

Abstract
p53 activation prevents the proliferation of genetically unstable cells. Conversely, p53 antagonism by its transcriptional target, the E3 ubiquitin ligase MDM2, is critical for the viability of unstressed, cycling cells. We demonstrate that MDM2 induces the degradation of p53 in both the nucleus and the cytoplasm. As p53 and MDM2 accumulate in the nuclei of stressed cells, we investigated mechanisms enabling p53 activation despite the high MDM2 levels generated during a DNA-damage response. We show that DNA damage destabilized MDM2 by a mechanism involving damage-activated kinases and MDM2 auto-ubiquitination. p53 was stable and transcriptionally active when MDM2 was unstable, but became unstable and inactive as the damage response waned and MDM2 stabilized. Importantly, blocking MDM2 destabilization in DNA-damaged cells prevented p53 target gene activation. Our data reveal that controlled MDM2 degradation is an important new step in p53 regulation.
Keywords: E3 ubiquitin ligase, HDM2, MDM2, p53, PI3K-related protein kinase
Introduction
The p53 tumor suppressor is a transcription factor that is activated by diverse genotoxic and non-genotoxic stresses. Upon exposure to these stresses, p53 is stabilized, accumulates in the nucleus, and forms tetramers capable of binding DNA. Stresses can induce post-translational modifications, such as phosphorylation in the N-terminal transactivation domain by ATM and ATR, which may subsequently enable association with and acetylation by transcriptional co-activators such as PCAF and p300/CBP (Appella and Anderson, 2001; Wahl and Carr, 2001). Once activated, p53 regulates the expression of genes that arrest the cell cycle (e.g., p21) or induce apoptosis (bax, puma, perp, etc.), thereby minimizing the emergence and proliferation of genetically unstable cells. p53 is mutated in the majority of human tumors, emphasizing its importance in controlling cell division and survival.
p53 must be inhibited in unstressed cells, as its inappropriate activation can induce cell cycle arrest, premature senescence, or death (Bottger et al, 1997; Blaydes and Wynford-Thomas, 1998; Mendrysa et al, 2003). MDM2 is a key negative regulator that inhibits p53 by binding its N-terminal transactivation domain, thereby blocking its access to the basal transcription machinery (Momand et al, 1992; Oliner et al, 1993) and preventing C-terminal acetylation by PCAF and p300/CBP (Kobet et al, 2000; Jin et al, 2002). MDM2 is also a RING domain-dependent E3 ubiquitin ligase that ubiquitinates multiple C-terminal lysines on p53, leading to its proteasomal degradation (Michael and Oren, 2003). The importance of MDM2 in p53 regulation is evidenced by the embryonic lethality of MDM2 knockout mice, presumably due to unrestrained p53 activity (Jones et al, 1995; Montes de Oca Luna et al, 1995). Conversely, many tumors with wild-type p53 exhibit supra-physiological levels of MDM2, highlighting the importance of attenuating MDM2 activity to prevent tumorigenesis (Momand et al, 1998).
MDM2-mediated nuclear export has been proposed to be required for p53 degradation. MDM2 overexpression results in an apparent increase in cytoplasmic p53 (Boyd et al, 2000; Geyer et al, 2000; Lohrum et al, 2001), and a recent study has been interpreted to indicate that cytoplasmic but not nuclear p53 can be ubiquitinated (O'Keefe et al, 2003). However, other studies indicate that p53 can be exported without MDM2 binding (Stommel et al, 1999), and that a p53 nuclear export signal (NES) mutant is degraded despite its constitutive nuclear localization (Geyer et al, 2000; Lohrum et al, 2001; Xirodimas et al, 2001; Shirangi et al, 2002). p53 cytoplasmic degradation is also hard to reconcile with p53 nuclear export requiring 3–8 h (Stommel et al, 1999; Henderson and Eleftheriou, 2000), while its half-life is about 20 min. Therefore, the extent to which nuclear export contributes to p53 regulation requires further analysis.
As MDM2 is a p53 target gene, many stresses have the paradoxical effects of simultaneously increasing p53 abundance and elevating MDM2 expression. Therefore, to ensure that p53 can regulate transcription, it is critical that the abundant nuclear MDM2 be prevented from inactivating p53 in stressed cells. One proposed mechanism for inhibiting MDM2 is through ARF, which stabilizes p53 by antagonizing MDM2 ubiquitin ligase activity (Michael and Oren, 2003). ARF is induced by oncogene overexpression, modulates the response to DNA damage, and is frequently mutated in cancer cell lines expressing wild-type p53 (Sherr, 2001). However, as ARF is unnecessary for p53 activation in some tissues (Tolbert et al, 2002), alternative mechanisms of MDM2 inhibition must exist.
N-terminal phosphorylation of p53 is a second mechanism by which MDM2 activity might be compromised. Stress-induced N-terminal phosphorylation sites lie within and near the MDM2-binding domain, and thus may influence its association with p53. Threonine 18 or serine 20 phosphorylation prevents MDM2 binding to p53 peptides in vitro (Chehab et al, 1999; Craig et al, 1999; Sakaguchi et al, 2000). However, full-length p53 constructs mutated at these residues are still stabilized in stressed, transfected cells (Ashcroft et al, 1999; Blattner et al, 1999). In addition, endogenous p53 mutated at the mouse equivalent of serine 20 has no measurable defects in p53 activity or stability (Wu et al, 2002). Thus, other mechanisms for inhibiting MDM2 interaction besides N-terminal phosphorylation are likely to exist.
We present a new mechanism of p53 activation involving DNA-damage-induced degradation of MDM2. MDM2-mediated p53 degradation occurred in the nucleus without prior nuclear-cytoplasmic shuttling. This raises the problem of how p53 can be activated in the nucleus in the presence of high levels of MDM2, as occurs in a stress response. We show that MDM2 is destabilized by DNA damage in a process requiring stress-activated PI 3-kinase family members. Importantly, we observed that MDM2 destabilization coincided with the duration of p53 transcriptional activation, and that blocking MDM2 degradation via proteasome inhibition prevented p53 transactivation in cells with DNA damage. Because the half-life of MDM2 in DNA-damaged cells was as short as 5 min, we suggest that interaction between MDM2 and p53 may occur only infrequently, since under damaging conditions MDM2 may spend most of its existence in processes related to its own ubiquitination and proteasome-mediated degradation.
Results
Proteasome inhibition leads to nuclear accumulation of p53
The hypothesis that p53 is degraded by cytoplasmic proteasomes predicts that proteasome inhibition should increase cytoplasmic p53. We analyzed the subcellular localization of endogenous p53 and HDM2 (human MDM2) in normal human fibroblasts (WS1) treated with inhibitors of the three major proteolytic activities of the proteasome (epoxomicin, NLVS, and MG132 (PI); Figure 1A). The subcellular localization of p53 and HDM2 was quantified by calculating nuclear-to-cytoplasmic (N/C) pixel intensity ratios from images of immunofluorescently stained cells (Figure 1B; see Materials and methods). In untreated cells, p53 and HDM2 staining was faint but predominantly nuclear (Figures 1A and B). Interestingly, while PI treatment increased both p53 and HDM2 protein levels predominantly in the nucleus (Figures 1A–C), it neither activated p53 target gene expression nor resulted in p53 phosphorylation at S15 or acetylation at K382 (Figures 1C and D). A possible explanation for the failure of PIs to activate p53 despite increasing p53 abundance is discussed below (see Figure 7). These data reveal that a significant effect of proteasome inhibition is to increase p53 and HDM2 nuclear abundance, suggesting that the nucleus might be an important site for their degradation.
Figure 1.
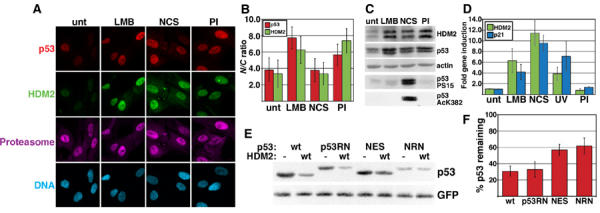
p53 can be degraded in the nucleus. (A) p53, HDM2, and proteasome subcellular localization in normal human fibroblasts (WS1). Cells were left untreated (unt), or treated with 20 nM LMB for 6 h, 640 ng/ml NCS for 4 h, or PIs for 4 h, and stained with antibodies to p53, HDM2, and the proteasome. (B) p53 and HDM2 nuclear:cytoplasmic (N/C) ratios for immunofluorescently stained WS1. See Materials and methods for determination of p53 and HDM2 N/C ratios. p53 ratios are depicted by gray bars and HDM2 ratios by white bars. Values represent the mean (±s.d.) of >30 cells. (C) Immunoblots of p53, HDM2, p53 phosphorylated S15 (PS15), and p53 acetylated lysine 382 (AcK382). WS1 cells were treated as in (A), then immunoblotted with antibodies to p53 (DO-1), HDM2 (IF2), and actin. The membrane was stripped and re-probed with an antibody to p53 PS15. Separate samples were lysed in acetylation lysis buffer, then stained with an antibody to AcK382. (D) p53 target gene induction. cDNA was harvested from WS1 that were untreated; LMB, NCS, or PI-treated (see (A)); or UV irradiated. HDM2 (white bars) or p21 (gray bars) gene induction was measured by real-time quantitative PCR. Induction is expressed as fold induction relative to untreated. Error bars indicate the 95% CI of ≥3 experiments. (E) Degradation of cytoplasmic p53 by HDM2. 2KO or SaOS-2 cells (data not shown) were transfected with plasmids containing luciferase, GFP, 150 ng p53 (wt, p53RN, NES, or NRN), and 1.2 μg HDM2 (wt) or empty vector (pcDNA3, (−)). Equal luciferase units of transfected cell lysates were immunoblotted with antibodies to p53 or GFP. (F) Densitometric analysis of p53 Western blots. Percent p53 remaining was calculated as the ratio of p53 co-transfected with wt HDM2 to p53, with empty vector normalized to GFP. Bars represent the mean band intensity±1 s.d. for >3 experiments.
Figure 7.
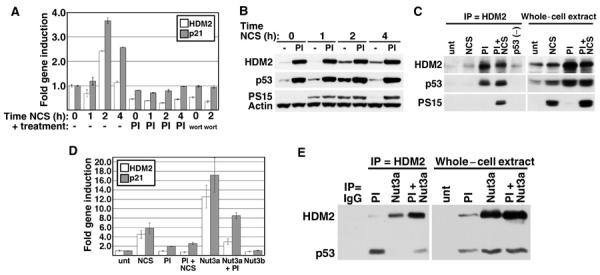
Proteasome inhibition prevents p53 activation by DNA damage. (A) HDM2 and p21 gene induction in PI-treated cells. WS1 cells were left untreated (bar 1), or treated with 50 ng/ml NCS for 1, 2, or 4 h (bars 2–4), PI alone for 4 h (bar 5), PI followed by concurrent treatment with NCS for 1, 2, or 4 h (bars 6–8), wortmannin (wort) alone for 30 min (bar 9), or wortmannin followed by concurrent treatment with NCS for 2 h (bar 10). RNA was harvested and subjected to real-time quantitative PCR as in Figure 1. Bars indicate the 95% CI of the mean induction of HDM2 (white) or p21 (gray). (B) p53 S15 phosphorylation status in cells treated with PI and NCS. WS1 cells were treated as in (A), then lysates were run on a 4–12% gradient gel and immunoblotted with antibodies to HDM2, phosphorylated p53 S15 (PS15), and actin, then stripped and re-probed with an antibody to unmodified p53 (DO-1). (C) HDM2/p53 association in PI and NCS-treated cells. One mg of WS1 or SaOS-2 (p53 (−)) whole-cell extract was immunoprecipitated with an HDM2 antibody (IF2, or SMP14 or 4B2, data not shown), then immunoblotted with antibodies to HDM2 and p53 (Ab-7). The blot was stripped and re-probed with p53 S15 antibody. (D) HDM2 and p21 gene induction in Nutlin-treated cells. WS1 cells were incubated in 10 μM Nutlin-3a (Nut3a) or 3b for 4 h, then PI for 4 h, then 50 ng/ml NCS for 3 h. Gene induction was analyzed as in (A). (E) HDM2/p53 association in Nutlin-treated cells. WS1 cells were treated as in (D), then 700 μg of whole-cell extract was immunoprecipitated with antibodies to HDM2 (IF2) or nonspecific IgG and immunoblotted with antibodies to HDM2 and p53 as in (C).
We also compared the subcellular distribution of p53 and HDM2 using other conditions reported to increase p53 abundance. Leptomycin B (LMB), an antibiotic that blocks nuclear export, increased the nuclear localization of both p53 and HDM2 two-fold, consistent with previous reports (Freedman and Levine, 1998; Kudo et al, 1998; Lain et al, 1999). Although LMB activated p53 as observed previously, we found that this occurred without S15 phosphorylation or K382 acetylation (Figures 1C and D; Lain et al, 1999; Menendez et al, 2003). One explanation for the ability of LMB to activate p53 is that it diminishes binding by HDM2 (Lain et al, 1999). While previous studies showing that LMB stabilizes nuclear p53 were interpreted as evidence that p53 nuclear export is required for its degradation, the observation that LMB activates p53 indicates that this drug has pleiotropic effects and thus experiments using it should be interpreted with caution.
We also analyzed the effects of the radiomimetic drug neocarzinostatin (NCS; Povirk, 1996). NCS increased p53 and HDM2 protein levels to the same extent in the nucleus and cytoplasm, and activated p53-dependent transcription similarly to LMB (Figures 1A–D). The proteasome localized to both compartments and none of the treatments changed its subcellular distribution (Figure 1A; data not shown).
p53 is degraded in the nucleus without nuclear-cytoplasmic shuttling
The above data suggest that p53 degradation might occur in the nucleus. We found that the levels of both wt p53 and an NES mutant decreased significantly in the presence of co-transfected HDM2, but the NES mutant was degraded only about half as well as wt p53 (Figures 1E and F). The p53 NES mutant is both constitutively nuclear and dimeric, while wt p53 is mainly tetrameric (Stommel et al, 1999). Therefore, we determined whether the attenuated degradation of the NES mutant was due to its nuclear localization or from its altered quaternary structure. We added a potent viral NES derived from HTLV Rex to the C-terminus of the p53 NES mutant to generate NRN, which is dimeric and cytoplasmic (see Supplementary Figure 1). Like the NES mutant, NRN was about two-fold more resistant to HDM2-mediated degradation than wt p53 (Figures 1E and F). A wt p53 construct with the attached Rex NES (p53RN, see Supplementary Figure 1) was degraded to the same extent as p53 without the exogenous NES, indicating that cytoplasmic localization does not enhance p53 degradation. Therefore, quaternary structure appears to be more important than subcellular localization for HDM2-mediated p53 degradation, and HDM2 can degrade p53 in either the nucleus or the cytoplasm.
It is possible that wt p53 and the NES mutant were re-localized to the cytoplasm and then degraded by co-expressed HDM2. Supplementary Figure 1 shows that wt p53, the NES mutant, and a mutant defective in HDM2 binding (Lin et al, 1994), all remained predominantly nuclear when PIs were added to cells co-transfected with HDM2, even though this treatment led to a four-fold increase in p53 levels and an accumulation of ubiquitinated species (data not shown). In addition, p53RN and NRN each remained strongly cytoplasmic in the presence or absence of PIs. Together, these results suggest that p53 degradation can occur in either the nucleus or the cytoplasm, without a prior requirement for nuclear-cytoplasmic shuttling.
HDM2 is unstable in DNA-damaged cells
HDM2 degrades nuclear p53 (Figure 1) and binds p53 on DNA (Jin et al, 2002), indicating that the nucleus is the major site where HDM2 antagonizes p53. As many stresses stabilize and activate p53 while concurrently increasing nuclear HDM2, p53 activation can only occur if stress-dependent mechanisms prevent HDM2 from inhibiting p53. Since N-terminal phosphorylations seem inadequate to explain p53 stabilization and transcriptional activation in stressed cells, we determined whether additional mechanisms mitigate the effects of HDM2 following DNA damage.
In unstressed U2OS cells co-transfected with wt p53 and HDM2, we detected HDM2 in only 33% of the cells expressing nuclear p53 (Figures 2A and B). In contrast, proteasome inhibition or conditions that prevented p53 ubiquitination increased the number of cells co-expressing these proteins to >80% (Figures 2A and B; data not shown). The difference in co-expression frequency between transfected U2OS and normal human fibroblasts (Figure 1) or SaOS-2 (Supplementary Figure 1) is likely because U2OS cells are ARF null, which enhances p53 degradation (data not shown). Although DNA damage prevents the degradation of p53 by HDM2, NCS did not increase the fraction of co-transfected cells expressing both proteins (Figures 2A and B). Interestingly, the low incidence of p53 and HDM2 co-expression resulted from a decrease in the stability of transfected HDM2 after DNA damage, which was accompanied by a corresponding increase in the stability of endogenous p53 (Figures 2C and D).
Figure 2.
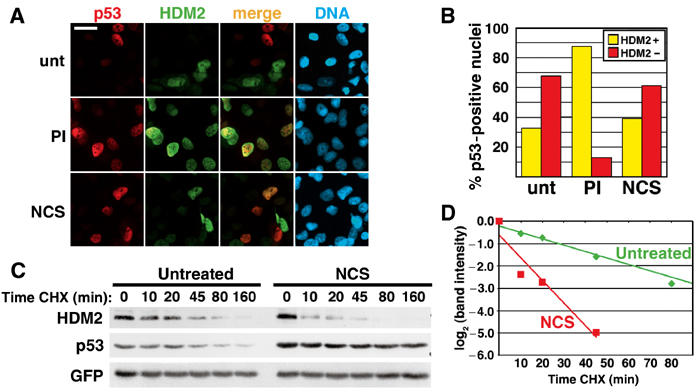
DNA damage destabilizes HDM2. (A) p53 and HDM2 immunofluorescence staining in transfected U2OS. Cells were co-transfected with 200 ng p53 and 1.6 μg HDM2, and left untreated (unt) or treated with PI or 640 ng/ml NCS for 4 h. Yellow cells in the ‘merge' panels indicate cells expressing both p53 and HDM2. Bar=20 μm. (B) Quantitation of p53 and HDM2 nuclear co-incidence. Cells with nuclear p53 staining were counted for the presence (HDM2+, yellow bars) or absence (HDM2−, red bars) of HDM2 co-expression, and displayed as a percentage of the total number of p53-positive cells. Greater than 347 cells were counted for each condition. (C) Half-life of HDM2 in transfected cells. U2OS cells were transfected with 2 μg HDM2 and 125 ng GFP. Transfected cells were treated with 640 ng/ml NCS for 4 h, then concurrently with carrier (t=0) or cycloheximide (CHX) for the indicated times. This figure is representative of >3 experiments. (D) Quantitation of HDM2 destabilization in (C). HDM2 band intensity was normalized to GFP, then normalized to the t=0 controls. Each decrease of one unit of log2 (band intensity) is equivalent to one half-life.
The steady-state levels of HDM2 protein increase in NCS-treated human fibroblasts due to p53-mediated transcriptional induction of endogenous HDM2. This results in cells in which HDM2 and p53 are at high levels in the nucleus simultaneously (see Figure 1). Presumably, endogenous HDM2 and p53 can both be readily detected in stressed normal cells due to stress-activated pathways that prevent p53 degradation. We asked whether rapid HDM2 turnover might also contribute to p53 regulation in DNA-damaged normal human fibroblasts. Figures 3A and B show that while NCS treatment increased HDM2 steady-state protein levels, the half-life of HDM2 decreased about seven-fold. The shorter HDM2 half-life coincided with an increase in p53 half-life, and was observed with two widely different doses of NCS (640 ng/ml, Figure 3A; 50 ng/ml, Figures 4 and 6). HDM2 destabilization is likely a general response to DNA damage since UV (Figures 3C and D) and the DNA alkylator BCNU (Figure 3E) also reduced HDM2 half-life. These experiments reveal that one effect of DNA damage is to decrease HDM2 stability, suggesting an additional mechanism of activating p53 through selective elimination of its negative regulator.
Figure 3.
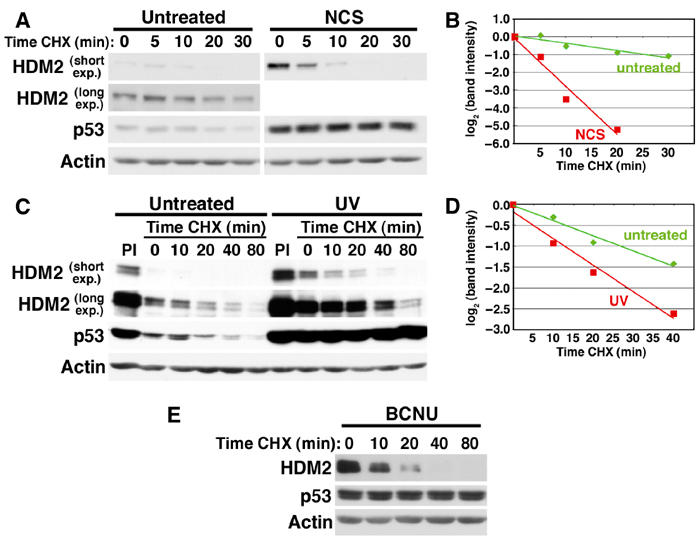
Multiple stresses decrease HDM2 half-life in normal human fibroblasts. (A) HDM2 half-life in NCS-treated WS1 cells. WS1 were left untreated or treated with 640 ng/ml NCS for 4 h. Protein synthesis was inhibited by concurrent treatment with CHX for the indicated times. Steady-state levels of HDM2 are higher in stressed cells due to increased p53-dependent gene activation; therefore, for comparison of the half-lives of HDM2 with and without DNA damage, short and long film exposures (short exp. and long exp.) are shown. This figure is representative of >3 experiments. (B) Quantitation of NCS-induced HDM2 destabilization in (A). HDM2 band intensity was normalized to actin, then expressed relative to the t=0 controls. Each decrease of one unit of log2 (band intensity) is equivalent to one half-life. (C) HDM2 half-life in UV-irradiated WS1 cells. WS1 were left untreated or exposed to 26 J/m2 UVC irradiation 16 h before incubation with CHX for the indicated times. PIs were added to UV-irradiated cells 12 h after UV exposure, then cells were harvested 4 h later. (D) Quantitation of UV-induced HDM2 destabilization in (C). The blot was scanned and analyzed as in (B). (E) HDM2 half-life in BCNU-treated WS1 cells. WS1 were left untreated (data not shown) or incubated with 100 μM BCNU 8 h prior to incubation with CHX for the indicated times.
Figure 4.
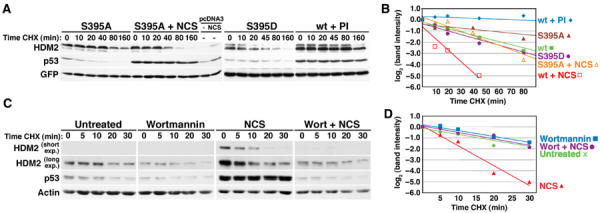
HDM2 destabilization is regulated by PI 3-kinase related kinases. (A) Half-life of HDM2 ATM phosphorylation site mutants. U2OS cells were transfected with 2 μg of HDM2 (S395A, S395D, or wt) or pcDNA3 and 125 ng GFP. Transfected cells were treated with 640 ng/ml NCS or PI for 4 h, then CHX for the indicated times. Different exposures of each transfection are shown to facilitate comparison of HDM2 stability. (B) Quantitation of HDM2 destabilization in (A) and Figure 2C. HDM2 band intensity was normalized to GFP, then normalized to the t=0 controls. (C) Half-life of HDM2 in wortmannin-treated cells. Normal human fibroblasts (WS1) were left untreated or were treated with 50 μM wortmannin for 30 min, 50 ng/ml NCS for 2 h, or wortmannin, then NCS. Carrier (t=0) or CHX was added for the indicated times. This figure is representative of three experiments. (D) Quantitation of HDM2 destabilization in (C). HDM2 band intensity was normalized to actin, then normalized to the t=0 controls. Each decrease of one unit of log2 (band intensity) is equivalent to one half-life.
Figure 6.
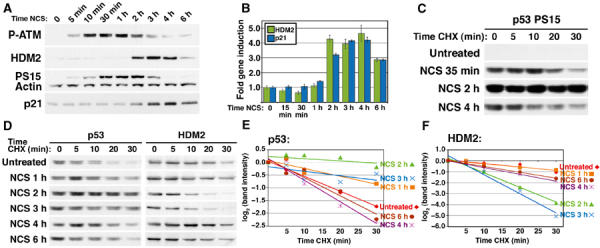
HDM2 destabilization correlates temporally with p53 transcriptional activity. (A) Timecourse of ATM and p53 phosphorylation. WS1 cells were treated with 50 ng/ml NCS for the indicated times. Lysates was separated on a 3–8% Tris–acetate gel and Western blots were probed with antibodies to p53-phosphorylated S15 (PS15), HDM2, phosphorylated ATM (P-ATM), and actin. A second 4–12% Tris–glycine gel was immunoblotted and probed with antibodies to p21 and actin (data not shown). (B) Timecourse of p53 target gene activation in NCS-treated WS1 cells. Cells were treated with 50 ng/ml NCS for the indicated times, and real-time quantitative PCR was performed with primers to HDM2 (white) and p21 (gray). Bars represent one s.d. of mean gene induction normalized to untreated. (C) Half-life of phosphorylated S15 p53 at early and late treatments of NCS. WS1 cells were treated with 50 ng/ml NCS and CHX for the indicated times, then Western blotted with antibodies to phosphorylated p53 S15 and actin (data not shown). To compare the steady-state p53 levels, this image was compiled from one film exposure. (D) Timecourse of p53 and HDM2 stability. WS1 cells were treated with 50 ng/ml NCS and CHX for the indicated times, then lysates were immunoblotted with antibodies to unmodified p53 (DO-1), HDM2, and actin (data not shown). This figure is representative of three experiments. As p53 and HDM2 steady-state levels varied at different times of NCS treatment, images from films exposed for different time periods are shown to better compare p53 and HDM2 half-lives (see Figures 4 and 5). (E) Quantitation of p53 half-life in (D). p53 band intensity was normalized to actin (data not shown), then normalized to the t=0 controls. Each decrease of one unit of log2 (band intensity) is equivalent to one half-life. (F) Quantitation of HDM2 half-life in (D). HDM2 half-life was quantitated as for p53.
Stress-activated PI 3-kinase family members regulate HDM2 stability
Because ATM phosphorylates HDM2 (Michael and Oren, 2003) and is activated by NCS, we examined whether ATM regulates HDM2 stability. HDM2 in an ATM mutant cell line was still partially destabilized after DNA damage (data not shown) and mutation of the HDM2 S395 ATM phosphorylation site to aspartate only marginally destabilized it in unstressed cells (Figures 4A and B). Conversely, an HDM2 S395A construct was partially stabilized in unstressed cells and was destabilized by DNA damage about half as well as wt HDM2 (Figures 4A and B, compare with Figure 2C). These data suggest that multiple kinases and phosphorylation sites might be required for DNA-damage-dependent HDM2 destabilization. We therefore analyzed HDM2 half-life in cells given wortmannin prior to DNA damage, as this drug inhibits the activity of multiple DNA-damage-activated PI 3-kinase family members including ATM, ATR, and DNA-PK (Sarkaria et al, 1998). Interestingly, this drug completely abrogated HDM2 destabilization after NCS (Figures 4C and D). Wortmannin pretreatment also blocked the damage-induced increase in p53 abundance (Figure 4C) and prevented transcription of HDM2 and p21 RNA in NCS-treated cells (see Figure 7A), suggesting that damage-dependent destabilization of HDM2 might be important for these activities. Wortmannin inhibits kinases in addition to those involved in the damage response, including PI 3-kinase, which increases HDM2 levels via Akt (Ashcroft et al, 2002). However, because the half-life of HDM2 in cells treated with wortmannin alone was the same as that in untreated cells (Figures 4C and D), we infer that the effects of wortmannin on HDM2 stability after NCS treatment are likely to be explained by its inhibition of damage-activated kinases.
The HDM2 RING domain is required for damage-induced destabilization
HDM2 is a RING-domain-dependent E3 ubiquitin ligase that mediates the ubiquitination of both p53 and itself (Fang et al, 2000; Honda and Yasuda, 2000). We therefore determined whether the DNA-damage-induced destabilization of HDM2 was dependent on its intrinsic ubiquitin ligase activity by analyzing the stability of an HDM2 RING domain mutant (HDM2 C464A) in stressed cells. The C464A mutant was much more stable than wt HDM2 in unstressed transfected U2OS cells (compare the untreated lanes in Figure 5 to untreated in Figure 2C). Moreover, the RING domain mutant was not destabilized in cells treated with NCS (Figure 5). These data suggest that HDM2 auto-ubiquitination controls its stability in unstressed cells, and this process is accelerated by stress-activated PI 3-kinase signaling pathways in cells sustaining DNA damage.
Figure 5.
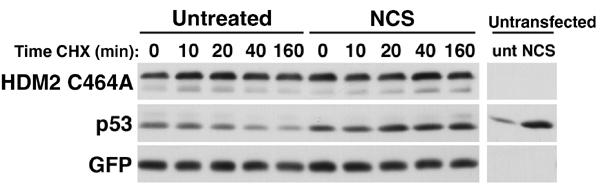
The HDM2 RING domain is required for DNA-damage-induced destabilization. U2OS cells were transfected with 2 μg of HDM2 RING domain mutant C464A and 125 ng GFP. Transfected cells were treated with 640 ng/ml NCS, then CHX for the indicated times. Untransfected cells were left untreated or incubated in NCS.
The timing of p53 transactivation coincides with HDM2 destabilization
DNA-damage signaling to p53 typically results in S15 phosphorylation by activated ATM or ATR, followed by p53 protein stabilization and target gene activation (Appella and Anderson, 2001). We therefore evaluated the kinetics of p53 activation and HDM2 destabilization with regard to these parameters. ATM was activated within minutes of NCS treatment, as indicated by its phosphorylation at S1981 (P-ATM; Figure 6A; Bakkenist and Kastan, 2003). However, this form declined after 2–3 h, possibly indicating a resolution of the DNA damage response at the low NCS dose used. Increased p21 and HDM2 RNA levels were first measurable between 1 and 2 h after NCS addition and declined after 4–6 h (Figure 6B). p53 stabilization occurred with kinetics similar to transactivation: p53 became stable between 1 and 2 h after NCS addition, but its half-life decreased again by 4–6 h, the time at which target gene activation waned (Figures 6D and E). Interestingly, the half-life of HDM2 inversely correlated with p53 transcriptional activity and stability: between 1 and 2 h after drug addition, HDM2 stability decreased, while p53 transcriptional activity and stability increased (Figures 6B and D–F). Furthermore, HDM2 stability approached that of the unstressed cells by 4–6 h, just as p53 became unstable and less transcriptionally active (see Figure 8 for discussion). In contrast, S15 phosphorylation did not correlate with either p53 transcriptional activity or stability. Not only was p53 phosphorylated at this site more than 1 h prior to the onset of p21 and HDM2 gene induction (Figures 6A and B), this modified form was unstable early and late in the damage response (Figures 6C and 7B). These data indicate that S15 phosphorylation is not sufficient for p53 activation, while showing that HDM2 destabilization correlates with p53 transcriptional activity.
Figure 8.
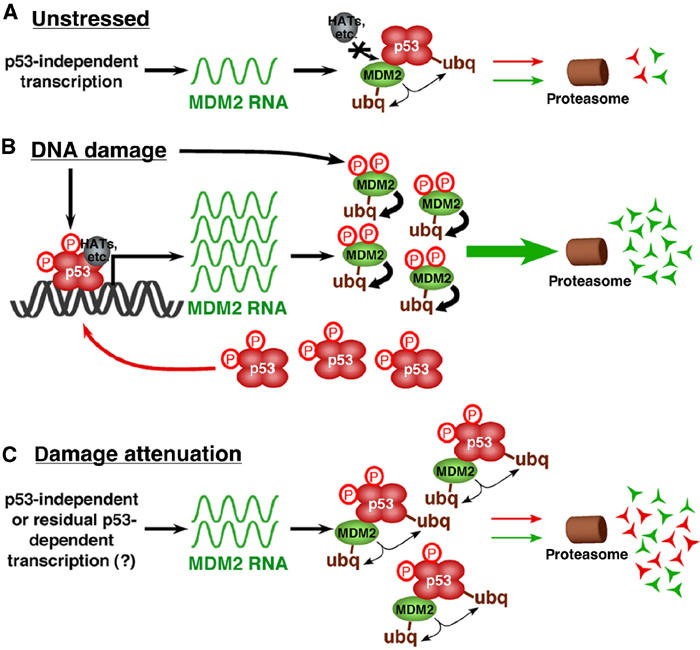
A model for regulated MDM2 destabilization in DNA-damaged cells. (A) In unstressed cells, growth and survival factors activate MDM2 transcription, maintaining basal levels of MDM2 RNA and protein. The resultant MDM2 protein keeps p53 inactive by (1) preventing p53 transactivation by blocking its access to transcriptional co-activators and (2) ubiquitinating the p53 C-terminus leading to its proteasome-mediated degradation. MDM2 auto-ubiquitination keeps its steady-state protein levels low. (B) In DNA-damaged cells, stress-dependent kinases modify MDM2, resulting in its accelerated auto-ubiquitination and proteasome-mediated degradation. MDM2 destabilization frees modified p53 to initiate transcription of high levels of MDM2, as well as genes involved in cell cycle arrest and apoptosis. However, persistent DNA-damage signaling prevents the newly transcribed and translated MDM2 from antagonizing p53 by inducing the rapid degradation of MDM2 protein. This process results in high levels of stable and active p53 that is maintained concurrently with high levels of unstable and ineffective MDM2. (C) Once the DNA damage is repaired, damage kinase signaling abates and re-stabilized MDM2 can bind and inactivate the p53 that had previously accumulated to high levels. Thus, DNA-damage-induced p53-dependent transactivation of MDM2 might provide the cell with a pool of MDM2 that can eliminate active p53 at later times when the stress is alleviated.
Blocking protein degradation prevents p53 transactivation
The data presented above suggest that HDM2 destabilization plays a role in p53 target gene activation. If this is correct, then stabilizing HDM2 during a stress should prevent p53 activity. We therefore blocked HDM2 degradation with PIs prior to adding NCS, then assayed for p53 transcriptional activity in normal human fibroblasts. Figure 7A shows that pretreatment with PIs prevented the NCS-mediated induction of p21 and HDM2 gene expression as effectively as wortmannin. The inhibition of p53 transactivation by PIs was not due to a reduction of damage-induced phosphorylation, as the level of S15-phosphorylated p53 was as high in PI+NCS-treated cells as it was in cells treated with NCS alone (Figure 7B). As HDM2 steady-state protein levels increased dramatically in cells treated with both PIs and NCS (Figure 7B), the inhibition of p53 transcriptional activity in proteasome-inhibited DNA-damaged cells might be due to enhanced negative regulation by over-abundant HDM2. We therefore measured the ability of HDM2 to bind p53 in cells treated with PI+NCS. Figure 7C shows that while p53 did not co-immunoprecipitate with HDM2 in cells treated with NCS alone, HDM2-bound p53 was readily detected in cells in which protein degradation was blocked prior to NCS treatment. Interestingly, this HDM2-bound p53 was phosphorylated at S15, suggesting that the instability of this modified form at early and late times of NCS treatment is due to its association with a relatively stable HDM2 (see Figure 6C). We also treated the cells with Nutlin-3a, a small molecule that blocks the binding of HDM2 to p53 (Vassilev et al, 2004), prior to adding PIs. Nutlin-3a treatment alone was sufficient to upregulate p53 target gene transcription and this activation was only modestly decreased by pretreatment with PIs, although it remained as high as that observed with NCS (Figure 7D). In contrast, Nutlin-3b, an inactive enantiomer of Nutlin-3a, had no effect on p53 activity. We next determined whether the modest decrease in p53 activity in Nutlin-3a+PI-treated cells was due to an increase in HDM2 binding in cells in which HDM2 protein was stabilized. Figure 7E shows that while Nutlin-3a alone completely blocked the binding of HDM2 to p53, concurrent treatment with PI led to a slight increase in HDM2/p53 association, consistent with the slight decrease in p53 transcriptional activity. Taken together, these data show that rapid HDM2 turnover provides an important mechanism by which the inhibition of p53 transcriptional activity is alleviated in a stress response.
Discussion
Since p53 activation can lead to permanent cell cycle arrest or apoptosis, the appropriate regulation of p53 activity is critical, but the mechanisms by which this is achieved have been challenging to elucidate. Here, we show that the regulation of MDM2 stability provides an important and previously unrecognized mechanism for controlling p53 transcriptional responses.
Our data address the contributions of several proposed mechanisms of p53 regulation. Our analyses of endogenous p53 in normal human fibroblasts and transfected p53 in rodent and human cell lines failed to find evidence that MDM2-mediated ubiquitination of p53 led to its nuclear export and cytoplasmic degradation (Figure 1 and Supplementary Figure 1). On the contrary, we found that MDM2 could degrade p53 in the nucleus. Consistent with this, a recent study showed that a cytoplasmic p53 NLS mutant was ubiquitinated but not degraded, while appending a dominant NLS induced nuclear localization and restored its degradation (O'Keefe et al, 2003). Nuclear degradation of p53 has been reported by other groups (Geyer et al, 2000; Lohrum et al, 2001; Xirodimas et al, 2001; Shirangi et al, 2002); however, its importance in p53 control was not emphasized, probably because the p53 NES mutant is degraded less efficiently than wt p53. This is most likely because p53 NES mutants are monomeric or dimeric, and therefore bind MDM2 inefficiently and are ubiquitinated less well than wt p53 (Mateu and Fersht, 1998; Maki, 1999; Stommel et al, 1999; O'Keefe et al, 2003). However, the dimeric NES mutant that we studied was degraded equally in the nucleus and the cytoplasm (Figure 1), implying that neither subcellular localization nor nuclear-cytoplasmic shuttling is critical in the control of p53 stability. This reconciles the seemingly inconsistent observations that p53 half-life is only about 20–30 min, yet its nuclear export takes between 3 and 8 h.
p53 and MDM2 are largely nuclear proteins, and p53 transcriptionally activates mdm2 in response to many stresses. Therefore, the ability of MDM2 to degrade p53 in the nucleus raises the question of how sustained p53 activation can ever be achieved. Many studies have emphasized the importance of p53 N-terminal phosphorylation in blocking MDM2 binding. However, neither in vivo nor transfection-based systems have revealed a compelling role for these modifications in the control of p53 activity or abundance (Ashcroft et al, 1999; Blattner et al, 1999; Wu et al, 2002). In addition, many agents that activate p53, such as LMB, deferoxamine, actinomycin D, and hypoxia, do so without inducing the stereotypical phosphorylations generated after ionizing or UV radiation (Figure 1; Ashcroft et al, 2000; Koumenis et al, 2001; Menendez et al, 2003). Moreover, mdm2 deficiency or agents that interfere with MDM2 binding activate p53 in cells that are otherwise unstressed (Jones et al, 1995; Montes de Oca Luna et al, 1995; Bottger et al, 1997; Blaydes and Wynford-Thomas, 1998; Vassilev et al, 2004). We observed that p53 S15 was phosphorylated more than 1 h prior to the onset of transcriptional activity, and that S15-phosphorylated p53 was unstable early and late in the damage response (Figure 6). Together, these data imply that N-terminal phosphorylations are not always necessary and are probably not sufficient for p53 activation, and they emphasize the importance of alternative mechanisms that affect the access of MDM2 to p53.
Our data lead us to propose that regulated MDM2 turnover is an important and novel mechanism for effective control of p53 function (Figure 8). We demonstrate that DNA-damaging agents rapidly destabilize MDM2, and that MDM2 destabilization correlated with, and was necessary for, p53 transcriptional activation. MDM2 destabilization requires the activity of DNA-damage-dependent PI 3-kinase family members, most likely through MDM2 phosphorylation at multiple sites (Figure 4; Michael and Oren, 2003). Others have observed that diverse stresses can reduce MDM2 levels through both transcriptional and post-translational mechanisms (Alarcon et al, 1999; Ashcroft et al, 2000; Inoue et al, 2001; Wang et al, 2002), resulting in an increase in p53 levels. By contrast, our data explain how p53 can be active under the more commonly observed conditions in which MDM2 transcription and protein abundance are high. We observed that p53 induced the expression of mdm2 RNA in DNA-damaged cells, resulting in increased MDM2 protein levels (Figure 1). However, it is important to note that steady-state protein levels are a balance of new transcription and translation versus protein degradation. The half-life of MDM2 in DNA-damaged cells is as short as 5 min (Figures 2, 3 and 4). Consequently, MDM2 might be prevented from accessing p53 because it spends most of its existence engaged in processes related to its own ubiquitination and proteasome-mediated degradation. It is reasonable that such degradation occurs in the nucleus as MDM2 is largely nuclear, proteasome inhibition increases its nuclear abundance (Figure 1 and Supplementary Figure 1), and it has a weak NES (Henderson and Eleftheriou, 2000; Lohrum et al, 2001), but a contribution of cytoplasmic degradation cannot be excluded. Previous work indicates that the cell is exquisitely sensitive to MDM2 levels, as a modest decrease in MDM2 in hypomorphic mice led to increased p53-dependent apoptosis in lymphatic and epithelial tissues and decreased proliferation of MEFs (Mendrysa et al, 2003). In addition, MDM2 haploinsufficiency in an Eμ-myc transgenic mouse made p53 more effective in blocking tumorigenesis (Alt et al, 2003). This additive effect occurred even though myc overexpression alone is sufficient to induce p53 activity and N-terminal modifications consistent with a stress response (Vafa et al, 2002). These observations indicate that subtle changes in MDM2 levels may be sufficient to impact p53 activity dramatically.
We found that blocking protein degradation prior to damage induction led to MDM2 stabilization and prevented p53 transactivation, even though p53 was phosphorylated at S15 (Figure 7). This inhibition is unlikely to be due to a global inhibition of transcription, as gene expression profiling has revealed no such effect (Zimmermann et al, 2000). PIs likely prevent p53 transactivation by stabilizing the binding of MDM2 to the p53 transactivation domain, as this treatment enhanced p53/MDM2 association in DNA-damaged cells (Figure 7C). In addition, preventing MDM2 binding by pretreatment with Nutlin-3a mitigated the effects of PIs on p53 transactivation (Figures 7D and E; Vassilev et al, 2004). These observations, along with the dramatic increase in MDM2 levels observed in the PI-treated cells, suggest that controlled degradation of MDM2 following a stress is an important component of p53 activation.
The destabilization of MDM2 in DNA-damaged cells requires its own ubiquitin ligase activity (Figure 5). Interestingly, MDM2 is one of many E3 ubiquitin ligases that catalyze the ubiquitination of both their substrates and themselves (Fang et al, 2000; Honda and Yasuda, 2000). Because E3s mediate their own degradation, mechanisms of attenuating this function must exist to ensure that short-lived E3s are sufficiently stable to gain access to their substrates. These mechanisms are poorly understood thus far. The presence of a mechanism by which MDM2 is specifically destabilized implies the existence of a stress-dependent switch between MDM2-mediated degradation of p53 and itself. This switch may lie within its RING domain, as an MDM2 construct with a heterologous RING domain can catalyze its own ubiquitination but not that of p53 (Fang et al, 2000). Moreover, the RING-domain-binding protein MDMX may be involved in preventing MDM2 auto-ubiquitination, since it stabilizes MDM2 (Sharp et al, 1999) and enhances p53 degradation in transfected cells (Gu et al, 2002). The destabilization of MDM2 by DNA-damage signaling might provide a general model for regulating the activity of E3 ubiquitin ligases by switching their substrate specificity. As so many cancers are derived from cells that inactivate wild-type p53 through the overexpression of MDM2, this switch may represent a novel point of intervention with chemotherapeutic agents.
Materials and methods
Cell culture, transfections, and drug treatments
SaOS-2 (human osteosarcoma, p53−/−), U2OS (human osteosarcoma, p14ARF silenced/p53 wt), and WS1 cells (primary normal human skin fibroblasts) were from ATCC—2KO cells (p53−/−/mdm2−/− mouse embryonic fibroblasts) and were provided by A Levine. WS1 cells were used at population doublings <25. See supplementary information for cell culture and transfection conditions. Leptomycin B (LMB) and neocarzinostatin (NCS) were provided by M Yoshida and R Shultz (Drug Synthesis & Chemistry Branch, National Cancer Institute). The PI cocktail consisted of 10 μM MG-132, 100 nM epoxomicin, and 6 μg/ml NLVS (Calbiochem) in Figure 1, or MG-132 and epoxomicin alone in the remaining figures. Cycloheximide (USBiological) was resuspended in ethanol and used at 100 μg/ml—0.1% ethanol was used for 5 or 10 min as carrier in the half-life experiments. Cells were given 26 J/m2 UVC irradiation (254 nm) at a dose rate of 4.4 J/m2/s. BCNU (Sigma) was resuspended in ethanol immediately before use. Wortmannin (Sigma) was used at 50 μM for 30 min prior to NCS treatments. Nutlin-3a and b were provided by L Vassilev and were used at 10 μM for 11 h.
Construction and mutagenesis of expression plasmids
pEGFP-N1 was purchased from Clontech. See supplementary data for cloning strategies for p53 NES, pcDNAp53, pcDNA22/23, p53RN, NRN, and HDM2 S395D. Wt HDM2 (CHDM1B), HDM2 C464A, HDM2 S395A, and CMX-luciferase were provided by A Levine, K Vousden, M Oren, and R Evans.
Real-time quantitative PCR
Real-time quantitative PCR was performed on an ABI PRISM 7700 Sequence Detection System. See supplementary data for cycling conditions and primer sequences.
Immunofluorescence staining and quantitation
Fixed cells were stained with primary antibodies to p53 (CM1, D Lane), HDM2 (2A9 and 5B10, Calbiochem), and 20S Proteasome (PW8265, Affiniti Research Products). See supplementary data for staining procedure. Images were captured on an epifluorescence microscope using equal exposure times for all images for each fluor. Nuclear/cytoplasmic ratios were determined by calculating total pixel intensity in a circle 4.25 μm in diameter in the nucleus and the cytoplasm using Openlab 3.0 software. The background was determined by calculating pixel intensity in a 4.25 μm circle in cell-free areas in each field, and then subtracting this from the nuclear and cytoplasmic measurements for each cell in the field within each channel. N/C ratios were calculated as invlog ∑log (N/C)/n.
Immunoprecipitations, immunoblotting, and quantitation
See supplementary data for immunoprecipitation, Western blotting, and quantitation procedures. Western blots were probed with antibodies to unmodified p53 (DO-1 or Ab-7 (Calbiochem)), HDM2 (IF2, 4B2 (Calbiochem), and SMP14 (Santa Cruz)), p53 phospho-serine 15 (Cell Signaling Technologies), p53 acetylated lysine 382 (Oncogene), α-actin (Sigma), GFP (Chemicon), p21 (C-19, Santa Cruz), or ATM phospho-serine 1981 (Rockland, Inc.).
Supplementary Material
Supplementary Figure 1
Supplementary Material
Acknowledgments
We thank Tony Hunter, Matthew Weitzman, Jan Karlseder, Franck Toledo, Mark Wade, and Ee Tsin Wong for thoughtful readings of the manuscript. We also thank John Kolman for assistance with quantitative PCR, Minoru Yoshida for providing Leptomycin B, and Lyubomir Vassilev for providing Nutlin-3. This work was supported by grants from the NIH (CA61449), the G Harold and Leila Y Mathers Charitable Foundation, and the HA & Mary K Chapman Charitable Trust to GMW. This material is based upon work supported under a National Science Foundation Graduate Fellowship awarded to JMS.
References
- Alarcon R, Koumenis C, Geyer RK, Maki CG, Giaccia AJ (1999) Hypoxia induces p53 accumulation through MDM2 down-regulation and inhibition of E6-mediated degradation. Cancer Res 59: 6046–6051 [PubMed] [Google Scholar]
- Alt JR, Greiner TC, Cleveland JL, Eischen CM (2003) Mdm2 haplo-insufficiency profoundly inhibits Myc-induced lymphomagenesis. EMBO J 22: 1442–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appella E, Anderson CW (2001) Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem 268: 2764–2772 [DOI] [PubMed] [Google Scholar]
- Ashcroft M, Kubbutat MH, Vousden KH (1999) Regulation of p53 function and stability by phosphorylation. Mol Cell Biol 19: 1751–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft M, Ludwig RL, Woods DB, Copeland TD, Weber HO, MacRae EJ, Vousden KH (2002) Phosphorylation of HDM2 by Akt. Oncogene 21: 1955–1962 [DOI] [PubMed] [Google Scholar]
- Ashcroft M, Taya Y, Vousden KH (2000) Stress signals utilize multiple pathways to stabilize p53. Mol Cell Biol 20: 3224–3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421: 499–506 [DOI] [PubMed] [Google Scholar]
- Blattner C, Tobiasch E, Litfen M, Rahmsdorf HJ, Herrlich P (1999) DNA damage induced p53 stabilization: no indication for an involvement of p53 phosphorylation. Oncogene 18: 1723–1732 [DOI] [PubMed] [Google Scholar]
- Blaydes JP, Wynford-Thomas D (1998) The proliferation of normal human fibroblasts is dependent upon negative regulation of p53 function by mdm2. Oncogene 16: 3317–3322 [DOI] [PubMed] [Google Scholar]
- Bottger A, Bottger V, Sparks A, Liu WL, Howard SF, Lane DP (1997) Design of a synthetic Mdm2-binding mini protein that activates the p53 response in vivo. Curr Biol 7: 860–869 [DOI] [PubMed] [Google Scholar]
- Boyd SD, Tsai KY, Jacks T (2000) An intact HDM2 RING-finger domain is required for nuclear exclusion of p53. Nat Cell Biol 2: 563–568 [DOI] [PubMed] [Google Scholar]
- Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD (1999) Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc Natl Acad Sci USA 96: 13777–13782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AL, Burch L, Vojtesek B, Mikutowska J, Thompson A, Hupp TR (1999) Novel phosphorylation sites of human tumour suppressor protein p53 at Ser20 and Thr18 that disrupt the binding of mdm2 (mouse double minute 2) protein are modified in human cancers. Biochem J 342: 133–141 [PMC free article] [PubMed] [Google Scholar]
- Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM (2000) Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem 275: 8945–8951 [DOI] [PubMed] [Google Scholar]
- Freedman DA, Levine AJ (1998) Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol Cell Biol 18: 7288–7293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer RK, Yu ZK, Maki CG (2000) The MDM2 RING-finger domain is required to promote p53 nuclear export. Nat Cell Biol 2: 569–573 [DOI] [PubMed] [Google Scholar]
- Gu J, Kawai H, Nie L, Kitao H, Wiederschain D, Jochemsen AG, Parant J, Lozano G, Yuan ZM (2002) Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J Biol Chem 277: 19251–19254 [DOI] [PubMed] [Google Scholar]
- Henderson BR, Eleftheriou A (2000) A comparison of the activity, sequence specificity, and CRM1-dependence of different nuclear export signals. Exp Cell Res 256: 213–224 [DOI] [PubMed] [Google Scholar]
- Honda R, Yasuda H (2000) Activity of MDM2, a ubiquitin ligase, toward p53 or itself is dependent on the RING finger domain of the ligase. Oncogene 19: 1473–1476 [DOI] [PubMed] [Google Scholar]
- Inoue T, Geyer RK, Yu ZK, Maki CG (2001) Downregulation of MDM2 stabilizes p53 by inhibiting p53 ubiquitination in response to specific alkylating agents. FEBS Lett 490: 196–201 [DOI] [PubMed] [Google Scholar]
- Jin Y, Zeng SX, Dai MS, Yang XJ, Lu H (2002) MDM2 inhibits PCAF (p300/CREB-binding protein-associated factor)-mediated p53 acetylation. J Biol Chem 277: 30838–30843 [DOI] [PubMed] [Google Scholar]
- Jones SN, Roe AE, Donehower LA, Bradley A (1995) Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378: 206–208 [DOI] [PubMed] [Google Scholar]
- Kobet E, Zeng X, Zhu Y, Keller D, Lu H (2000) MDM2 inhibits p300-mediated p53 acetylation and activation by forming a ternary complex with the two proteins. Proc Natl Acad Sci USA 97: 12547–12552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koumenis C, Alarcon R, Hammond E, Sutphin P, Hoffman W, Murphy M, Derr J, Taya Y, Lowe SW, Kastan M, Giaccia A (2001) Regulation of p53 by hypoxia: dissociation of transcriptional repression and apoptosis from p53-dependent transactivation. Mol Cell Biol 21: 1297–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo N, Wolff B, Sekimoto T, Schreiner EP, Yoneda Y, Yanagida M, Horinouchi S, Yoshida M (1998) Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp Cell Res 242: 540–547 [DOI] [PubMed] [Google Scholar]
- Lain S, Midgley C, Sparks A, Lane EB, Lane DP (1999) An inhibitor of nuclear export activates the p53 response and induces the localization of Hdm2 and p53 to U1A-positive nuclear bodies associated with the PODs. Exp Cell Res 248: 457–472 [DOI] [PubMed] [Google Scholar]
- Lin J, Chen J, Elenbaas B, Levine AJ (1994) Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev 8: 1235–1246 [DOI] [PubMed] [Google Scholar]
- Lohrum MA, Woods DB, Ludwig RL, Balint E, Vousden KH (2001) C-terminal ubiquitination of p53 contributes to nuclear export. Mol Cell Biol 21: 8521–8532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki CG (1999) Oligomerization is required for p53 to be efficiently ubiquitinated by MDM2. J Biol Chem 274: 16531–16535 [DOI] [PubMed] [Google Scholar]
- Mateu MG, Fersht AR (1998) Nine hydrophobic side chains are key determinants of the thermodynamic stability and oligomerization status of tumour suppressor p53 tetramerization domain. EMBO J 17: 2748–2758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendrysa SM, McElwee MK, Michalowski J, O'Leary KA, Young KM, Perry ME (2003) mdm2 Is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol Cell Biol 23: 462–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez S, Higgins M, Berkson RG, Edling C, Lane DP, Lain S (2003) Nuclear export inhibitor leptomycin B induces the appearance of novel forms of human Mdm2 protein. Br J Cancer 88: 636–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael D, Oren M (2003) The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol 13: 49–58 [DOI] [PubMed] [Google Scholar]
- Momand J, Jung D, Wilczynski S, Niland J (1998) The MDM2 gene amplification database. Nucleic Acids Res 26: 3453–3459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ (1992) The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69: 1237–1245 [DOI] [PubMed] [Google Scholar]
- Montes de Oca Luna R, Wagner DS, Lozano G (1995) Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378: 203–206 [DOI] [PubMed] [Google Scholar]
- O'Keefe K, Li H, Zhang Y (2003) Nucleocytoplasmic shuttling of p53 is essential for MDM2-mediated cytoplasmic degradation but not ubiquitination. Mol Cell Biol 23: 6396–6405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B (1993) Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 362: 857–860 [DOI] [PubMed] [Google Scholar]
- Povirk LF (1996) DNA damage and mutagenesis by radiomimetic DNA-cleaving agents: bleomycin, neocarzinostatin and other enediynes. Mutat Res 355: 71–89 [DOI] [PubMed] [Google Scholar]
- Sakaguchi K, Saito S, Higashimoto Y, Roy S, Anderson CW, Appella E (2000) Damage-mediated phosphorylation of human p53 threonine 18 through a cascade mediated by a casein 1-like kinase. Effect on Mdm2 binding. J Biol Chem 275: 9278–9283 [DOI] [PubMed] [Google Scholar]
- Sarkaria JN, Tibbetts RS, Busby EC, Kennedy AP, Hill DE, Abraham RT (1998) Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res 58: 4375–4382 [PubMed] [Google Scholar]
- Sharp DA, Kratowicz SA, Sank MJ, George DL (1999) Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J Biol Chem 274: 38189–38196 [DOI] [PubMed] [Google Scholar]
- Sherr CJ (2001) The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol 2: 731–737 [DOI] [PubMed] [Google Scholar]
- Shirangi TR, Zaika A, Moll UM (2002) Nuclear degradation of p53 occurs during down-regulation of the p53 response after DNA damage. FASEB J 16: 420–422 [DOI] [PubMed] [Google Scholar]
- Stommel JM, Marchenko ND, Jimenez GS, Moll UM, Hope TJ, Wahl GM (1999) A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J 18: 1660–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolbert D, Lu X, Yin C, Tantama M, Van Dyke T (2002) p19(ARF) is dispensable for oncogenic stress-induced p53-mediated apoptosis and tumor suppression in vivo. Mol Cell Biol 22: 370–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, Wahl GM (2002) c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell 9: 1031–1044 [DOI] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303: 844–848 [DOI] [PubMed] [Google Scholar]
- Wahl GM, Carr AM (2001) The evolution of diverse biological responses to DNA damage: insights from yeast and p53. Nat Cell Biol 3: E277–E286 [DOI] [PubMed] [Google Scholar]
- Wang X, Michael D, de Murcia G, Oren M (2002) p53 Activation by nitric oxide involves down-regulation of Mdm2. J Biol Chem 277: 15697–15702 [DOI] [PubMed] [Google Scholar]
- Wu Z, Earle J, Saito S, Anderson CW, Appella E, Xu Y (2002) Mutation of mouse p53 Ser23 and the response to DNA damage. Mol Cell Biol 22: 2441–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xirodimas DP, Stephen CW, Lane DP (2001) Cocompartmentalization of p53 and Mdm2 is a major determinant for Mdm2-mediated degradation of p53. Exp Cell Res 270: 66–77 [DOI] [PubMed] [Google Scholar]
- Zimmermann J, Erdmann D, Lalande I, Grossenbacher R, Noorani M, Furst P (2000) Proteasome inhibitor induced gene expression profiles reveal overexpression of transcriptional regulators ATF3, GADD153 and MAD1. Oncogene 19: 2913–2920 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Material