanchi I, Peinado H, Kassen D, Hussenet T, Metzger D, Chambon P, et al. Cutaneous cancer stem cell maintenance is dependent on β-catenin signaling. Nature. 2008;452:650–653. doi: 10.1038/nature06835. [DOI] [PubMed] [Google Scholar]
- 36.Ku TK, Nguyen DC, Karaman M, Gill P, Hacia JG, Crowe DL. Loss of p53 expression correlates with metastatic phenotype and transcriptional profile in a new mouse model of head and neck cancer. Mol Cancer Res. 2007;5:351–362. doi: 10.1158/1541-7786.MCR-06-0238. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y, Lyle S, Yang Z, Cotsarelis G. Keratin 15 promoter targets putative epithelial stem cells in the hair follicle bulge. J Invest Derm. 2003;121:963–968. doi: 10.1046/j.1523-1747.2003.12600.x. [DOI] [PubMed] [Google Scholar]
- 38.Tumbar T, Guasch G, Greco V, Blanpain C, Lowry WE, Rendl M, Fuchs E. Defining the epithelial stem cell niche in the skin. Science. 2004;303:359–363. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ito M, Liu Y, Yang Z, Nguyen J, Liang F, Morris RJ, Cotsarelis G. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nature Med. 2005;11:1351–1354. doi: 10.1038/nm1328. [DOI] [PubMed] [Google Scholar]
- 40.Savage SA, Alter BP. Dyskeratosis congenita. Hematol Oncol Clinics N Am. 2009;23:215–231. doi: 10.1016/j.hoc.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trempus CS, Morris RJ, Ehinger M, Elmore A, Bortner CD, Ito M, et al. CD34 expression by hair follicle stem cells is required for skin tumor development in mice. Cancer Res. 2007;67:4173–4181. doi: 10.1158/0008-5472.CAN-06-3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Denchi EL, Celli G, de Lange T. Hepatocytes with extensive telomere deprotection and fusion remain viable and regenerate liver mass through endoreduplication. Genes Dev. 2006;20:2648–2653. doi: 10.1101/gad.1453606. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Capper R, Britt-Compton B, Tankimanova M, Rowson J, Letsolo B, Man S, et al. The nature of telomere fusion and a definition of the critical telomere length in human cells. Genes Dev. 2007;21:2495–2508. doi: 10.1101/gad.439107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA. Telomere dysfunction promotes nonreciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641–645. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- 45.Kusumbe AP, Bapat SA. Cancer stem cells and aneuploid populations within developing tumors are the major determinants of tumor dormancy. Cancer Res. 2009;69:9245–9253. doi: 10.1158/0008-5472.CAN-09-2802. [DOI] [PubMed] [Google Scholar]
- 46.Liang Y, Zhong Z, Huang Y, Deng W, Cao J, Tsao G, et al. Stem like cancer cells are inducible by increasing genomic instability in cancer cells. J Biol Chem. 2010;285:4931–4940. doi: 10.1074/jbc.M109.048397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martinez P, Thanasoula M, Munoz P, Liao C, Tejera A, McNees C, et al. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009;3:2060–2075. doi: 10.1101/gad.543509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blanpain C, Lowry WE, Geoghegan A, Polak L, Fuchs E. Self renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell. 2004;118:635–648. doi: 10.1016/j.cell.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 49.Clayton E, Doupe DP, Klein AM, Winton DJ, Simons BD, Jones PH. A single type of progenitor cell maintains normal epidermis. Nature. 2007;446:185–189. doi: 10.1038/nature05574. [DOI] [PubMed] [Google Scholar]
- 50.Snippert HJ, Haegebarth A, Kasper M, Jaks V, van Es JH, Barker N, et al. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science. 2010;327:1385–1389. doi: 10.1126/science.1184733. [DOI] [PubMed] [Google Scholar]