

Abstract
Abasic sites are common DNA lesions, which are strong blocks to replicative polymerases and are potentially mutagenic when bypassed. We report here the 2.8 Å structure of the bacteriophage RB69 replicative DNA polymerase attempting to process an abasic site analog. Four different complexes were captured in the crystal asymmetric unit: two have DNA in the polymerase active site whereas the other two molecules are in the exonuclease mode. When compared to complexes with undamaged DNA, the DNA surrounding the abasic site reveals distinct changes suggesting why the lesion is so poorly bypassed: the DNA in the polymerase active site has not translocated and is therefore stalled, precluding extension. All four molecules exhibit conformations that differ from the previously published structures. The polymerase incorporates dAMP across the lesion under crystallization conditions, indicating that the different conformations observed in the crystal may be part of the active site switching reaction pathway.
Keywords: abasic site, DNA lesion, replicative DNA polymerase, structure
Introduction
Abasic sites are the most frequent DNA lesion encountered by cells; roughly 10 000 are formed per human cell per day (Lindahl, 1993). These sites of base loss primarily arise through depurination, although they are also produced by hydroxyl radical attack on the sugar residue releasing the free base (Breen and Murphy, 1995). Sites of base loss are also intermediates in the base excision repair process initiated by monofunctional DNA glycosylases (Wallace, 1997). The interaction between abasic sites and DNA polymerases has been studied for some time both in vitro and in cells (for reviews, see Loeb and Preston, 1986; Evans et al, 1993; Hatahet and Wallace, 1997). In general, abasic sites are strong blocks to in vitro DNA synthesis, and in the presence of proofreading termination sites are observed one base prior to the site of base loss. Both replicative and repair polymerases are blocked at abasic sites, and sequence context (Hatahet et al, 1999) and processivity (Shibutani et al, 1997) have been shown to affect the extent of bypass synthesis. In keeping with the in vitro studies, abasic sites are lethal in vivo when introduced specifically into biologically active DNAs (Schaaper and Loeb, 1981; Evans et al, 1993). When abasic sites are bypassed in vitro or in vivo, A is preferentially inserted followed by G or T, depending on the sequence context and the polymerase used, thus resulting in mutations (Loeb and Preston, 1986). Even the DNA polymerases of the Y family, whose purpose is to bypass DNA damage, are assisted by polymerase accessory proteins or additional polymerases to bypass efficiently sites of base loss (for reviews, see Sutton and Walker, 2001; Prakash and Prakash, 2002; Yang, 2003).
DNA polymerases are found in several distinct families and are generally classified as replicative or repair polymerases. Several structures of replicative DNA polymerases have recently been solved, including those in family B from bacteriophage RB69 (Wang et al, 1997; Shamoo and Steitz, 1999; Franklin et al, 2001), and from several archaeal organisms (Hopfner et al, 1999; Zhao et al, 1999; Rodriguez et al, 2000; Hashimoto et al, 2001). The structure of a family A replicative polymerase, T7 DNA polymerase, is also known (Doublié et al, 1998). Structures of a number of repair polymerases of the A, X and Y families have been solved as apoenzymes or in complex with undamaged DNA (for reviews, see Brautigam and Steitz, 1998; Patel and Loeb, 2001; Goodman, 2002; Yang, 2003). DNA polymerases share a common architecture resembling a cupped right hand (Steitz, 1993) where the thumb associates with the minor groove of duplex DNA, the palm contains the conserved carboxylate residues involved in catalysis of the nucleotidyl transfer reaction (Delarue et al, 1990), and conserved residues in the fingers domain bind the incoming nucleotide (Brautigam and Steitz, 1998; Kunkel and Bebenek, 2000; Beard and Wilson, 2003). An open–closed model has been proposed in which binding of a dNTP to the fingers domain in the open form leads to a conformational change where, upon closure of the fingers toward the palm, the incipient nucleotide and template base fit tightly into a compact binding pocket within the polymerase active site (Sawaya et al, 1997; Doublié et al, 1999; Steitz, 1999).
To date, there are only two crystal structures of DNA polymerases bound to lesion-containing DNA. The structure of DNA polymerase β has been solved as a ternary complex with DNA containing an oxidative lesion, 8-oxoguanine, and dCTP (Krahn et al, 2003), whereas Dpo4 was crystallized in complex with a cis-syn thymine dimer (Ling et al, 2001, 2003). In order to elucidate the lesion/polymerase interactions that make abasic sites such strong replication blocks in polymerases of other families, we set out to solve the structure of a replicative DNA polymerase in the presence of a primer/template DNA containing tetrahydrofuran (furan, or F), a stable abasic site analog. We chose RB69 gp43, a replicative DNA polymerase from the B family, and a close relative of T4 DNA polymerase, because of the wealth of biochemical and structural information available for this polymerase. Three RB69 gp43 structures have been reported including the apoenzyme (apo; Wang et al, 1997), and liganded structures with DNA in the polymerase active site (pol; Franklin et al, 2001) or in the exonuclease active site (exo; Shamoo and Steitz, 1999). Thus, once we determined the structure of RB69 gp43 in complex with DNA containing an abasic site, we would be able to compare it to the available open (apo and exo) and closed (pol) structures.
In the work presented here, we show that the asymmetric unit of the RB69 gp43 crystals in complex with abasic site-containing DNA contains no fewer than four different polymerase molecules: two have DNA in the polymerase active site (Pol1, Pol2), and in one the DNA is located in the exonuclease active site (Exo1). In the fourth molecule (Exo2), where the DNA is not visible in either active site, the polymerase conformation is globally similar to that of Exo1. The DNA polymerase is active and can incorporate dAMP across from furan under crystallization conditions, suggesting that the observed complexes are part of the reaction pathway between polymerase and exonuclease active sites. Comparison with the previously solved structures of RB69 gp43 revealed that the polymerase was captured in at least three new conformations, which are clearly distinct from the reported open (apo; Wang et al, 1997; exo; Shamoo and Steitz, 1999) and closed conformations (pol; Franklin et al, 2001). In addition, when compared to complexes with undamaged DNA, the DNA around the abasic site reveals distinct conformational changes, suggesting why this lesion is so poorly bypassed.
Results and discussion
Primer extension
T4 DNA polymerase is known to incorporate preferentially dAMP opposite abasic sites (Goodman et al, 1993; Hatahet et al, 1999; Berdis, 2001). NMR structures of nucleosides opposite tetrahydrofuran have shown that dAMP is stacked in an intrahelical conformation whereas dGMP can exist in both intra- and extrahelical conformations. Pyrimidines, on the other hand, stack poorly and are extrahelical (Cuniasse et al, 1987, 1990). This observation was confirmed in our primer extension assay with RB69 gp43 and furan-containing DNA. All four nucleotides were tested individually under identical reaction conditions. RB69 gp43 incorporated dAMP preferentially opposite the abasic site (Figure 1A) and, to a lesser extent, dGMP. Incorporation of the pyrimidines, as well as dideoxy-ATP, was undetectable in our assay.
Figure 1.
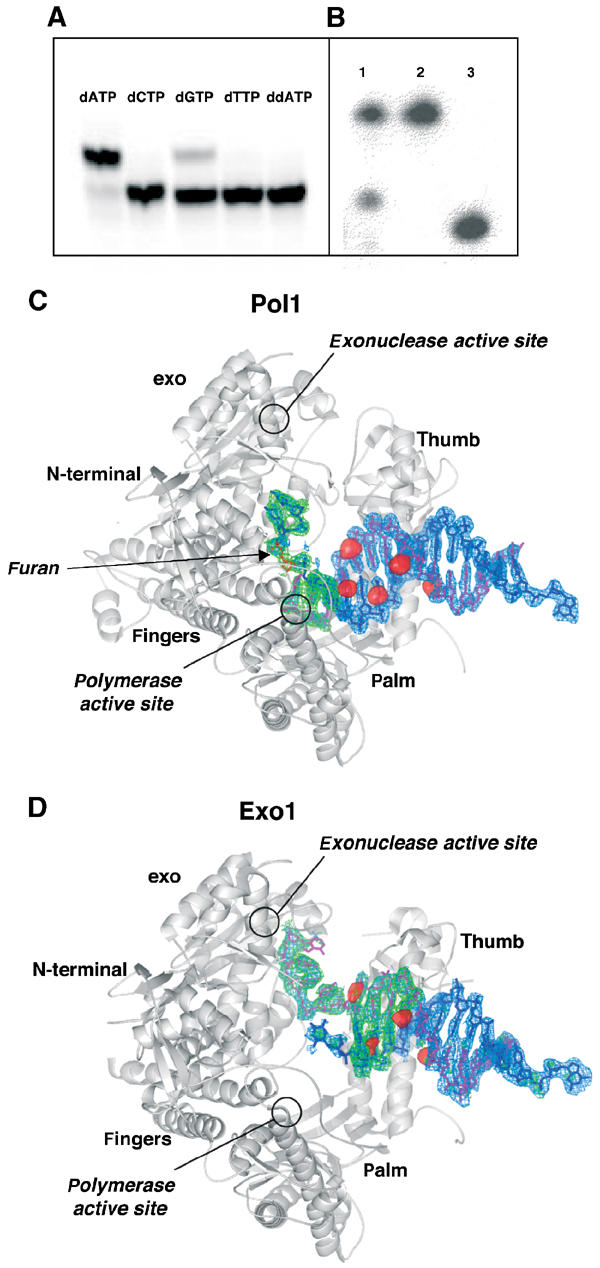
Crystallization of RB69 gp43 exo− with furan-containing DNA. (A) RB69 gp43 exo− is able to extend a primer by incorporating dAMP, and to a lesser extent dGMP, opposite an abasic site. No measurable incorporation of the pyrimidines was detected nor was incorporation of ddATP. (B) dATP is incorporated by RB69 gp43 exo− under crystallization conditions. The primer has been extended by one base and there is no evidence of residual exonuclease activity (lane 1: dissolved crystal; lane 2: template; lane 3: primer). (C) Molecule with DNA in the polymerase active site (Pol1). An Fo–Fc map (blue) calculated with DNA omitted and contoured to 3σ and an Fo–Fc simulated annealing omit map (green) with a 10 Å sphere omitted around the abasic site and contoured to 3.5σ are superimposed on the protein model. The red surfaces represent peaks in the anomalous difference Fourier map calculated from the brominated DNA data contoured at 5σ. The primer strand is shown in magenta and the abasic site in red. (D) Molecule with DNA in the exonuclease active site (Exo1). All maps are as those in (C) with the exception that the simulated annealing omit maps were calculated with a 10 Å sphere omitted around both the 3′ end of the primer and the 5′ end of the template. Note that the 5′ end of the template becomes disordered once it disassociates from the primer, and thus the furan is not visible in this structure nor is the 5′ single-stranded template. The DNA sequence used in co-crystallization is described in Materials and methods.
Crystallization and structure determination
We have succeeded in crystallizing full-length exonuclease-deficient RB69 gp43 in the presence of furan-containing DNA, dATP and Mg2+, its natural metal cofactor. Under our crystallization conditions, dAMP was incorporated across from the furan (Figure 1B) and the primer DNA was not extended past the lesion, resulting in a binary complex, which was expected based on solution studies using T4 DNA polymerase (Takeshita et al, 1987; Hatahet et al, 1999; Berdis, 2001). We note that the DNA did not translocate after dAMP incorporation and thus there is no room to accommodate an incoming nucleotide triphosphate. In fact, our attempts to crystallize the polymerase with furan-containing DNA, dATP and the next nucleotide to be incorporated (dCTP) have failed. The binary complex crystals belong to a monoclinic space group (P21) with four molecules per asymmetric unit. Data sets were collected on several derivatized crystals, including crystals grown with brominated DNA or the selenomethionyl protein variant and heavy atom-soaked crystals. The structure was solved at 2.8 Å resolution using a combination of molecular replacement and multiwavelength anomalous diffraction (MAD) methods (see Materials and methods). The model is currently refined to a free R factor=29.1% and a crystallographic R factor=24.6% with good stereochemistry. The data collection and refinement statistics are reported in Table I.
Table 1.
Data collection and refinement statistics
| Data collection | Se-Remote (A) | Se-Edge (A) | Se-Peak (A) | Se-Remote (B) | Se-Edge (B) | Se-Peak (B) |
|---|---|---|---|---|---|---|
| Wavelength (Å) | 0.9500 | 0.9790 | 0.9788 | 0.9500 | 0.9789 | 0.9787 |
| Resolution (Å) | 50–2.9 | 50–2.85 | 50–2.8 | 40–3.15 | 40–3.0 | 40–3.1 |
| Number of reflections | ||||||
| Measured | 422 352 | 441 284 | 471 659 | 357 523 | 398 046 | 354 495 |
| Unique | 118 308 | 123 652 | 130 580 | 98 495 | 108 359 | 101 746 |
| Redundancy | 3.6 | 3.6 | 3.6 | 3.7 | 3.6 | 3.6 |
| Completeness (%)a | 99.8 (98.6) | 99.3 (94.5) | 99.8 (98.7) | 99.0 (94.1) | 99.4 (96.3) | 99.5 (97.4) |
| Rmerge (%)b | 7.8 | 8.0 | 8.5 | 11.2 | 9.1 | 10.2 |
| I/σ | 15.55 (2.5) | 14.33 (2.3) | 12.79 (2.9) | 11.37 (2.3) | 13.36 (2.2) | 11.79 (2.4) |
| RCullis c | 0.84 | 0.73 | 0.76 | 0.87 | 0.75 | 0.78 |
| Phasing power | 0.74 | 1.27 | 1.27 | 0.68 | 0.97 | 1.00 |
| Data collection |
Hg-peak |
Pt-peak |
Br-peak |
|||
| Wavelength (Å) | 1.0055 | 1.0711 | 0.9109 | Overall FOMd 0.55/0.88 | ||
| Resolution (Å) | 40–2.9 | 50–3.15 | 40–2.9 | Refinement (against data set Se-peak (A)) | ||
| Number of reflections | Number of atoms | |||||
| Measured | 443 832 | 327 667 | 442 194 | Protein (non-hydrogen) | 29 586 | |
| DNA | 2240 | |||||
| Unique | 118 013 | 90 372 | 118 856 | Water | 213 | |
| Redundancy | 3.8 | 3.6 | 3.7 | Rwork (%)e | 24.6 | |
| Completeness (%) | 99.7 (99.2) | 98.5 (89.8) | 99.2 (98.5) | Rfree (%)e | 29.1 | |
| Rmerge (%) | 6.2 | 7.1 | 8.7 | r.m.s.d. bond distances (Å) | 0.0073 | |
| I/σ | 21.1 (3.3) | 16.2 (2.1) | 14.3 (2.0) | r.m.s.d. bond angles (Å) | 1.22 | |
| Phasing power | 0.08 | 0.19 | 0.04 | |||
| Riso f (%) | 12.4 | 22.5 | 22.5 | |||
| |
Pol1 |
Pol2 |
Exo1 |
Exo2 |
||
| Ramachandran plot | ||||||
| Core region (%) | 84.2 | 84.8 | 74.3 | 63.3 | ||
| Allowed (%) | 14.1 | 14.4 | 22.3 | 33.3 | ||
| Generously allowed (%) | 1.6 | 0.7 | 2.7 | 2.7 | ||
| Disallowed (%) | 0.1 | 0.1 | 0.7 | 0.7 | ||
| Average B factor all atoms (Å2) | ||||||
| Protein only | 49.7 | 42.9 | 65.5 | 95.0 | ||
| DNA only |
54.4 |
89.6 |
96.7 |
110.7 |
|
|
| aThe numbers in parentheses are the statistics for the highest resolution shell. | ||||||
| bRmerge=∑∣I−〈I〉∣/∑I, where 〈I〉 is the average intensity from multiple observations of symmetry-related reflections. | ||||||
| cRCullis=[〈(LOC)2〉]1/2 (〈∣ΔF∣2〉)1/2, where LOC is the lack-of-closure error. | ||||||
| dFigure of merit, before and after density modification. | ||||||
| eRwork and Rfree=∑h∥∣Fo∣−∣Fc∥/∑h∣Fo∣, where Fo and Fc are the observed and calculated structure factor amplitudes. Rfree was calculated with 10% of the reflections not used in refinement. | ||||||
| fRiso=∑∣FPH−FP∣/∑∣FP∣, where FP is the observed structure factor amplitude for the reference data set (Se-peak (A)) and FPH is the observed structure factor amplitude for the heavy atom derivatives. | ||||||
Crystallographic snapshots of different DNA conformations
From the beginning of model building, it was clear that we had crystallized RB69 gp43 in complex with DNA in at least two different conformations. The four molecules in the asymmetric unit did not, however, appear to exist as a pair of open molecules and a pair of closed molecules. In fact, the differences among the molecules were large enough to preclude the use of noncrystallographic symmetry averaging during refinement. It was also clear that we had not captured any of the molecules in the previously observed closed polymerizing mode, as all of the finger domains were found in the open position seen in the apo (Wang et al, 1997) and editing forms (Shamoo and Steitz, 1999) of the protein. The DNA, however, was found in the polymerase active site of two of the molecules (Pol1 and Pol2) and in the exonuclease active site of the third (Exo1). The DNA location in Pol1 (Figure 1C), Pol2 and Exo1 (Figure 1D) was confirmed by the overlay on the thymines of bromine peaks from anomalous difference Fourier maps, as well as by calculating simulated annealing omit maps in the vicinity of the furan. The fourth molecule (Exo2) showed clear density for only part of the DNA duplex. This observation is consistent with the fact that the temperature factors for this molecule are substantially higher than those of the other three molecules in the asymmetric unit (Table I).
DNA in the polymerase active site
Pol1 and Pol2 have DNA located within the polymerase active site but they differ in both DNA geometry and the relative positions of protein domains. In both molecules, the DNA is B-form throughout, similar to the ternary complex of RB69 DNA polymerase complexed with undamaged DNA (pol; Franklin et al, 2001). In family A polymerases, in contrast, the DNA in the vicinity of the active site is A-form, which is characterized by a wider, shallower minor groove (Doublié et al, 1998; Li et al, 1998). In Pol1 and Pol2, the primer terminates with the incorporated dAMP directly across from the furan and still in the active site, as the DNA has not translocated past the catalytic aspartates, D411, D621 and D623. In the previously reported ternary complex, two Ca2+ ions are ligated by D411, D623 and the triphosphate tail of the incoming nucleotide (Franklin et al, 2001). In our 2.8 Å resolution electron density map, no magnesium ions are seen in the vicinity of the catalytic aspartates or phosphate oxygens. This is not unexpected, considering that we have captured a binary complex. The geometry of the DNA in the active site is distorted and remarkably different from that seen in the ternary complex (see discussion below).
The interactions between the polymerase and furan-containing DNA are shown in Figure 2. In Pol1, the furan moiety stacks against G568 of the P helix of the fingers domain. The two unpaired bases at the 5′ end of the template are excluded from the active site via a sharp bend in the phosphate backbone (Figure 3A). F359 of the N-terminal domain packs against the base immediately 5′ of the furan, while the 5′-terminal base stacks against I253 of a β-hairpin from the exonuclease domain. R260 of the same hairpin loop is 4 Å from the 5′ OH group of the terminal residue, while K251 is 3.2 Å from the terminal nucleotide. The same β-hairpin has been implicated in the transition between exonuclease and polymerase active sites (see discussion below; Reha-Krantz et al, 1998). E219 is not part of the hairpin loop but comes in close contact with the terminal nucleobase of the template (Figures 2A and 3A). The palm residues contact the sugar phosphate backbone of the DNA. The thumb domain contacts the DNA primer and template strands further away from the polymerase active site, primarily via interactions with the phosphate backbone (Figure 2A).
Figure 2.
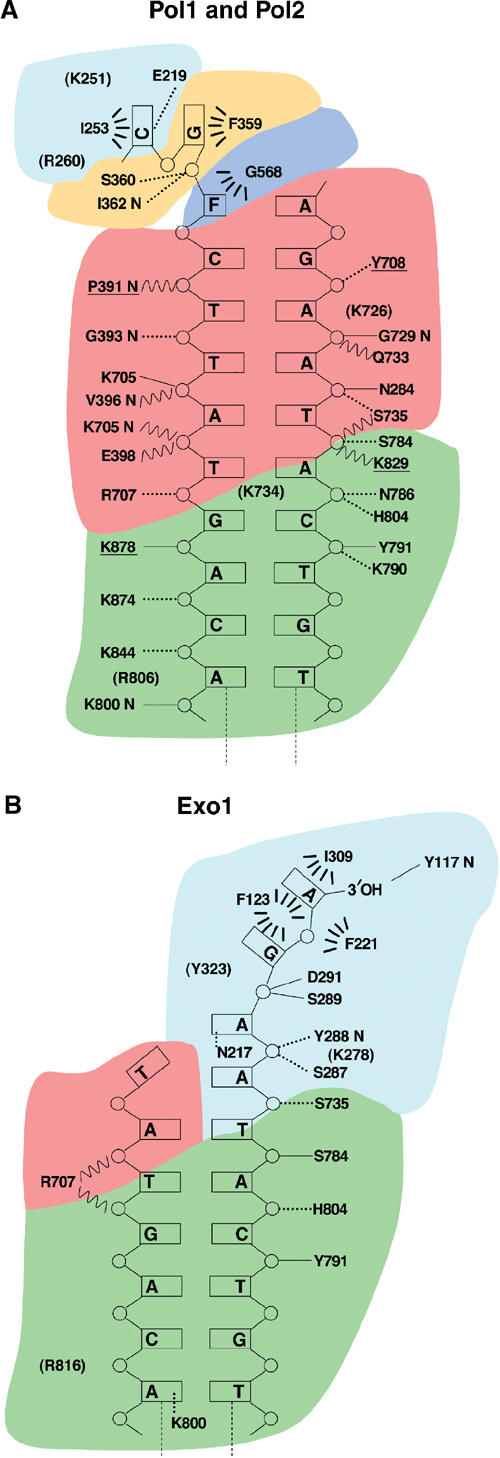
Protein/DNA contacts in polymerizing and editing conformations. (A) Polymerase mode. Hydrogen bond contacts less than 2.8 Å are represented by solid lines and those greater than 2.8 and less than 3.5 Å are shown with dashed lines. Residues in parentheses represent charge interactions longer than 3.5 Å. Wavy lines represent water-mediated contacts. Hydrophobic interactions are symbolized by dashed arcs. Contacts mediated by a nitrogen main-chain atom are indicated by the suffix ‘N'. Protein residues are colored according to their domain location: exonuclease (cyan), N-terminal domain (orange), palm (red), thumb (green) and fingers (blue). Underlined residues are contacts in Pol1 that are absent in Pol2. (B) Exonuclease mode. All descriptions as above.
Figure 3.
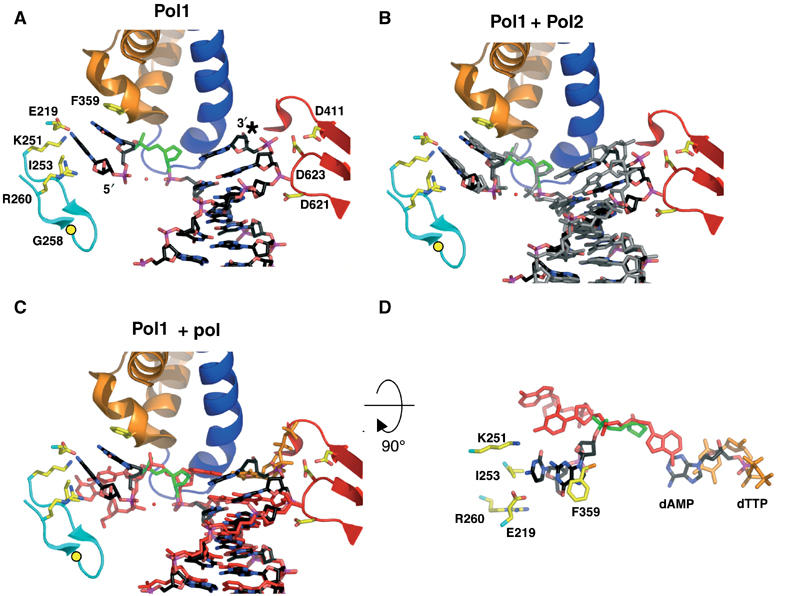
Furan-containing DNA in the polymerase active site is distorted and differs significantly from normal DNA. (A) The polymerase active site of Pol1. The protein backbone is colored according to the different protein domains: palm (red), fingers (blue), exonuclease (cyan) and N-terminal (orange). The furan moiety is in green and the dAMP incorporated opposite the furan is marked with an asterisk. Gly258 is shown as a yellow circle and the red sphere is a modeled water molecule. (B) DNA from Pol2 (gray) is superimposed on the Pol1 model after alignment via their palm domains (residues 383–468 and 573–729). (C) Superposition of DNA from the ternary complex (red; Franklin et al, 2001) with Pol1 (black) shows marked differences in the DNA conformation. Note that the last base pair in the Pol1 binary complex superimposes with the ultimate base pair in the ternary complex, composed of the incoming dTTP (orange) and its template base, which implies that the DNA has not translocated in the Pol1 binary complex. (D) The DNA from (C) is rotated 90° toward the reader and with most protein residues removed from view for clarity. The trajectory of the 5′ template DNA from Pol1 (black) differs from that seen in the ternary complex (red). As in (C), the furan moiety from Pol1 is shown in green and the incoming dTTP from the ternary complex model in orange.
The DNA in Pol2 globally superimposes on the DNA from Pol1 (Figure 3B) but it is further from the protein than the DNA in Pol1. The phosphate backbone in Pol2 has moved 4 Å further away from the catalytic aspartates and the disassociation from the protein extends further down the DNA helix. In particular, contacts with the phosphate oxygens one base pair upstream from the A:furan mismatch are present in Pol1, but are lost in Pol2. The furans in both molecules are superimposable, and the 5′ template in Pol2 binds the β-hairpin loop in the same manner as Pol1 (Figure 3B).
The interactions between Exo1 and the lesion-containing DNA are shown in Figure 2B. The 3′ end of the primer is bound in the exonuclease site. Very few of the interactions from the residues in the palm and thumb described for Pol1 in Figure 2A are retained in Exo1.
Comparison with the ternary polymerizing complex
A superposition of the DNA duplex in Pol1 with that of the closed ternary complex (Franklin et al, 2001) reveals some marked differences in the orientation of the bases near the active site (Figure 3C). The mispaired A:furan and the four base pairs adjacent to it are tilted and twist away from the corresponding base pair in the ternary complex, culminating in a 30° angle between the incorporated dAMP in Pol1 and the incoming dTTP in the ternary complex (Franklin et al, 2001). The DNA in the polymerase active site has not translocated and is therefore stalled, precluding extension past the lesion and thereby providing a structural basis for the inability of the polymerase to extend beyond an abasic site (Reineks and Berdis, 2003). We note that the DNA geometry in the active site does not, however, shed light on the polymerase's preferential incorporation of A over G opposite furan.
The template strand of Pol1 superimposes globally with that of the ternary complex, except for the unpaired bases at the 5′ end of the template. As shown in Figure 3D, the angular difference between the unpaired template bases is about 46°. While the primer strand is within the polymerase active site as seen in the ternary complex, the template strand takes on a conformation more akin to that seen in the template strand of the editing complex (Shamoo and Steitz, 1999), implying that the DNA seen in Pol1 and Pol2 is actually in a conformation that is intermediate between a bona fide pol and exo conformation. This observation also holds when the minor groove interactions are compared. Interactions between conserved polymerase residues and O2 and N3, the ‘universal' hydrogen bond acceptors in the minor groove (Seeman et al, 1976), have been posited to be part of a mechanism aimed at detecting mismatches (Doublié et al, 1998; Li et al, 1998; Franklin et al, 2001). In the event of a mispair, one of the bases projects into the major groove (Hunter et al, 1986), thereby disrupting interactions with the polymerase, which can ultimately lead to the switch to the exonuclease binding site. In the ternary complex, K706 hydrogen bonds with O2 of the nucleobase 5′ to the primer terminus and Y567 makes water-mediated contacts with O4′ of the ribose of the template base in the penultimate base pair as well as with N3 of the template G of the terminal base pair (Figure 4A). These amino acids are highly conserved in the family B of polymerases, and mutations decrease the polymerase activity, probably because of the disruption of DNA binding (Blasco et al, 1995). These minor groove contacts are maintained provided the geometry of the base pair is Watson–Crick (Seeman et al, 1976). In Pol1, these contacts are lost (Figure 4B). The projection of the bases into the major groove in response to the A:furan mismatch disrupts these contacts, leaving the protein with no minor groove contacts immediately upstream from the mismatch. Interestingly in Pol2, the DNA moves even further away from the protein (Figure 4C). Although Pol1 and Pol2 clearly have DNA in the polymerase active site, the DNA conformation is a hybrid between pol and exo, implying that we have captured two different DNA conformations on the path to the exonuclease active site. Interactions between the thumb and exonuclease domains of Pol1 and Pol2 allow the DNA to remain in the polymerase active site, despite the loss of minor groove contacts. Such interactions are absent in Exo1 and Exo2, which allow the thumb domains to accommodate the switch to the exonuclease active site (see below).
Figure 4.
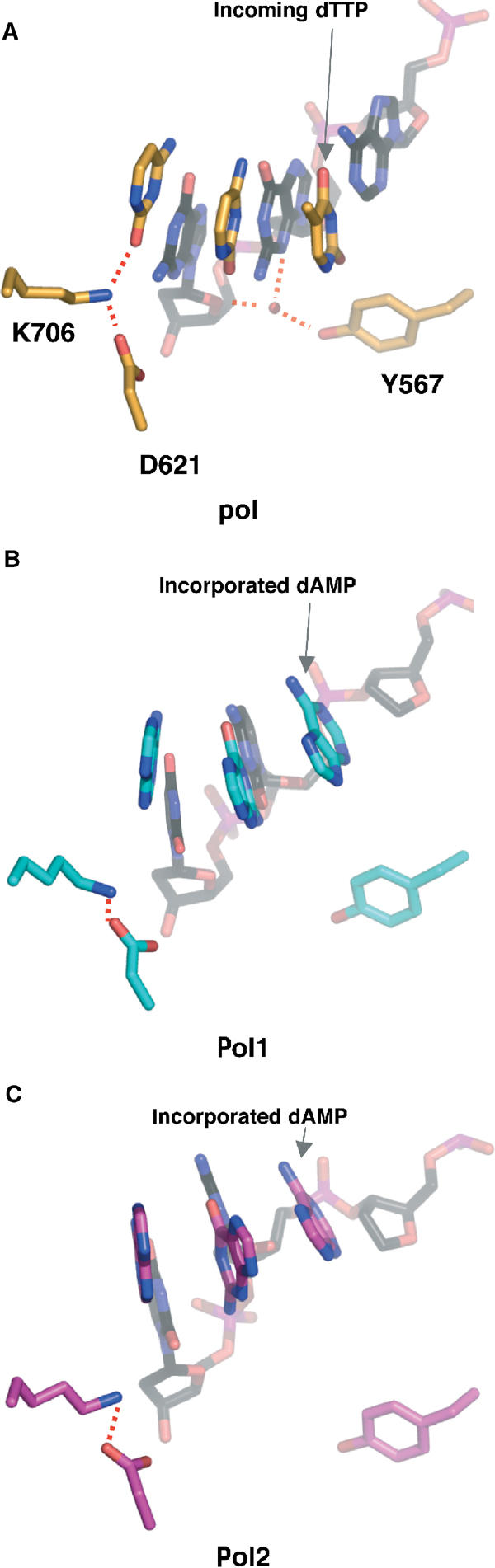
Minor groove interactions are lost in furan-containing DNA. (A) Minor groove interactions in the active site of ternary complex (1IG9; Franklin et al, 2001). Hydrogen bonds are shown as dashed red lines. For clarity only the bases of the primer strand are shown. (B) Pol1, in the same orientation as the ternary complex in (A). All minor groove interactions seen in the ternary complex are lost. (C) Pol2, in the same orientation as in (A) and (B). The DNA shifts further away from the palm domain.
DNA in the exonuclease active site
In addition to having two distinct protein complexes containing DNA in the polymerase active site, we have also captured a polymerase with DNA in the exonuclease active site. In this structure, the three bases at the 3′ end of the primer, including the incorporated dAMP, have unwound from their template pairs and flipped up into the exonuclease active site (Figure 5A). The five unpaired bases at the 5′ end of the template (including the furan) are disordered. Interestingly, the tip of the β-hairpin loop (residues 253–261), which forms a close association with the single-stranded template in Pol1 and Pol2, is disordered in Exo1 (Figure 5B) with no visible electron density (see discussion below).
Figure 5.
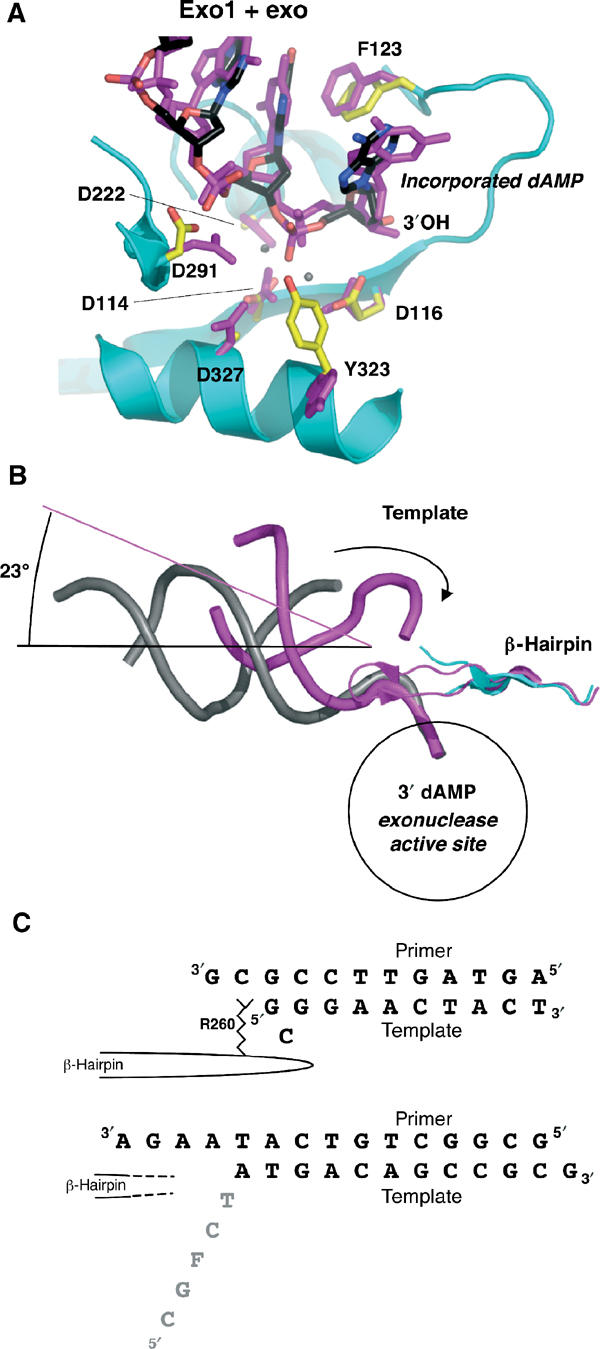
DNA in the exonuclease active site. (A) Exo1 model (protein: cyan; DNA: black) aligned with exo complex (magenta, with calcium ions shown as gray spheres; Shamoo and Steitz, 1999). The incorporated dAMP opposite the furan at the primer terminus is flipped into the exonuclease domain. The position of the single-stranded DNA and its contacts with the protein are similar between the model with undamaged DNA and that with furan-containing DNA. (B) Same alignment as in (A), showing the entire length of the DNA backbone (Exo1 in gray, exo model in magenta). The template (indicated by the arrow) in the exo model is in contact with the β-hairpin loop. In Exo1, the unpaired portion of the template is disordered. The β-hairpin loop is in a similar position in both models, but the tip of the loop is disordered in Exo1. The helical axes are shown to indicate the angular difference between the two DNA molecules. (C) Comparison of the DNA sequences in the exo model (Shamoo and Steitz, 1999) top sequence, and Exo1, bottom sequence. In the exo model, the Arg260 in the β-hairpin loop stacks against the template strand, which forms a G–G base pair and a bulged, unpaired C. In contrast, the tip of the β-hairpin loop is disordered in Exo1, and so is the single-stranded template (gray).
The fourth molecule, Exo2, is considered to be in the editing mode based on overall conformational similarities with Exo1. Clear density exists for the 3′ template overhang and six adjacent base pairs. As the DNA gets closer to the core of the polymerase, however, the quality of the electron density worsens, which precluded building DNA into either polymerase or exonuclease sites, suggesting that the DNA in this molecule is not tightly bound by the protein. This observation is consistent with the fact that there were no bromine peaks in difference Fourier maps for this molecule. The entire model for Exo2 suffered from poor density and refines with much higher B factors than the other three molecules. Nevertheless, it is clear that the overall structure of Exo2 is much more similar to Exo1 than to the polymerizing forms of Pol1 and Pol2.
Comparison with exo model
The exonuclease active site of Exo1 is globally similar to the previously reported editing model (exo) (Shamoo and Steitz, 1999) and the exonuclease fragment of T4 gp43 (Wang et al, 1996). There are three bases of single-stranded DNA of the primer strand in the exonuclease active site, which is consistent with the observation that two to three bases of DNA need to be melted for exonuclease activity (Cowart et al, 1989; Reddy et al, 1992). There are, however, two key differences between Exo1 and exo. Superposition of the two models via their exonuclease domains shows similar positioning of the primer terminus within the hydrophobic pocket of the exonuclease active site (Figure 5A). The duplex DNA, however, does not take the same path out of the molecule. The duplex DNA in the exo model (Shamoo and Steitz, 1999) continues on a straight line out of the molecule, whereas an angular difference of 23° is measured when compared to the trajectory of the DNA in the Exo1 model (Figure 5B). In the exo model (Shamoo and Steitz, 1999) three base pairs are denatured as the primer 3′ end enters the exonuclease active site and Arg260 of the aforementioned β-hairpin stacks against the terminal template base (Figure 5C). Interestingly, in the exo model, the terminal template base forms a G–G base pair with the third primer base, while the adjacent template base, C, bulges out of the duplex. In contrast, the primer in Exo1 has separated from the template by four base pairs and as a result, the single-stranded template DNA is disordered.
Concerted polymerase domain movements between polymerase and exonuclease modes
In order to produce an active complex, DNA polymerases have been posited to undergo coupled conformational changes (Johnson, 1993; Kunkel and Bebenek, 2000; Beard and Wilson, 2003). In the unliganded form, the protein assumes an open conformation with the thumb and finger domains angled away from the palm of the polymerase. Binding of duplex DNA and the correct nucleotide leads to the rotation of the fingers domain toward the palm, accompanied by a rotation of the thumb domain toward the DNA duplex. These structural changes have been observed previously in DNA polymerases across several families (Doublié et al, 1999). The switch into the editing mode in RB69 gp43 requires opening of the fingers and rotation of the thumb away from the palm (Shamoo and Steitz, 1999). The RB69 gp43 structures complexed to DNA reported previously were obtained with undamaged, normally paired DNA (Shamoo and Steitz, 1999; Franklin et al, 2001). The use of an abasic site analog to create a mismatch has allowed us to capture for the first time, in a single crystal, what we believe to be four distinct protein conformational states between the polymerizing and editing modes of RB69 DNA polymerase.
Alignment of the known ternary (Franklin et al, 2001) and editing (Shamoo and Steitz, 1999) complexes and all four molecules in our structure along their palm domains (residues 383–468 and 573–729) revealed significant conformational changes, as shown in the r.m.s. distances calculated for each domain (Figure 6A). Switching from the ternary complex to Pol1 involves a 36° rotation of the fingers domain from the closed to open conformation (Figure 6B). The β-hairpin rotates by 28° downward toward the palm to bind the template DNA, while the entire exonuclease domain moves by an average of 3 Å. The fingers rotation is the largest difference in the distance matrix (17 Å), followed by differences in the thumb domain (Figure 6A). The thumb can be divided into two subdomains, the base that connects to the palm and the tip that associates with the phosphodiester backbone and minor groove of the DNA duplex (Figure 6C). As the protein switches the primer into the exonuclease domain, the thumb tip rotates up and away from the palm by 7–11 Å, as seen in Exo1. The contacts to the DNA duplex still visible in Exo1 and Exo2 are in identical positions in Pol1, indicating that the DNA need not translocate in order to switch from the polymerase to the exonuclease active sites (Figures 2B and 6C). A comparison with the previously reported editing model shows that the thumb in Exo1 is opened even further than previously reported. A concerted motion of the exonuclease and thumb domains is observed as the primer DNA is switched from the polymerizing to editing mode (Figure 6D).
Figure 6.
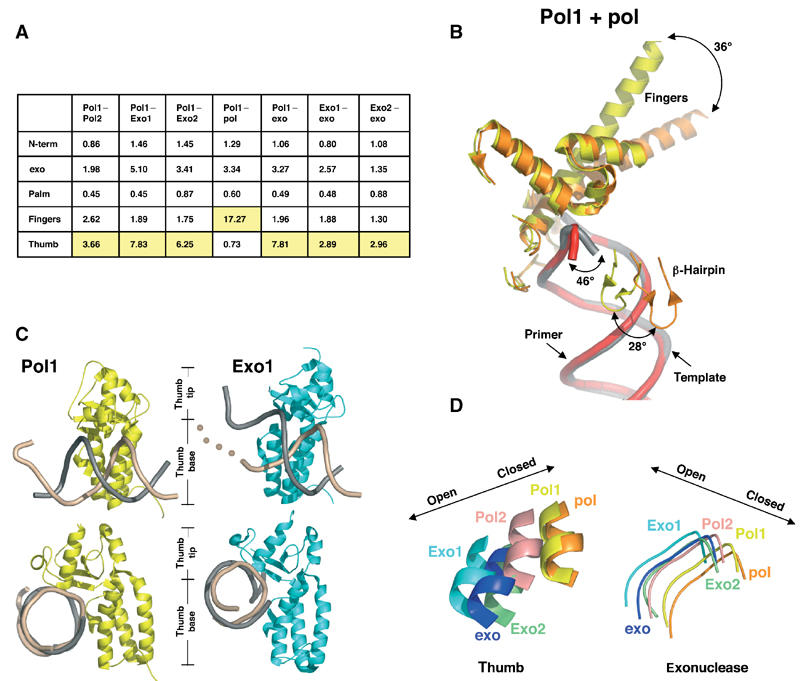
Protein domain movements. (A) Table of root mean square (r.m.s.) deviations (Å). The largest deviations between pairs of conformations are highlighted in yellow. Molecules were aligned using the palm residues (383–468 and 573–729). (B) Protein movements between Pol1 (yellow, with DNA in gray) and ternary complex (orange, with DNA in red). The largest protein movement (36°) is seen in the fingers domain. (C) Thumb position in Pol1 and Exo1. The thumb domains from Pol1 (yellow) and Exo1 (cyan) contact template (beige) and primer (gray) DNA. The beige dots illustrate that the template DNA in Exo1 has no visible density once it unwinds from the primer. Most of the contacts between the thumb and the DNA are maintained between the polymerizing and editing modes, suggesting that the DNA has not translocated during the switch from the polymerase to exonuclease active sites. The bottom set of images have been oriented to look down the DNA helical axis to show the increased opening of the thumb tip in the Exo1 model. (D) Coordinated motion of the thumb and exonuclease domains. All molecules have been aligned via the palm domain (residues 383–468 and 573–729). A loop from the exonuclease domain (residues 120–129) and a portion of helix W from the thumb domain (residues 809–815) are shown. Movement of these domains to the left represents a more open conformation as described for the apo and exo models, and movement to the right represents a closed conformation as described for the ternary complex. A coordination between exonuclease and thumb movements exists, such that as the thumb relaxes its grip the exonuclease domain shifts closer to the thumb.
Polymerase to exonuclease active site switch
During polymerization, the primer DNA is elongated at a rate of several hundred bases per second in processive polymerases (Carroll and Benkovic, 1990; Johnson, 1993). Elongation of a mismatched primer terminus, on the other hand, is extremely slow (reviewed by Goodman et al, 1993), which gives the polymerase the opportunity to switch the primer 3′ end from the polymerase to the exonuclease active site, a reaction that is normally substantially slower than polymerization, with an apparent rate of 4 s−1 (Capson et al, 1992; Marquez and Reha-Krantz, 1996). The distance between the two catalytic centers in polymerases of the A and B families is similar, about 30–40 Å, but their exonuclease domains lie on opposite sides of the palm domains (Wang et al, 1997), suggesting that the mechanisms they employ for active site switching might differ.
When we compared the 5′ end of the DNA template in our Pol1 and Pol2 structures to that of the ternary complex, we found that the tip of the β-hairpin loop (residues 253–261) contacts the single-stranded template in Pol1 and Pol2 (Figure 3), an interaction not seen in the ternary complex (Franklin et al, 2001). The β-hairpin residues 250–260 have previously been shown to disrupt DNA base pairing in order to allow the single-stranded primer to enter the exonuclease site. Arg260 in particular disrupts Watson–Crick base pairing by stacking against the paired 5′ end of the template strand (Figure 5C) (Wang et al, 1996; Shamoo and Steitz, 1999). In our Exo1 model, on the other hand, the tip of the loop is disordered, indicating movement (Figures 5B and C). The close contact of the hairpin residues to the single-stranded template in Pol1 and Pol2, but not in the ternary complex, taken together with the observation that the loop is disordered in our Exo1 model, suggests that this loop accompanies DNA during the switch from the polymerase to the exonuclease active site. This is consistent with a proposed proofreading pathway in which β-hairpin residues 250–260 play a part in strand separation and transfer of the primer strand from the polymerase active site to form a pre-exonuclease complex (Reha-Krantz et al, 1998). In particular, mutating G255 in T4 gp43 (G258 in RB69; Figure 3) to serine has been shown to decrease the rate of movement of the primer terminus from the polymerase to the exonuclease active site by about 10-fold (Marquez and Reha-Krantz, 1996; Baker and Reha-Krantz, 1998). Taken together, these results suggest that the different hairpin conformations captured in our crystal are therefore likely to be found on the active site switching pathway.
Conclusions
The unique ability to compare four different protein–DNA complexes to the previously reported ternary (Franklin et al, 2001) and editing (Shamoo and Steitz, 1999) models, combined with a wealth of biochemical and mutational data, indicates that we may have captured snapshots of a DNA molecule on the path between the polymerase to the exonuclease active sites. In addition, the distortion in the DNA in the vicinity of the furan and the fact that it has not translocated readily explains why this DNA polymerase does not extend past this lesion.
Materials and methods
Deoxyadenosine triphosphate (100 mM solution) was purchased from Amersham Biosciences, PEG 2000 monomethylether from Hampton Research, and all other chemicals from Fisher or Sigma. Oligonucleotide primer (5′-GCGGCTGTCATAAG-3′) and template 5′-CGFCTTATGACAGCCGC-3′, F=tetrahydrofuran) were synthesized by Oligos Etc. and gel purified. The underlined thymines were replaced with 5-bromo-uracil in the crystals grown with brominated DNA. The template has an unpaired 3′ guanine overhang, which was previously found to be essential for growing well-diffracting crystals (Franklin et al, 2001).
Protein purification
Plasmid expressing exonuclease-deficient RB69 DNA polymerase (D222A, D327A) was obtained from Dr SW Morrical (University of Vermont), originally from Dr JD Karam (Tulane University). RB69 gp43 exo− was expressed in BL21(DE3) cells and grown for 4 h after induction at 37°C in LB medium. The protein was purified using a three-column procedure: DEAE, phosphocellulose and DNA cellulose. Frozen cells were lysed by sonication. After centrifugation, the lysate supernatant was dialyzed and loaded onto a DE52 column (Whatman) and eluted with a 20–200 mM NaCl gradient in 20 mM Tris pH 7.5. Pooled fractions were dialyzed into low-salt phosphocellulose buffer (10% glycerol, 1 mM EDTA, 2–10 mM β-mercaptoethanol, 10 mM KCl and 20 mM potassium phosphate pH 6.0), loaded on a phosphocellulose column and eluted with 10–500 mM KCl, in 20 mM potassium phosphate buffer pH 6.0. Fractions containing protein were dialyzed and loaded on a DNA cellulose column (Sigma) using the same buffers as for the phosphocellulose column. Fractions were pooled and dialyzed into 10 mM HEPES (pH 7.5), 50 mM NaCl and 1 mM DTT. The selenomethionyl protein was produced by inhibiting methionine biosynthesis in Escherichia coli (Doublié, 1997) and purified as above.
Primer extension
Fluorescently labeled primer/template DNA was mixed in a buffer containing the same reagents as the crystallization buffer minus magnesium along with 2 mM dNTP. Enzyme (10 nM) was added to the DNA/buffer and mixed for 20 s. Magnesium acetate (10 nM) was added to start the reaction, which was then stopped by quenching in a solution of 95% formamide and 10 nM EDTA after incubation for 5 min at 25°C. Primer extension in grown crystals was observed by radio-labeling the DNA from dissolved crystals. The washed crystals were dissolved in polynucleotide kinase reaction buffer and heated to 90°C. 32P dATP and polynucleotide kinase were added to the mixture and allowed to react for 30 min at 37°C. All oligonucleotides were separated on 16% polyacrylamide gels and were imaged on a Bio-Rad Fx imager system.
Crystallization
The polymerase was mixed in an equimolar ratio with DNA and 2 mM dATP. Hanging drops were made by mixing 1 μl of the reaction mix with 1 μl of reservoir solution (8% w/v PEG 2000 monomethyl ether, 100 mM MgSO4, 100 mM sodium acetate, 100 mM HEPES pH 7.0, 2 mM β-mercaptoethanol and 15% glycerol) and were equilibrated against 1 ml of reservoir solution. The crystallization conditions were found using an incomplete factorial (Carter and Carter, 1979), as the reported conditions (Franklin et al, 2001) failed to produce crystals. Monoclinic crystals grew to a maximum of 200 μm in the longest dimension in space group P21 with unit cell parameters a=133.02 Å, b=123.21 Å, c=165.62 Å and β=95.8°. There are four molecules per asymmetric unit, with a solvent content of 47%. The crystals were flash cooled directly from the crystallization drop into liquid nitrogen.
Data collection
The availability of models of RB69 gp43 in the apo form (Wang et al, 1997) as well as with DNA in both the polymerizing (Franklin et al, 2001) and editing modes (Shamoo and Steitz, 1999) suggested that molecular replacement might be the fastest way to attain phase information. Initial results proved fruitless as no program could provide a solution that packed four molecules properly in the asymmetric unit. To obtain phases, then, we set out to perform MAD and SAD experiments from a variety of heavy atom derivatives. X-ray data were collected at 100 K on a Brandeis B1.3 CCD detector at the X-12C beamline of the National Synchrotron Light Source (Upton, NY). Two complete selenomethionyl MAD data sets were collected at the inflection, peak and high-energy remote wavelengths from two separate crystals to a resolution of 2.8 Å. Data were also collected at the peak wavelength for the mercury (ethyl mercury phosphate)- and platinum (K2PtCl4)-soaked crystals, and a crystal grown with brominated DNA. The data sets were processed and scaled using Denzo and Scalepack (Otwinowski and Minor, 1997). Data collection statistics are summarized in Table I.
Structure determination and refinement
Unless indicated otherwise, all of the structure determination and refinement steps were performed using CNS (Brünger et al, 1998). Initial attempts to locate the selenium atoms from the MAD data were unsuccessful, but a molecular replacement solution was eventually found using the program BEAST (Read, 2001) as implemented in CCP4i (Potterton et al, 2003). This program allowed the use of both the closed (PDB 1IG9; Franklin et al, 2001) and open (PDB 1CLQ; Shamoo and Steitz, 1999) forms of the enzyme. Alternating between the two search models stripped of all nonprotein atoms led to a solution for three of the expected four molecules in the asymmetric unit.
Phasing with the three molecules followed by density modification resulted in a map with clear solvent boundary around the four molecules, and evidence of secondary structure elements, but the overall quality was not high enough for model building. The molecular replacement phases, however, were used to calculate anomalous difference Fourier maps for each of the heavy atom derivatives, which produced clear peaks for four mercury, four platinum, 14 of the expected 20 bromines and 85 of the expected 100 selenium atoms in the asymmetric unit. Refinement and phasing with the known selenium atoms followed by density modification produced a map showing interpretable density for most residues as well as for the DNA in three of the four molecules. The highest quality map was produced by combining all the MAD and SAD phase sets prior to density modification.
The model was built using the program O (Jones et al, 1991). A set of 20 models was generated by simulated annealing, from which residues that best fit the experimental density map were selected and spliced together by the program Maxenstein (MA Rould, personal communication). Iterative rounds of simulated annealing in CNS (Brünger et al, 1998) and model building were performed and the free R factor was monitored at each step. Refinement statistics are found in Table I. The quality of the protein models was assessed with Procheck (Laskowski et al, 1993). The only amino-acid residue in the disallowed region of the Ramachandran plot in molecules Pol1 and Pol2 is T622. This residue lies between the two catalytic aspartates (D621 and D623) in the polymerase active site, and its geometry is similarly distorted in the ternary complex (Franklin et al, 2001). All outliers in Exo1 and Exo2 are away from the active sites of the polymerase. Although the B factors for DNA are high, they are similar to those previously reported for liganded RB69 gp43: 〈B〉=57.6 Å2 for pol (Franklin et al, 2001) and 98.6 Å2 for exo (Shamoo and Steitz, 1999). We note that these values are significantly higher than the average B factors reported for well-resolved structures of polymerase/DNA complexes: ∼23 Å2 for T7 DNA polymerase (Doublié et al, 1998) and 30–45 Å2 for a thermostable Bacillus DNA polymerase (Johnson et al, 2003). Coordinates and experimental diffraction amplitudes have been deposited in the Protein Data Bank with accession number 1RV2 and will be available immediately upon publication.
Illustrations
All figures, except Figure 2, were generated with PyMol (DeLano, 2002).
Acknowledgments
We thank Wendy Cooper for help with protein purification, Drs Jeffrey Bond, Scott Morrical, Linda Reha-Kranz and Bob Melamede for helpful discussions and Drs Morrical and John Burke for critically reading the manuscript. We are grateful to Dr Mark A Rould for his help with data collection and refinement, the use of his unpublished programs and for stimulating discussions. We thank Dr Robert Sweet for access to X12C (NSLS), a beamline funded by NIH's National Center for Research Resources and DOE's Office of Biological and Environmental Research. This work was supported by NIH R01 CA52040 (to SSW) awarded by the National Cancer Institute and by an award to the University of Vermont under the Howard Hughes Medical Institute Biomedical Research Support Program for Medical Schools. SD is a Pew Scholar in the Biomedical Sciences.
References
- Baker RP, Reha-Krantz LJ (1998) Identification of a transient excision intermediate at the crossroads between DNA polymerase extension and proofreading pathways. Proc Natl Acad Sci USA 95: 3507–3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard WA, Wilson SH (2003) Structural insights into the origins of DNA polymerase fidelity. Structure (Camb) 11: 489–496 [DOI] [PubMed] [Google Scholar]
- Berdis AJ (2001) Dynamics of translesion DNA synthesis catalyzed by the bacteriophage T4 exonuclease-deficient DNA polymerase. Biochemistry 40: 7180–7191 [DOI] [PubMed] [Google Scholar]
- Blasco MA, Mendez J, Lazaro JM, Blanco L, Salas M (1995) Primer terminus stabilization at the phi 29 DNA polymerase active site. Mutational analysis of conserved motif KXY. J Biol Chem 270: 2735–2740 [DOI] [PubMed] [Google Scholar]
- Brautigam CA, Steitz TA (1998) Structural and functional insights provided by crystal structures of DNA polymerases and their substrate complexes. Curr Opin Struct Biol 8: 54–63 [DOI] [PubMed] [Google Scholar]
- Breen AP, Murphy JA (1995) Reactions of oxyl radicals with DNA. Free Radic Biol Med 18: 1033–1077 [DOI] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Capson TL, Peliska JA, Kaboord BF, Frey MW, Lively C, Dahlberg M, Benkovic SJ (1992) Kinetic characterization of the polymerase and exonuclease activities of the gene 43 protein of bacteriophage T4. Biochemistry 31: 10984–10994 [DOI] [PubMed] [Google Scholar]
- Carroll SS, Benkovic SJ (1990) Mechanistic aspects of DNA polymerases: Escherichia coli DNA polymerase I (Klenow fragment) as a paradigm. Chem Rev 90: 1291–1307 [Google Scholar]
- Carter CW Jr, Carter CW (1979) Protein crystallization using incomplete factorial experiments. J Biol Chem 254: 12219–12223 [PubMed] [Google Scholar]
- Cowart M, Gibson KJ, Allen DJ, Benkovic SJ (1989) DNA substrate structural requirements for the exonuclease and polymerase activities of procaryotic and phage DNA polymerases. Biochemistry 28: 1975–1983 [DOI] [PubMed] [Google Scholar]
- Cuniasse P, Fazakerley GV, Guschlbauer W, Kaplan BE, Sowers LC (1990) The abasic site as a challenge to DNA polymerase. A nuclear magnetic resonance study of G, C and T opposite a model abasic site. J Mol Biol 213: 303–314 [DOI] [PubMed] [Google Scholar]
- Cuniasse P, Sowers LC, Eritja R, Kaplan B, Goodman MF, Cognet JA, LeBret M, Guschlbauer W, Fazakerley GV (1987) An abasic site in DNA. Solution conformation determined by proton NMR and molecular mechanics calculations. Nucleic Acids Res 15: 8003–8022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL (2002) The PyMol Molecular Graphics System. San Carlos, CA, USA [Google Scholar]
- Delarue M, Poch O, Tordo N, Moras D, Argos P (1990) An attempt to unify the structure of polymerases. Protein Eng 3: 461–467 [DOI] [PubMed] [Google Scholar]
- Doublié S (1997) Preparation of selenomethionyl proteins for phase determination. Methods Enzymol 276: 523–530 [PubMed] [Google Scholar]
- Doublié S, Sawaya MR, Ellenberger T (1999) An open and closed case for all polymerases. Struct Fold Des 7: R31–R35 [DOI] [PubMed] [Google Scholar]
- Doublié S, Tabor S, Long AM, Richardson CC, Ellenberger T (1998) Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature 391: 251–258 [DOI] [PubMed] [Google Scholar]
- Evans J, Maccabee M, Hatahet Z, Courcelle J, Bockrath R, Ide H, Wallace S (1993) Thymine ring saturation and fragmentation products: lesion bypass, misinsertion and implications for mutagenesis. Mutat Res 299: 147–156 [DOI] [PubMed] [Google Scholar]
- Franklin MC, Wang J, Steitz TA (2001) Structure of the replicating complex of a pol alpha family DNA polymerase. Cell 105: 657–667 [DOI] [PubMed] [Google Scholar]
- Goodman MF (2002) Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu Rev Biochem 71: 17–50 [DOI] [PubMed] [Google Scholar]
- Goodman MF, Creighton S, Bloom LB, Petruska J (1993) Biochemical basis of DNA replication fidelity. Crit Rev Biochem Mol Biol 28: 83–126 [DOI] [PubMed] [Google Scholar]
- Hashimoto H, Nishioka M, Fujiwara S, Takagi M, Imanaka T, Inoue T, Kai Y (2001) Crystal structure of DNA polymerase from hyperthermophilic archaeon Pyrococcus kodakaraensis KOD1. J Mol Biol 306: 469–477 [DOI] [PubMed] [Google Scholar]
- Hatahet Z, Wallace S (1997) Translesion DNA Synthesis. In DNA Damage and Repair, Vol. 1: DNA Repair in Prokaryotes and Lower Eukaryotes, Nickoloff J, Hoekstra M (eds) pp 229–262. Totowa, NJ: Humana Press Inc. [Google Scholar]
- Hatahet Z, Zhou M, Reha-Krantz LJ, Ide H, Morrical SW, Wallace SS (1999) In vitro selection of sequence contexts which enhance bypass of abasic sites and tetrahydrofuran by T4 DNA polymerase holoenzyme. J Mol Biol 286: 1045–1057 [DOI] [PubMed] [Google Scholar]
- Hopfner KP, Eichinger A, Engh RA, Laue F, Ankenbauer W, Huber R, Angerer B (1999) Crystal structure of a thermostable type B DNA polymerase from Thermococcus gorgonarius. Proc Natl Acad Sci USA 96: 3600–3605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter WN, Brown T, Anand NN, Kennard O (1986) Structure of an adenine–cytosine base pair in DNA and its implications for mismatch repair. Nature 320: 552–555 [DOI] [PubMed] [Google Scholar]
- Johnson KA (1993) Conformational coupling in DNA polymerase fidelity. Annu Rev Biochem 62: 685–713 [DOI] [PubMed] [Google Scholar]
- Johnson SJ, Taylor JS, Beese LS (2003) Processive DNA synthesis observed in a polymerase crystal suggests a mechanism for the prevention of frameshift mutations. Proc Natl Acad Sci USA 100: 3895–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard (1991) Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Krahn JM, Beard WA, Miller H, Grollman AP, Wilson SH (2003) Structure of DNA polymerase Beta with the mutagenic DNA lesion 8-oxodeoxyguanine reveals structural insights into its coding potential. Structure (Camb) 11: 121–127 [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Bebenek K (2000) DNA replication fidelity. Annu Rev Biochem 69: 497–529 [DOI] [PubMed] [Google Scholar]
- Laskowski R, MacArthur M, Moss D, Thornton J (1993) PROCHECK: a program to check the sterochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Li Y, Korolev S, Waksman G (1998) Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J 17: 7514–7525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T (1993) Instability and decay of the primary structure of DNA. Nature 362: 709–715 [DOI] [PubMed] [Google Scholar]
- Ling H, Boudsocq F, Plosky BS, Woodgate R, Yang W (2003) Replication of a cis-syn thymine dimer at atomic resolution. Nature 424: 1083–1087 [DOI] [PubMed] [Google Scholar]
- Ling H, Boudsocq F, Woodgate R, Yang W (2001) Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell 107: 91–102 [DOI] [PubMed] [Google Scholar]
- Loeb LA, Preston BD (1986) Mutagenesis by apurinic/apyrimidinic sites. Annu Rev Genet 20: 201–230 [DOI] [PubMed] [Google Scholar]
- Marquez LA, Reha-Krantz LJ (1996) Using 2-aminopurine fluorescence and mutational analysis to demonstrate an active role of bacteriophage T4 DNA polymerase in strand separation required for 3′ → 5′-exonuclease activity. J Biol Chem 271: 28903–28911 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Patel PH, Loeb LA (2001) Getting a grip on how DNA polymerases function. Nat Struct Biol 8: 656–659 [DOI] [PubMed] [Google Scholar]
- Potterton E, Briggs P, Turkenburg M, Dodson E (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr D 59: 1131–1137 [DOI] [PubMed] [Google Scholar]
- Prakash S, Prakash L (2002) Translesion DNA synthesis in eukaryotes: a one- or two-polymerase affair. Genes Dev 16: 1872–1883 [DOI] [PubMed] [Google Scholar]
- Read RJ (2001) Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr D D57: 1373–1382 [DOI] [PubMed] [Google Scholar]
- Reddy MK, Weitzel SE, von Hippel PH (1992) Processive proofreading is intrinsic to T4 DNA polymerase. J Biol Chem 267: 14157–14166 [PubMed] [Google Scholar]
- Reha-Krantz LJ, Marquez LA, Elisseeva E, Baker RP, Bloom LB, Dunford HB, Goodman MF (1998) The proofreading pathway of bacteriophage T4 DNA polymerase. J Biol Chem 273: 22969–22976 [DOI] [PubMed] [Google Scholar]
- Reineks EZ, Berdis AJ (2003) Evaluating the effects of enhanced processivity and metal ions on translesion DNA replication catalyzed by the bacteriophage T4 DNA polymerase. J Mol Biol 328: 1027–1045 [DOI] [PubMed] [Google Scholar]
- Rodriguez AC, Park HW, Mao C, Beese LS (2000) Crystal structure of a pol alpha family DNA polymerase from the hyperthermophilic archaeon Thermococcus sp. 9 degrees N-7. J Mol Biol 299: 447–462 [DOI] [PubMed] [Google Scholar]
- Sawaya MR, Prasad R, Wilson SH, Kraut J, Pelletier H (1997) Crystal structures of human DNA polymerase beta complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry 36: 11205–11215 [DOI] [PubMed] [Google Scholar]
- Schaaper RM, Loeb LA (1981) Depurination causes mutations in SOS-induced cells. Proc Natl Acad Sci USA 78: 1773–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman NC, Rosenberg JM, Rich A (1976) Sequence-specific recognition of double helical nucleic acids by proteins. Proc Natl Acad Sci USA 73: 804–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamoo Y, Steitz TA (1999) Building a replisome from interacting pieces: sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell 99: 155–166 [DOI] [PubMed] [Google Scholar]
- Shibutani S, Takeshita M, Grollman A (1997) Translesional synthesis on DNA templates containing a single abasic site. A mechanistic study of the ‘A rule'. J Biol Chem 272: 13916–13922 [DOI] [PubMed] [Google Scholar]
- Steitz TA (1993) DNA- and RNA-dependent DNA polymerases. Curr Opin Struct Biol 3: 31–38 [Google Scholar]
- Steitz TA (1999) DNA polymerases: structural diversity and common mechanisms. J Biol Chem 274: 17395–17398 [DOI] [PubMed] [Google Scholar]
- Sutton MD, Walker GC (2001) Managing DNA polymerases: coordinating DNA replication, DNA repair, and DNA recombination. Proc Natl Acad Sci USA 98: 8342–8349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita M, Chang CN, Johnson F, Will S, Grollman AP (1987) Oligodeoxynucleotides containing synthetic abasic sites. Model substrates for DNA polymerases and apurinic/apyrimidinic endonucleases. J Biol Chem 262: 10171–10179 [PubMed] [Google Scholar]
- Wallace SS (1997) Oxidative Damage to DNA and Its Repair. In Oxidative Stress and the Molecular Biology of Antioxidant Defenses, Scandalios J (ed) pp 49–90. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Wang J, Sattar AK, Wang CC, Karam JD, Konigsberg WH, Steitz TA (1997) Crystal structure of a pol alpha family replication DNA polymerase from bacteriophage RB69. Cell 89: 1087–1099 [DOI] [PubMed] [Google Scholar]
- Wang J, Yu P, Lin TC, Konigsberg WH, Steitz TA (1996) Crystal structures of an NH2-terminal fragment of T4 DNA polymerase and its complexes with single-stranded DNA and with divalent metal ions. Biochemistry 35: 8110–8119 [DOI] [PubMed] [Google Scholar]
- Yang W (2003) Damage repair DNA polymerases Y. Curr Opin Struct Biol 13: 23–30 [DOI] [PubMed] [Google Scholar]
- Zhao Y, Jeruzalmi D, Moarefi I, Leighton L, Lasken R, Kuriyan J (1999) Crystal structure of an archaebacterial DNA polymerase. Struct Fold Des 7: 1189–1199 [DOI] [PubMed] [Google Scholar]