

Abstract
Saccharomonospora cyanea Runmao et al. 1988 is a member of the genus Saccharomonospora in the family Pseudonocardiaceae that is moderately well characterized at the genome level thus far. Members of the genus Saccharomonospora are of interest because they originate from diverse habitats, such as soil, leaf litter, manure, compost, surface of peat, moist, over-heated grain, and ocean sediment, where they probably play a role in the primary degradation of plant material by attacking hemicellulose. Species of the genus Saccharomonospora are usually Gram-positive, non-acid fast, and are classified among the actinomycetes. S. cyanea is characterized by a dark blue (= cyan blue) aerial mycelium. After S. viridis, S. azurea, and S. marina, S. cyanea is only the fourth member in the genus for which a completely sequenced (non-contiguous finished draft status) type strain genome will be published. Here we describe the features of this organism, together with the draft genome sequence, and annotation. The 5,408,301 bp long chromosome with its 5,139 protein-coding and 57 RNA genes was sequenced as part of the DOE funded Community Sequencing Program (CSP) 2010 at the Joint Genome Institute (JGI).
Keywords: draft genome, aerobic, chemoheterotrophic, Gram-positive, vegetative and aerial mycelia, spore-forming, non-motile, soil bacterium, Pseudonocardiaceae, CSP 2010
Introduction
Strain NA-134T (= DSM 44106 = ATCC 43724 = NBRC 14841) is the type strain of the species Saccharomonospora cyanea [1], one out of currently nine members in the genus Saccharomonospora [2]. The strain was originally isolated from a soil sample collected from Guangyun, Sichuan, China [1]. The genus name Saccharomonospora was derived from the Greek words for sakchâr, sugar, monos, single or solitary, and spora, a seed or spore, meaning the sugar(-containing) single-spored (organism) [3]; the species epithet was derived from the Latin adjective cyanea, dark blue, referring to the color of the aerial mycelium [1]. S. cyanea and the other type strains of the genus Saccharomonospora were selected for genome sequencing in a DOE Community Sequencing Project (CSP 312) at Joint Genome Institute (JGI), because members of the genus (which originate from diverse habitats such as soil, leaf litter, manure, compost, surface of peat, moist, over-heated grain and ocean sediment) are supposed to play a role in the primary degradation of plant material by attacking hemicellulose. This expectation was underpinned by the results of the analysis of the genome of S. viridis [4], one of the recently sequenced GEBA genomes [5]. The S. viridis genome, which was the first sequenced genome from a member of the genus Saccharomonospora, contained an unusually large number of genes, 24, for glycosyl hydrolases (GH) belonging to 14 GH families, which were identified in the Carbon Active Enzyme Database [6]. Hydrolysis of cellulose and starch were also reported for other members of the genus (that are included in CSP 312), such as S. marina [7], S. halophila [8], S. saliphila [9], S. paurometabolica [10], and S. xinjiangensis [11]. Here we present a summary classification and a set of features for S. marina NA-134T, together with the description of the genomic sequencing and annotation.
Classification and features
A representative genomic 16S rRNA sequence of strain NA-134T was compared using NCBI BLAST [12,13] under default settings (e.g., considering only the high-scoring segment pairs (HSPs) from the best 250 hits) with the most recent release of the Greengenes database [14] and the relative frequencies of taxa and keywords (reduced to their stem [15]) were determined, weighted by BLAST scores. The most frequently occurring genera were Saccharomonospora (72.4%), Prauserella (11.3%), Kibdelosporangium (6.7%), Amycolatopsis (4.3%) and Actinopolyspora (3.0%) (101 hits in total). Regarding the two hits to sequences from members of the species, the average identity within HSPs was 99.9%, whereas the average coverage by HSPs was 99.8%. Regarding the 47 hits to sequences from other members of the genus, the average identity within HSPs was 97.1%, whereas the average coverage by HSPs was 98.5%. Among all other species, the one yielding the highest score was S. xinjiangensis (AJ306300), which corresponded to an identity of 98.7% and an HSP coverage of 100%. (Note that the Greengenes database uses the INSDC (= EMBL/NCBI/DDBJ) annotation, which is not an authoritative source for nomenclature or classification.) The highest-scoring environmental sequence was FN667150 ('stages composting process full scale municipal waste compost clone FS1575'), which showed an identity of 99.6% and an HSP coverage of 97.9%. The most frequently occurring keywords within the labels of all environmental samples which yielded hits were 'skin' (26.6%), 'nare' (10.5%), 'fossa' (5.3%), 'forearm, volar' (4.5%) and 'human' (4.4%) (149 hits in total), and show no fit to the habitats from which the validly named members of the genus were isolated. The most frequently occurring keywords within the labels of those environmental samples which yielded hits of a higher score than the highest scoring species were 'compost' (25.0%) and 'full, municip, process, scale, stage, wast' (12.5%) (1 hit in total).
Figure 1 shows the phylogenetic neighborhood of S. cyanea in a 16S rRNA based tree. The sequences of the three identical 16S rRNA gene copies in the genome differ by one nucleotide from the previously published 16S rRNA sequence (Z38018).
Figure 1.

Phylogenetic tree highlighting the position of S. cyanea relative to the type strains of the other species within the family Pseudonocardiaceae. The tree was inferred from 1,371 aligned characters [16,17] of the 16S rRNA gene sequence under the maximum likelihood (ML) criterion [18]. Rooting was done initially using the midpoint method [19] and then checked for its agreement with the current classification (Table 1). The branches are scaled in terms of the expected number of substitutions per site. Numbers adjacent to the branches are support values from 600 ML bootstrap replicates [20] (left) and from 1,000 maximum-parsimony bootstrap replicates [21] (right) if larger than 60%. Lineages with type strain genome sequencing projects registered in GOLD [22] are labeled with one asterisk, those also listed as 'Complete and Published' with two asterisks [4,23,24] (S. azurea [25] and S. marina [26] miss their second asterisk due to very recent publication). Actinopolyspora iraqiensis Ruan et al. 1994 was ignored in the tree, because a proposal for the transfer of this species to the genus Saccharomonospora [27] was recently rejected on formal criteria [3].
Table 1. Classification and general features of S. cyanea NA-134T according to the MIGS recommendations [28] published by the Genome Standards Consortium [29].
| MIGS ID | Property | Term | Evidence code |
|---|---|---|---|
| Current classification | Domain Bacteria | TAS [30] | |
| Phylum Actinobacteria | TAS [31] | ||
| Class Actinobacteria | TAS [32] | ||
| Subclass Actinobacteridae | TAS [32,33] | ||
| Order Actinomycetales | TAS [32-35] | ||
| Suborder Pseudonocardineae | TAS [32,33,36] | ||
| Family Pseudonocardiaceae | TAS [36] [32,33,36] [32,33,36] [[36] [32,33,36-38] | ||
| Genus Saccharomonospora | TAS [34,39] | ||
| Species Saccharomonospora cyanea | TAS [1] | ||
| Type-strain NA-134 | TAS [1] | ||
| Gram stain | positive | NAS | |
| Cell shape | variable, substrate and aerial mycelia | TAS [1] | |
| Motility | non-motile | TAS [1] | |
| Sporulation | small, non-motile spores with warty surface; single and mostly from aerial mycelium | TAS [1] | |
| Temperature range | mesophile, 24-40°C | TAS [1] | |
| Optimum temperature | 28-37°C | TAS [1] | |
| Salinity | grows well in up to 10% (w/v) NaCl | TAS [1] | |
| MIGS-22 | Oxygen requirement | aerobic | TAS [1] |
| Carbon source | pentoses, hexoses, but not D-glucose | TAS [1] | |
| Energy metabolism | chemoheterotrophic | NAS | |
| MIGS-6 | Habitat | soil | TAS [1] |
| MIGS-15 | Biotic relationship | free living | TAS [1] |
| MIGS-14 | Pathogenicity | none | NAS |
| Biosafety level | 1 | TAS [40] | |
| MIGS-23.1 | Isolation | soil | TAS [1] |
| MIGS-4 | Geographic location | Guangyan City, Sichuan, China | TAS [1] |
| MIGS-5 | Sample collection time | 1988 or before | NAS |
| MIGS-4.1 | Latitude | 32.450 | TAS [1] |
| MIGS-4.2 | Longitude | 105.843 | TAS [1] |
| MIGS-4.3 | Depth | not reported | |
| MIGS-4.4 | Altitude | about 40 m | NAS |
Evidence codes - TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from of the Gene Ontology project [41].
Cells of strain NA-134T form an irregularly branched, non-fragmenting, vegetative mycelium of 0.2 to 0.4 μm diameter (Figure 2) [1]. The aerial mycelium had a diameter of 0.3 to 0.6 μm and formed more sessile spores than the substrate mycelium [1]. Spores are non-motile, small, and oval to ellipsoid with warty surface [1]. The growth range of strain NA-134T spans from 24°C to 40°C, with an optimum at 28°C to 37°C [1]. Strain NA-134T grows well in up to 10% NaCl, but not in 15% NaCl [1]. Substrates used by the strain are summarized in detail in the strain description [1].
Figure 2.

Scanning electron micrograph of S. cyanea NA-134T
Chemotaxonomy
The cell wall of strain NA-134T are of type IV, containing meso-diaminopimelic acid, with type A whole-cell sugar pattern (galactose and arabinose present) [1]. The fatty acids spectrum is dominated by saturated penta- to heptadecanoic acids: C17:1 cis-9 (17.0%), anteiso-C17:0 (13.0%), iso-C16:0 (12.0%), C17:0 (11.5%), C15:0 (7.0%), iso-C16:0 2OH (7.0%), C16:1 cis-9 (6.0%), C16:0 (5.0%), iso-C15:0 (3.0%), and anteiso-C15:0 (2.0%) [42].
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing as part of the DOE Joint Genome Institute Community Sequencing Program (CSP) 2010, CSP 312, “Whole genome type strain sequences of the genus Saccharomonospora – a taxonomically troubled genus with bioenergetic potential”. The genome project is deposited in the Genomes On Line Database [22] and the complete genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI) using state of the art sequencing technology [43]. A summary of the project information is shown in Table 2.
Table 2. Genome sequencing project information.
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Non-contiguous finished |
| MIGS-28 | Libraries used | Three genomic libraries: one 454 pyrosequence standard library, one 454 PE library (12 kb insert size), one Illumina library |
| MIGS-29 | Sequencing platforms | Illumina GAii, 454 GS FLX Titanium |
| MIGS-31.2 | Sequencing coverage | 1,005.1 × Illumina; 8.6 × pyrosequence |
| MIGS-30 | Assemblers | Newbler version 2.3, Velvet version 1.0.13, phrap version SPS - 4.24 |
| MIGS-32 | Gene calling method | Prodigal, GenePRIMP |
| INSDC ID | CM001440, AHLY00000000.1 | |
| GenBank Date of Release | February 3, 2012 | |
| GOLD ID | Gi07556 | |
| NCBI project ID | 61997 | |
| Database: IMG | 2508501013 | |
| MIGS-13 | Source material identifier | DSM 44106 |
| Project relevance | Bioenergy and phylogenetic diversity |
Growth conditions and DNA isolation
Strain NA-134T, DSM 44106, was grown in DSMZ medium 3 (Azotobacter Medium) [44] at 28°C. DNA was isolated from 0.5-1 g of cell paste using Jetflex Genomic DNA Purification Kit (GENOMED 600100) following the standard protocol as recommended by the manufacturer with the following modifications: extended cell lysis time (60 min.) with additional 30µl achromopeptidase, lysostaphin, mutanolysin; proteinase K was applied in 6-fold the supplier recommended amount for 60 min. at 58°C. The purity, quality and size of the bulk gDNA preparation were according to DOE-JGI guidelines and routine protocols by the DNA Bank Network [45]. DNA is available through the DNA Bank Network [46].
Genome sequencing and assembly
The genome was sequenced using a combination of Illumina and 454 sequencing platforms. All general aspects of library construction and sequencing can be found at the JGI website [47]. Pyrosequencing reads were assembled using the Newbler assembler (Roche). The initial Newbler assembly consisting of 148 contigs in one scaffold was converted into a phrap [48] assembly by making fake reads from the consensus, to collect the read pairs in the 454 paired end library. Illumina GAii sequencing data (5,624.1 Mb) were assembled with Velvet [49] and the consensus sequences were shredded into 1.5 kb overlapped fake reads and assembled together with the 454 data. The 454 draft assembly was based on 103.5 Mb 454 draft data and all of the 454 paired end data. Newbler parameters are -consed -a 50 -l 350 -g -m -ml 20. The Phred/Phrap/Consed software package [48] was used for sequence assembly and quality assessment in the subsequent finishing process. After the shotgun stage, reads were assembled with parallel phrap (High Performance Software, LLC). Possible mis-assemblies were corrected with gapResolution [47], Dupfinisher [50], or sequencing cloned bridging PCR fragments with subcloning. Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR primer walks (J.-F. Chang, unpublished). A total of 157 additional reactions were necessary to close gaps and to raise the quality of the finished sequence. Illumina reads were also used to correct potential base errors and increase consensus quality using a software Polisher developed at JGI [51]. The error rate of the completed genome sequence is less than 1 in 100,000. Together, the combination of the Illumina and 454 sequencing platforms provided 1,013.7 × coverage of the genome. The final assembly contained 366,256 pyrosequence and 71,412,890 Illumina reads.
Genome annotation
Genes were identified using Prodigal [52] as part of the DOE-JGI [53] genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [54]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) non-redundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [55].
Genome properties
The genome consists of a 5,408,301 bp long circular chromosome with a 69.7% G+C content (Table 3 and Figure 3). Of the 5,196 genes predicted, 5,139 were protein-coding genes, and 57 RNAs; 93 pseudogenes were also identified. The majority of the protein-coding genes (74.7%) were assigned a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.
Table 3. Genome Statistics.
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 5,408,301 | 100.00% |
| DNA coding region (bp) | 4,926,834 | 91.10% |
| DNA G+C content (bp) | 3,771,475 | 69.74% |
| Number of replicons | 1 | |
| Extrachromosomal elements | 0 | |
| Total genes | 5,196 | 100.00% |
| RNA genes | 57 | 1.10% |
| rRNA operons | 3 | |
| tRNA genes | 47 | 0.90% |
| Protein-coding genes | 5,139 | 98.90% |
| Pseudo genes | 93 | 1.79% |
| Genes with function prediction (proteins) | 3,880 | 74.67% |
| Genes in paralog clusters | 2,852 | 54.89% |
| Genes assigned to COGs | 3,834 | 73.79% |
| Genes assigned Pfam domains | 4,014 | 77.25% |
| Genes with signal peptides | 1,512 | 29.10% |
| Genes with transmembrane helices | 1,206 | 23.21% |
| CRISPR repeats | 0 |
Figure 3.

Graphical map of the chromosome. From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew (purple/olive).
Table 4. Number of genes associated with the general COG functional categories.
| Code | value | %age | Description |
|---|---|---|---|
| J | 184 | 4.3 | Translation, ribosomal structure and biogenesis |
| A | 1 | 0.0 | RNA processing and modification |
| K | 512 | 11.8 | Transcription |
| L | 182 | 4.2 | Replication, recombination and repair |
| B | 2 | 0.1 | Chromatin structure and dynamics |
| D | 34 | 0.8 | Cell cycle control, cell division, chromosome partitioning |
| Y | 0 | 0.0 | Nuclear structure |
| V | 79 | 1.8 | Defense mechanisms |
| T | 209 | 4.8 | Signal transduction mechanisms |
| M | 174 | 4.0 | Cell wall/membrane biogenesis |
| N | 5 | 0.1 | Cell motility |
| Z | 0 | 0.0 | Cytoskeleton |
| W | 0 | 0.0 | Extracellular structures |
| U | 35 | 0.8 | Intracellular trafficking and secretion, and vesicular transport |
| O | 130 | 3.0 | Post-translational modification, protein turnover, chaperones |
| C | 274 | 6.3 | Energy production and conversion |
| G | 327 | 7.6 | Carbohydrate transport and metabolism |
| E | 341 | 7.9 | Amino acid transport and metabolism |
| F | 96 | 2.2 | Nucleotide transport and metabolism |
| H | 202 | 4.7 | Coenzyme transport and metabolism |
| I | 213 | 4.9 | Lipid transport and metabolism |
| P | 210 | 4.9 | Inorganic ion transport and metabolism |
| Q | 196 | 4.5 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 588 | 13.6 | General function prediction only |
| S | 330 | 7.6 | Function unknown |
| - | 1,362 | 26.2 | Not in COGs |
Insights into the genome sequence
Comparative genomics
The phylum Actinobacteria is one of the most species-rich phyla in the domain Bacteria [31]. As of today the phylum contains the following ten orders, Acidimicrobiales, Actinomycetales, Bifidobacteriales, Coriobacteriales, Euzebyales, Gaiellales, Nitriliruptorales, Rubrobacterales, Solirubrobacterales, Thermoleophilales, with a total of 58 families [3]. Among these, the family Pseudonocardiaceae holds the genus Saccharomonospora, with 5 out of the 9 type strains for the member species having already completely sequenced genomes; the remaining 4 type strains have yet unpublished draft genome sequences according to the Genomes On Line Database (GOLD) [22].
Here we present a brief comparative genomics comparison of S. cyanea with a selection of its closest phylogenetic neighbors that have already published genome sequences (according to Figure 1): S. viridis [4], S. azurea [25] and S. marina [26].
The genomes of the four sequenced Saccharomonospora type strains differ significantly in their size, S. cyanea having 5.4 Mbp, S. viridis 4.3 Mbp, S. azurea 4.8 Mbp and S. marina 6.0 Mbp and their total number of genes, 5,196, 3,962, 4,530 and 5,784, respectively.
An estimate of the overall similarity between S. cyanea, on the one hand, and S. viridis, S. azurea and S. marina, on the other hand, was generated with the Genome-to-Genome Distance Calculator (GGDC) [56-58]. This system calculates the distances by comparing the genomes to obtain HSPs (high-scoring segment pairs) and interfering distances via a set of formulas (1, HSP length / total length; 2, identities / HSP length; 3, identities / total length). For convenience the GGDC also reports model-based DDH estimates along with their confidence intervals [58]. Table 5 shows the results of the pairwise comparison.
Table 5. Pairwise comparison of S. cyanea with S. viridis, S. azurea and S. marina using the GGDC (Genome-to-Genome Distance Calculator).
The comparison of S. cyanea with S. azurea reached the highest scores using the GGDC, 71% of the average of genome length are covered with HSPs. The identity within the HSPs was 85%, whereas the identity over the whole genome was 61%. The lowest similarity scores were observed in the comparison of S. cyanea with S. marina with only 28% of the average of both genome lengths covered with HSPs. The identity within these HSPs was 79%, whereas the identity over the whole genome was only 22%.
With regard to S. cyanea and S. azurea the corresponding DDH estimates were below the 70% threshold under formulas 1-3 throughout: 52.6% (±3), 28.6% (±3) and 45.4% (±3). The DDH estimates confidence intervals are given in parentheses as provided by [58]. The remaining pairings resulted in even smaller DDH estimates (data not shown).
As expected, those distances relating HSP coverage (formula 1) and number of identical base pairs within HSPs to total genome length (formula 3) are higher between S. cyanea and S. azurea than between S. cyanea and S. viridis or S. marina, respectively. That the distances relating the number of identical base pairs to total HSP length (formula 2) behave differently indicates that the genomic similarities between all four type strain genomes are strongly restricted to more conserved sequences, a kind of saturation phenomenon [56].
In order to further compare the genomes of S. cyanea, S. viridis, S. azurea and S. marina, correlation values (Pearson coefficient) according to the similarity on the level of COG category, pfam and TIGRfam were calculated (see Table 6). The highest correlation value (0.97) was reached for S. cyanea and S. azurea on the level of pfam data; the correlation values on the basis of COG and TIGRfam data were only slightly smaller with 0.96 and 0.93, respectively. As a correlation value of 1 indicates the highest correlation, we can find a very high correlation between the genomes of S. cyanea and S. azurea considering the above data [55].
Table 6. Pearson's correlation coefficients according to the similarity on the level of Pfam, COG category and TIGRfam (in this order and separated by slashes).
| S. cyanea | S. azurea | S. viridis | S. marina | |
|---|---|---|---|---|
| S. cyanea | 1.00 / 1.00 / 1.00 | - | - | - |
| S. azurea | 0.97 / 0.96 / 0.93 | 1.00 / 1.00 / 1.00 | - | - |
| S. viridis | 0.95 / 0.90 / 0.87 | 0.96 / 0.93 / 0.90 | 1.00 / 1.00 / 1.00 | - |
| S. marina | 0.93 / 0.90 / 0.86 | 0.93 / 0.90 / 0.87 | 0.94 / 0.90 / 0.83 | 1.00 / 1.00 / 1.00 |
The comparison of the number of genes belonging to the different COG categories revealed only small differences in the genomes of S. cyanea and S. azurea with 0.4% deviation between the same COG categories on average. A slightly higher fraction of genes belonging in the categories transcription (S. cyanea 11.8%, S. azurea 10.6%), carbohydrate metabolism (S. cyanea 7.6%, S. azurea 7.0%), secondary catabolism (S. cyanea 4.5%, S. azurea 4.1%), defense mechanisms (S. cyanea 1.8%, S. azurea 1.6%), inorganic ion transport and metabolism (S. cyanea 4.9%, S. azurea 4.7%) and lipid transport (S. cyanea 4.9%, S. azurea 4.8%) were identified in S. cyanea. The gene count in further COG categories such as cell cycle control, cell motility, cell biogenesis, lipid metabolism, secondary catabolism, post-translational modification and signal transduction was also slightly increased in S. cyanea but differed at most by 5 genes. In contrast, a slightly smaller fraction of genes belonging in the categories posttranslational modification (S. cyanea 3.0%, S. azurea 3.6%), coenzyme metabolism (S. cyanea 4.7%, S. azurea 5.2%), amino acid metabolism (S. cyanea 7.9%, S. azurea 8.4%), replication system (S. cyanea 4.2%, S. azurea 4.7%), translation (S. cyanea 4.3%, S. azurea 4.6%), signal transduction (S. cyanea 4.8%, S. azurea 5.1%), energy production/conversion (S. cyanea 6.3%, S. azurea 6.6%), nucleotide transport (S. cyanea 2.2%, S. azurea 2.4%) and cell wall biogenesis (S. cyanea 4.0%, S. azurea 4.2%) were identified in S. cyanea. The remaining COG categories intracellular transport, cell cycle control, cell motility and RNA modification differed by not more than a single gene.
The synteny dot plots in Figure 4 shows nucleotide-based comparisons of the genomes of S. cyanea vs. S. viridis, S. azurea and S. marina. In most parts of the genomes a high degree of similarity becomes visible with only a little amount of indels. There exists a pronounced collinearity between the four genomes.
Figure 4.
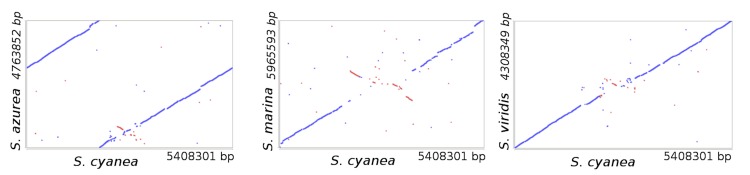
Synteny dot plot based on the genome sequences of S. cyanea vs. those of S. viridis, S. azurea and S. marina. Blue dots represent regions of similarity found on parallel strands and red dots show regions of similarity found on anti-parallel strands.
The Venn-diagram Figure 5 shows the number of shared genes between the completely sequenced and published genomes of Saccharomonospora type strains. All four genomes share a rather high fraction of 3,159 genes (59-74% of the genes, respectively) whereas only 247 (S. azurea, 5%) to 1,401 (S. marina, 26%) genes are unique for one genome in the genus. The genomes of S. cyanea and S. azurea contain the highest number (324) of pairwise shared genes, including many that encode hypothetical or unknown proteins (expectedly, due to the low level of functionally characterized genes in the genus), but also numerous transcriptional regulators (such as Sigma-70 and ATP-dependent transcriptional regulator) and transporters (such as TRAP transporters, arabinose efflux permeases, ABC-type sugar transport systems and Fe3+- transport systems, p-aminobenzoyl-glutamate transporter, 2-keto-3-deoxygluconate permease, Na+/H+ antiporter NhaD and related arsenite permeases, H+/gluconate symporter and related permeases). Surprisingly, these two genomes also share a suite of gas vesicle synthesis proteins.
Figure 5.

Venn-diagram depicting the intersections of protein sets (total numbers in parentheses) of S. marina, S. azurea, S. cyanea and S. viridis. The diagram was created with [59].
Acknowledgements
We would like to gratefully acknowledge the help of Evelyne-Marie Brambilla (DSMZ) for DNA extraction and quality control. The work conducted by the U.S. Department of Energy Joint Genome Institute was supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
References
- 1.Runmao H, Lin C, Guizhen W. Saccharomonospora cyanea sp. nov. Int J Syst Bacteriol 1988; 38:444-446 10.1099/00207713-38-4-444 [DOI] [Google Scholar]
- 2.Garrity G. NamesforLife. BrowserTool takes expertise out of the database and puts it right in the browser. Microbiol Today 2010; 37:9 [Google Scholar]
- 3.Euzéby JP. List of Bacterial Names with Standing in Nomenclature: a folder available on the internet. Int J Syst Bacteriol 1997; 47:590 10.1099/00207713-47-2-590 [DOI] [PubMed] [Google Scholar]
- 4.Pati A, Sikorski J, Nolan M, Lapidus A, Copeland A, Glavina Del Rio T, Lucas S, Chen F, Tice H, Pitluck S, et al. Complete genome sequence of Saccharomonospora viridis type strain (P101T). Stand Genomic Sci 2009; 1:141-149 10.4056/sigs.20263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven Genomic Encyclopaedia of Bacteria and Archaea. Nature 2009; 462:1056-1060 10.1038/nature08656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carbon Active Enzyme Database www.cazy.org
- 7.Liu Z, Li Y, Zheng LQ, Huang YJ, Li WJ. Saccharomonospora marina sp. nov., isolated from ocean sediment of the East China Sea. Int J Syst Evol Microbiol 2010; 60:1854-1857 10.1099/ijs.0.017038-0 [DOI] [PubMed] [Google Scholar]
- 8.Al-Zarban SS, Al-Musallam AA, Abbas I, Stackebrandt E, Kroppenstedt RM. Saccharomonospora halophila sp. nov., a novel halophilic actinomycete isolated from marsh soil in Kuwait. Int J Syst Evol Microbiol 2002; 52:555-558 [DOI] [PubMed] [Google Scholar]
- 9.Syed DG, Tang SK, Cai M, Zhi XY, Agasar D, Lee JC, Kim CJ, Jiang CL, Xu LH, Li WJ. Saccharomonospora saliphila sp. nov., a halophilic actinomycete from an Indian soil. Int J Syst Evol Microbiol 2008; 58:570-573 10.1099/ijs.0.65449-0 [DOI] [PubMed] [Google Scholar]
- 10.Li WJ, Tang SK, Stackebrandt E, Kroppenstedt RM, Schumann P, Xu LH, Jiang CL. Saccharomonospora paurometabolica sp. nov., a moderately halophilic actinomycete isolated from soil in China. Int J Syst Evol Microbiol 2003; 53:1591-1594 10.1099/ijs.0.02633-0 [DOI] [PubMed] [Google Scholar]
- 11.Jin X, Xu LH, Mao PH, Hseu TH, Jiang CL. De-scription of Saccharomonospora xinjiangensis sp. nov. based on chemical and molecular classifica-tion. Int J Syst Bacteriol 1998; 48:1095-1099 10.1099/00207713-48-4-1095 [DOI] [PubMed] [Google Scholar]
- 12.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Bascic local alignment search tool. J Mol Biol 1990; 215:403-410 [DOI] [PubMed] [Google Scholar]
- 13.Korf I, Yandell M, Bedell J. BLAST, O'Reilly, Sebastopol, 2003. [Google Scholar]
- 14.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006; 72:5069-5072 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porter MF. An algorithm for suffix stripping. Program: electronic library and information systems 1980; 14:130-137.
- 16.Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452-464 10.1093/bioinformatics/18.3.452 [DOI] [PubMed] [Google Scholar]
- 17.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17:540-552 10.1093/oxfordjournals.molbev.a026334 [DOI] [PubMed] [Google Scholar]
- 18.Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol 2008; 57:758-771 10.1080/10635150802429642 [DOI] [PubMed] [Google Scholar]
- 19.Hess PN, De Moraes Russo CA. An empirical test of the midpoint rooting method. Biol J Linn Soc Lond 2007; 92:669-674 10.1111/j.1095-8312.2007.00864.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? Lect Notes Comput Sci 2009; 5541:184-200 10.1007/978-3-642-02008-7_13 [DOI] [Google Scholar]
- 21.Swofford DL. PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4.0 b10. Sinauer Associates, Sunderland, 2002. [Google Scholar]
- 22.Pagani I, Liolios K, Jansson J, Chen IM, Smirnova T, Nosrat B, Markowitz VM, Kyrpides NC. The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2012; 40:D571-D579 10.1093/nar/gkr1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Land M, Lapidus A, Mayilraj S, Chen R, Copeland A, Glavina Del Rio T, Nolan M, Lucas S, Tice H, Cheng JF, et al. Complete genome sequence of Actinosynnema mirum type strain (101T). Stand Genomic Sci 2009; 1:46-53 10.4056/sigs.21137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liolios K, Sikorski J, Jando M, Lapidus A, Copeland A, Glavina Del Rio T, Nolan M, Lucas S, Tice H, Cheng JF, et al. Complete genome sequence of Thermobispora bispora type strain (R51T). Stand Genomic Sci 2010; 2:318-326 10.4056/sigs.962171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klenk HP, Held B, Lucas S, Lapidus A, Copeland A, Hammon N, Pitluck S, Goodwin LA, Han C, Tapia R, et al. Genome sequence of the soil bacterium Saccharomonospora azurea type strain (NA-128T). Stand Genomic Sci 2012; 6:220-229 10.4056/sigs.2635833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klenk HP, Lu M, Lucas S, Lapidus A, Copeland A, Pitluck S, Goodwin LA, Han C, Tapia R, Brambilla EM, et al. Genome sequence of the ocean sediment bacterium Saccharomonospora marina type strain (XMU15T). Stand Genomic Sci 2012; 6:265-275 10.4056/sigs.2655905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang SK, Wang Y, Klenk HP, Shi R, Lou K, Zhang YJ, Chen C, Ruan JS, Li WJ. Actinopolyspora alba sp. nov. and Actinopolyspora erythraea sp. nov., isolated from a salt field, and reclassification of Actinopolyspora iraqiensis Ruan et al 1994 as a heterotypic synonym of Saccharomonospora halophila. Int J Syst Evol Microbiol 2011; 61:1693-1698 10.1099/ijs.0.022319-0 [DOI] [PubMed] [Google Scholar]
- 28.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541-547 10.1038/nbt1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Field D, Amaral-Zettler L, Cochrane G, Cole JR, Dawyndt P, Garrity GM, Gilbert J, Glöckner FO, Hirschman L, Karsch-Mzrachi I, et al. PLoS Biol 2011; 9:e1001088 10.1371/journal.pbio.1001088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms. Proposal for the domains Archaea and Bacteria. Proc Natl Acad Sci USA 1990; 87:4576-4579 10.1073/pnas.87.12.4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garrity GM, Holt JG. The Road Map to the Manual. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 1, Springer, New York, 2001, p. 119-169. [Google Scholar]
- 32.Stackebrandt E, Rainey FA, Ward-Rainey NL. Proposal for a new hierarchic classification system, Actinobacteria classis nov. Int J Syst Bacteriol 1997; 47:479-491 10.1099/00207713-47-2-479 [DOI] [Google Scholar]
- 33.Zhi XY, Li WJ, Stackebrandt E. An update of the structure and 16S rRNA gene sequence-based definition of higher ranks of the class Actinobacteria, with the proposal of two new suborders and four new families and emended descriptions of the existing higher taxa. Int J Syst Evol Microbiol 2009; 59:589-608 10.1099/ijs.0.65780-0 [DOI] [PubMed] [Google Scholar]
- 34.Skerman VBD, McGowan V, Sneath PHA. Approved Lists of Bacterial Names. Int J Syst Bacteriol 1980; 30:225-420 10.1099/00207713-30-1-225 [DOI] [PubMed] [Google Scholar]
- 35.Buchanan RE. Studies in the nomenclature and classification of bacteria. II. The primary subdivisions of the Schizomycetes. J Bacteriol 1917; 2:155-164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Labeda DP, Goodfellow M, Chun J, Zhi X-Y, Li W-J. Reassessment of the systematics of the suborder Pseudonocardineae: transfer of the genera within the family Actinosynnemataceae Labeda and Kroppenstedt 2000 emend. Zhi et al 2009 into an emended family Pseudonocardiaceae Embley et al 1989 emend. Zhi et al 2009. Int J Syst Evol Microbiol 2011; 61:1259-1264 10.1099/ijs.0.024984-0 [DOI] [PubMed] [Google Scholar]
- 37.Validation List no. 29. Validation of the publication of new names and new combinations previously effectively published outside the IJSB. Int J Syst Bacteriol 1989; 39:205-206 10.1099/00207713-39-2-205 [DOI] [Google Scholar]
- 38.Embley MT, Smida J, Stackebrandt E. The phylogeny of mycolate-less wall chemotype IV Actinomycetes and description of Pseudonocardiaceae fam. nov. Syst Appl Microbiol 1988; 11:44-52 10.1016/S0723-2020(88)80047-X [DOI] [Google Scholar]
- 39.Nonomura H, Ohara Y. Distribution of actinomycetes in soil. X. New genus and species of monosporic actinomycetes in soil. J Ferment Technol 1971; 49:895-903 [Google Scholar]
- 40.BAuA. Classification of bacteria and archaea in risk groups. TRBA 466. p. 194. Bundesanstalt für Arbeitsschutz und Arbeitsmedizin, Germany. 2010. [Google Scholar]
- 41.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25-29 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Compendium W.http://www.dsmz.de/microorganisms/wink_pdf/DSM44106.pdf
- 43.Mavromatis K, Land ML, Brettin TS, Quest DJ, Copeland A, Clum A, Goodwin L, Woyke T, Lapidus A, Klenk HP, et al. The fast changing landscape of sequencing technologies and their impact on microbial genome assemblies and annotation. PLoS ONE 2012; 7:e48837 10.1371/journal.pone.0048837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.List of growth media used at DSMZ. http://www.dsmz.de/catalogues/catalogue-microorganisms/culture-technology/list-of-media-for-microorganisms.html
- 45.Zetzsche H, Klenk HP, Raubach MJ, Knebelsberger T, Gemeinholzer B. Comparison of methods and protocols for routine DNA extraction in the DNA Bank Network. In: Gradstein R, Klatt S, Normann F, Weigelt P Willmann R, Wilson R (eds) Systematics Universitätsverlag Göttingen, Göttingen, 2008 p. 354. [Google Scholar]
- 46.Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk HP, Güntsch A, Berendsohn WG, Wägele JW. The DNA Bank Network: the start from a German initiative. Biopreserv Biobank 2011; 9:51-55 10.1089/bio.2010.0029 [DOI] [PubMed] [Google Scholar]
- 47.The DOE Joint Genome Institute www.jgi.doe.gov
- 48.Phrap and Phred for Windows. MacOS, Linux, and Unix. www.phrap.com
- 49.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821-829 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han C, Chain P. Finishing repeat regions automatically with Dupfinisher. In: Proceeding of the 2006 international conference on bioinformatics & computational biology. Arabnia HR, Valafar H (eds), CSREA Press. June 26-29, 2006: 141-146. [Google Scholar]
- 51.Lapidus A, LaButti K, Foster B, Lowry S, Trong S, Goltsman E. POLISHER: An effective tool for using ultra short reads in microbial genome assembly and finishing. AGBT, Marco Island, FL, 2008. [Google Scholar]
- 52.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal Prokaryotic Dynamic Programming Genefinding Algorithm. BMC Bioinformatics 2010; 11:119 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mavromatis K, Ivanova NN, Chen IM, Szeto E, Markowitz VM, Kyrpides NC. The DOE-JGI Standard operating procedure for the annotations of microbial genomes. Stand Genomic Sci 2009; 1:63-67 10.4056/sigs.632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pati A, Ivanova N, Mikhailova N, Ovchinikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: A Gene Prediction Improvement Pipeline for microbial genomes. Nat Methods 2010; 7:455-457 10.1038/nmeth.1457 [DOI] [PubMed] [Google Scholar]
- 55.Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271-2278 10.1093/bioinformatics/btp393 [DOI] [PubMed] [Google Scholar]
- 56.Auch AF, von Jan M, Klenk HP, Göker M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci 2010; 2:117-134 10.4056/sigs.531120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Auch AF, Klenk HP, Göker M. Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand Genomic Sci 2010; 2:142-148 10.4056/sigs.541628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 2013; 14:60 10.1186/1471-2105-14-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen H, Boutros P. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 2011; 12:35 10.1186/1471-2105-12-35 [DOI] [PMC free article] [PubMed] [Google Scholar]