

Abstract
p47 is a major adaptor molecule of the cytosolic AAA ATPase p97. The principal role of the p97–p47 complex is in regulation of membrane fusion events. Mono-ubiquitin recognition by p47 has also been shown to be crucial in the p97–p47-mediated Golgi membrane fusion events. Here, we describe the high-resolution solution structures of the N-terminal UBA domain and the central domain (SEP) from p47. The p47 UBA domain has the characteristic three-helix bundle fold and forms a highly stable complex with ubiquitin. We report the interaction surfaces of the two proteins and present a structure for the p47 UBA–ubiquitin complex. The p47 SEP domain adopts a novel fold with a βββααβ secondary structure arrangement, where β4 pairs in a parallel fashion to β1. Based on biophysical studies, we demonstrate a clear propensity for the self-association of p47. Furthermore, p97 N binding abolishes p47 self-association, revealing the potential interaction surfaces for recognition of other domains within p97 or the substrate.
Keywords: NMR, p47, p97, SEP, UBA, UBX
Introduction
p47 was first identified in complex with the ATPase p97, in rat liver cytosol (Kondo et al, 1997). p97 belongs to the family of ATPases Associated with different cellular Activities (AAA) (Patel and Latterich, 1998) and utilises specific adaptor molecules in the regulation of various cellular pathways. Several p97 cofactors have been identified so far and include p47, the heterodimer Ufd1–Npl4, SVIP (small VCP/p97-interacting protein) and VCIP135 (Meyer et al, 2000; Uchiyama et al, 2002; Nagahama et al, 2003). p47 is required for the p97-regulated membrane reassembly of the endoplasmic reticulum (ER), the nuclear envelope and the Golgi apparatus (Acharya et al, 1995; Latterich et al, 1995; Rabouille et al, 1995; Kondo et al, 1997; Patel et al, 1998; Roy et al, 2000; Hetzer et al, 2001). It is thought to assist p97 in the disassociation of postfusion Golgi membrane surface receptor complexes, specifically syntaxin 5, using the energy from p97 ATP hydrolysis or binding, and prepare them for further rounds of membrane fusion (Rabouille et al, 1998). Interestingly, it has recently been reported that this process requires the recognition of mono-ubiquitin by the N-terminal UBA domain within p47 (Meyer et al, 2002). It is plausible that ubiquitin serves as a regulatory signal for the structural alteration and relocation of syntaxin 5 complexes. Furthermore, VCIP135 has been shown to bind to the p97/p47/syntaxin 5 complex and dissociate it during p97-catalysed ATP hydrolysis (Uchiyama et al, 2002).
An atomic detailed understanding of adaptor–p97 interactions and the mechanisms by which these molecules modify p97 function has yet to be characterised. A p97 monomer comprises three domains, an N-terminal domain (N) followed by two AAA domains, termed D1 and D2. Structural studies using crystallography and electron microscopy have shown that the D1 and D2 domains are responsible for the formation of a hexamer, while domain N is located at the perimeter of the hexameric ring and exhibits some degree of flexibility (Rockel et al, 1999; Rouiller et al, 2000; Zhang et al, 2000; Beuron et al, 2003). p47 can exist as a trimer and, in the presence of p97, it has been shown that one p47 trimer can form a highly stable complex with one hexamer of p97 (Kondo et al, 1997). In contrast to this observation, a cryo-electron microscopic (cryo-EM) study has suggested that, in the presence of ATP, six p47 molecules bind to the perimeter of the p97 hexamer ring, and that the six flexible p97 N domains become locked into a more rigid conformation (Rouiller et al, 2000).
In this study, we present high-resolution solution structures of the two remaining p47 domains. The UBA domain spans residues 1–45, adopts a characteristic three-helix bundle and forms a specific complex with ubiquitin. We also define the complete interaction surfaces and present a structural model for the p47 UBA–ubiquitin complex. The central domain spans residues 179–246 and it is present in all p47 sequences, the p47 homologue, shp1 from Saccharomyces cerevisiae (Zhang et al, 1995) and eyes closed gene (eyc) from Drosophila melanogaster (Sang and Ready, 2002). It was therefore recently named as the SEP domain, after shp1, eyc and p47 (SMART accession number SM00553; Schultz et al, 2000). The p47 SEP domain adopts a novel fold with a βββααβ secondary structure arrangement, where β4 packs parallel to β1. The SEP–UBX region of p47 shows a clear tendency for specific self-association that could be responsible for p47 trimerisation both in vivo and in vitro. In addition, the p47 SEP–UBX construct forms a stable complex with the p97 N domain and, at the same time, its self-association is completely abolished. This is particularly intriguing as it may have implications for the mechanism of p47-adapted p97 function.
Results and discussion
p47 is a 40.7 kDa protein comprising 370 amino-acid residues. For NMR studies and based on solubility characteristics, we dissected the protein into several constructs (Figure 1). According to secondary structure prediction, p47 contains a small α-helical domain at its N-terminus that has similarity to the UBA domain (Smart accession number SM00165; Schultz et al, 2000). Initial NMR analyses indicate that residues 1–45 of the p47(1–174) construct contain three helices, whereas line width, chemical shift and NOE data indicate that residues 46–174 are completely unstructured. The p47(171–370) fragment is composed of two predicted domains, the most C-terminal of which is the UBX domain (residues 286–370). The structure of this domain has been previously reported and adopts a ubiquitin-like fold (Buchberger et al, 2001; Yuan et al, 2001). Intriguingly, the central SEP domain from p47 comprising residues 179–246 has little sequence homology with other proteins and no known function.
Figure 1.
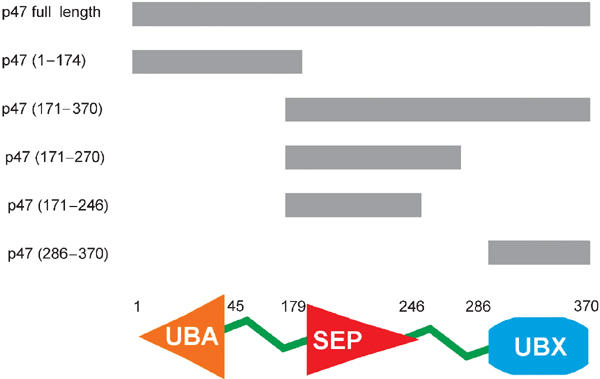
Schematic representation of the domain structure of full-length rat p47 and fragments used in this study.
Solution structure of the p47 UBA domain
The family of NMR structures for p47 UBA domain is shown in Figure 2A. The 10 final structures were selected from the 100 calculated based on agreement with the experimental data and overall structural quality, with no NOE violations greater than 0.5 Å and no torsion angle violations greater than 5°. Structure calculation statistics and structural quality evaluated using the program PROCHECK_NMR (Laskowski et al, 1996) are summarised in Table I. The p47 UBA domain adopts the characteristic UBA fold that comprises a compact three-helix bundle (see Figure 2B). The closest structure to p47 UBA as searched in the DALI structure database (Holm and Sander, 1995) is the crystal structure of the UBA domain from the elongation factor Tu (PDB code 1efu; Kawashima et al, 1996), with a z score of 5.1 and an RMSD of 1.7 Å over 41 equivalent Cα atoms.
Figure 2.
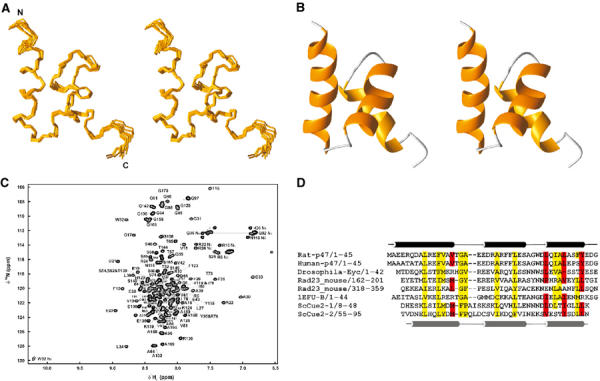
Solution structures of p47 UBA domain. (A) Cα traces representing the superimposition of the 10 refined p47 UBA domain structures. (B) Ribbon representation of p47 UBA domain for the orientation displayed in (A). (C) 1H–15N HSQC spectrum of the p47(1–174) fragment with UBA domain and unstructured region assignments. (D) Sequence alignment and topology for p47 UBA domain. The approximate location of secondary structure elements is indicated; helices are delineated as tubes. Principal residues at the UBA–ubiquitin interface are shaded red and conserved hydrophobic residues are highlighted in yellow.
Table 1.
Structural statistics
| p47 UBA domain | p47 SEP domain | |
|---|---|---|
| Number of experimental restraints (all) | 1011 | 1154 |
| NOE-derived | ||
| Intraresidue | 290 | 521 |
| Sequential | 231 | 240 |
| Medium range (∣i−j∣⩽4) | 201 | 77 |
| Long range (∣i−j∣>4) | 187 | 200 |
| Hydrogen bonds | 44 | 40 |
| Talos (φ/ψ) | 58 | 76 |
| RMSD from experimental restraints | ||
| Distance (Å) | 0.044±0.0024 | 0.046±0.0018 |
| Dihedral angle (deg) | 0.90±0.1526 | 0.632±0.159 |
| RMSD from idealized covalent geometry | ||
| Bonds (Å) | 0.0049±0.0003 | 0.0044±0.00011 |
| Angles (deg) | 0.60±0.022 | 0.543±0.016 |
| Energies (kcal/mol) | ||
| ENOE | 59.3±6.7 | 33.8±5.2 |
| Ebond | 9.0±2.2 | 5.86±1.3 |
| Eangle | 59.8±4.5 | 77.1±3.8 |
| Evdw | 42.5±4.1 | 71.5±4.6 |
| Coordinate RMSD (Å) | Residues 3–42 | Residues 11–65 |
| All backbone atoms | 0.17 | 0.566 |
| All heavy atoms | 0.58 | 1.196 |
| Ramachandran plota | ||
| Residues in most favoured regions (%) | 88.5 | 71.2 |
| Residues in additionally allowed regions (%) | 8.5 | 24.2 |
| Residues in generously allowed regions (%) | 2.9 | 4.3 |
| Residues in disallowed regions (%) |
0.0 |
0.2 |
| a Structural quality was evaluated using PROCHECK_NMR (Laskowski et al, 1996). | ||
The UBA domain is a common sequence motif that occurs 555 times in 457 proteins (Schultz et al, 2000), and is found in many proteins of the ubiquitination pathway, UV excision repair, cell-signalling pathways and cell-cycle control (Hofmann and Bucher, 1996). Specific examples of UBA-containing proteins include p62, the phosphotyrosine-independent ligand of the SH2 domain of p56(lck) (Vadlamudi et al, 1996), the DNA repair protein HHR23A (the human homologue of yeast Rad23A) (Withers-Ward et al, 2000) and the protein nuclear export factor 1 (Tap/NXF1) (Grant et al, 2003). It has been well documented that UBA domains are able to bind to ubiquitin or multi-ubiquitin chains (Bertolaet et al, 2001b; Chen et al, 2001; Wilkinson et al, 2001; Raasi and Pickart, 2003). The UBA domain from p47 binds specifically to mono-ubiquitin and prevents further extension of the ubiquitin chain (Meyer et al, 2002). A multiple sequence alignment of selected UBA domains is illustrated in Figure 2D. It is striking that a number of highly conserved hydrophobic residues form a hydrophobic surface patch that has been predicted to be a common binding site, possibly for ubiquitin (Mueller and Feigon, 2002; Grant et al, 2003), but a detailed atomic picture of this interaction has yet to be reported.
Structural studies of the p47 UBA–ubiquitin complex
Meyer et al (2002) have shown that the UBA domain from p47 is able to bind mono-ubiquitin preferentially and inhibit poly-ubiquitination. Since the high-resolution structure of a UBA–ubiquitin complex has yet to be reported, we performed NMR titration experiments in order to probe this. Figure 3A shows the sequence dependence of combined chemical shift deviations for the backbone 15N and 1HN resonances of 15N-p47 UBA and 15N-ubiquitin in the presence of ubiquitin and p47 UBA, respectively. A number of clustered amide resonances move or broaden upon addition of the binding partner, signifying a likely contact, while the majority of the spectrum remains unchanged. The ubiquitin-binding surface of p47 lies solely in the UBA domain, as no resonance within the unstructured linker is perturbed. Estimation of dissociation constant based on the tracking of peak movements is consistent with the literature studies of UBA–ubiquitin interactions (KD>10 μM) (Bertolaet et al, 2001).
Figure 3.
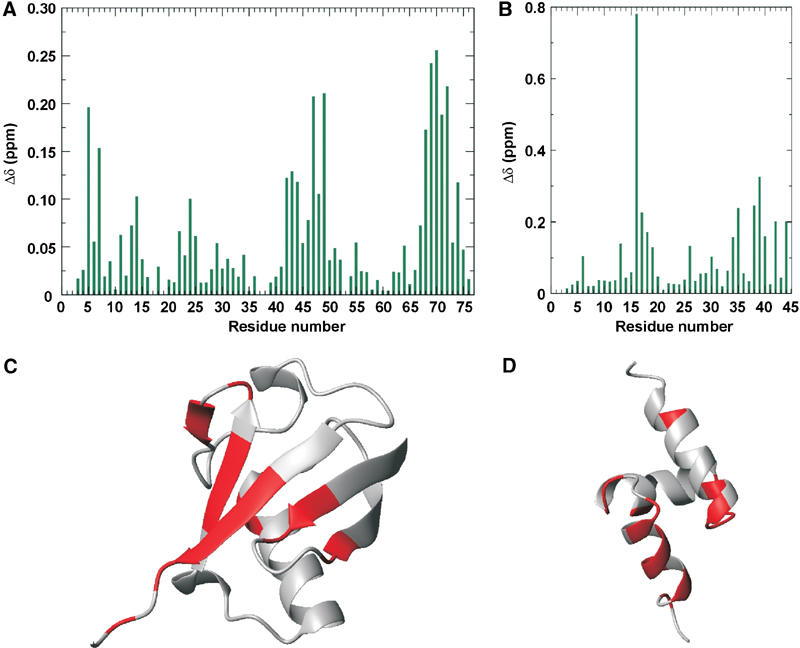
Identification of the mutual interaction surfaces for p47 UBA and ubiquitin. (A) Sequence dependence of the combined chemical shift deviations (Δδ) for the backbone 15N and 1HN resonances of 15N-ubiquitin in the presence of p47 UBA. (B) Sequence dependence of the combined chemical shift deviations (Δδ) for the backbone 15N and 1HN resonances of 15N-p47 UBA in the presence of ubiquitin. (C) Ribbon representation of ubiquitin, with residues experiencing greater than average combined 1H/15N chemical shift perturbations coloured red. (D) Ribbon representation of p47 UBA, with residues experiencing greater than average combined 1H/15N chemical shift perturbations coloured red.
The chemical shift perturbation studies delineated clear interaction surfaces in the p47 UBA–ubiquitin complex (shown in Figure 3), making this highly suitable for structure calculation using the HADDOCK approach (Dominguez et al, 2003). The family of final structures had the lowest intermolecular energy (−420.56 kcal/mol) and the highest buried surface area (1265 Å2). The average pairwise RMSDs for this cluster is 1.0±0.2 Å for the backbone and 1.4±0.3 Å for all heavy atoms (Figure 4). Analysis of the p47 UBA–ubiquitin interface reveals that α1, α3 and loop 1 within p47 UBA form the interface, with α3, being the principal contact face. In α3 hydrophobic side chains of L34, L38 and Y42 make numerous contacts with several ubiquitin side chains, including those of L8, R42, I44 G47, H68 and V70. Furthermore, mutation of the adjacent F41 in p47 eliminates ubiquitin binding (Meyer et al, 2002). The primary side chain from the helix α1/loop 1 region is V15, which interacts with a surface hydrophobic region delineated by L8, H68, I44 and V70 in ubiquitin.
Figure 4.
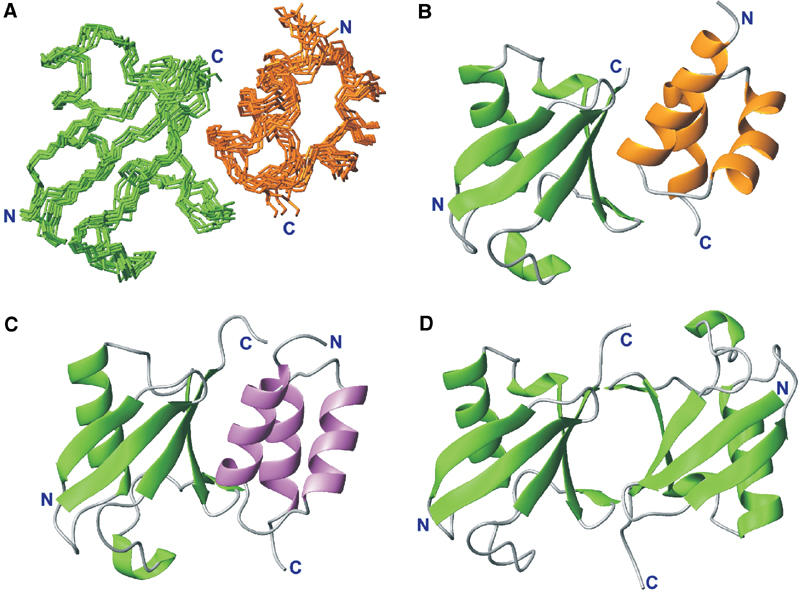
Comparison of p47 UBA–ubiquitin, CUE–ubiquitin and di-ubiquitin structures. (A) Cα traces representing the superimposition of the 10 refined p47 UBA–ubiquitin complex structures using the HADDOCK approach. p47 UBA is shown in gold and ubiquitin in green. (B) Ribbon representation of the p47 UBA–ubiquitin complex. (C) Ribbon representation of the structure of the yeast CUE2-1 domain–ubiquitin complex in the same orientation as in (B). The CUE domain is shown in purple (Kang et al, 2003; Shih et al, 2003). (D) Ribbon representation of the structure of Lys48-linked di-ubiquitin in the same orientation as in (B) (Cook et al, 1992, 1994; Cummings et al, 1995; Beal et al, 1998; Phillips et al, 2001; Varadan et al, 2002).
One of the most striking observations is that similar regions within ubiquitin and ubiquitin-like domains are implicated in the interaction within several related complexes, including CUE–ubiquitin (Kang et al, 2003; Prag et al, 2003; Shih et al, 2003) and intramolecular interactions within the multi-ubiquitin chains (Cook et al, 1992, 1994; Cummings et al, 1995; Beal et al, 1998; Phillips et al, 2001; Varadan et al, 2002). The CUE domain is a small protein that has a role in mono- and poly-ubiquitin recognition, as well as facilitating intramolecular mono-ubiquitination. The CUE domain forms a three-helix bundle that, despite no clear sequence homology, is reminiscent of the classic UBA fold (Figure 4C). The CUE domain binds ubiquitin via two principal motifs within the N- and C-terminal helices. These highly conserved, interfacial sequence motifs, namely the N-terminal MFP motif and the C-terminal di-leucine, could also be identified within some members of the UBA family, corresponding to MGF/Y and di-leucine (Kang et al, 2003; Prag et al, 2003; Shih et al, 2003). These residues lie at the heart of the CUE–ubiquitin interface and were postulated also to be involved in UBA–ubiquitin interactions. Although p47 UBA does not possess this precise family signature, VTGA replaces MGF/Y and the leucine repeat is exchanged for FY; our structural model for the p47 UBA–ubiquitin complex places these residues at the critical interface in an analogous fashion to the CUE–ubiquitin interaction. The average p47 UBA–ubiquitin structure superimposes within a backbone RMSD of 2.4 Å over 100 equivalent residues from the yeast CUE2-1 domain–ubiquitin complex (Figure 4C) (Kang et al, 2003). Furthermore, our model concurs with mutagenesis data, in which modification of the glycine in loop 1 and second leucine in di-leucine within UBA domains reduces ubiquitin binding (Bertolaet et al, 2001; Wilkinson et al, 2001). Our comparison of the CUE–ubiquitin and p47 UBA–ubiquitin complexes validates the proposal that UBA and CUE domains share a similar mode of ubiquitin recognition (Kang et al, 2003; Prag et al, 2003; Shih et al, 2003).
Also noteworthy is the similarity with the ubiquitin interface in K48-linked di-ubiquitin (Cook et al, 1992, 1994; Cummings et al, 1995; Beal et al, 1998; Phillips et al, 2001; Varadan et al, 2002). In the structure of K48-linked di-ubiquitin (Figure 4D), several hydrophobic (L8, I44 and V70) and basic residues (R42 and R72) are positioned at the interface in both crystal and solution forms. It can be seen that the nature of this interface overlaps with that of p47 UBA–ubiquitin, which provides a possible explanation for the inhibitory effect of p47 UBA in poly-ubiquitination of mono-ubiquitinated substrates (Meyer et al, 2002). In this scenario, p47 UBA could compete for this surface and block extension of the ubiquitin chain (Figure 4).
Solution structure of the p47 SEP domain
p47 SEP domain spans residues 179–246 (SMART accession number SM00553; Schultz et al, 2000) and its structure, presented here, is the first to be reported for this sequence family. The sequence alignment for the SEP domain family is shown in Figure 5D. In addition to p47 homologues and sequences of unknown function, a notable member of this family includes undulin 2 (Just et al, 1991). Undulin 2 is an extracellular matrix glycoprotein that comprises two fibronectin type III domains and a potential SEP domain at its C-terminus.
Figure 5.
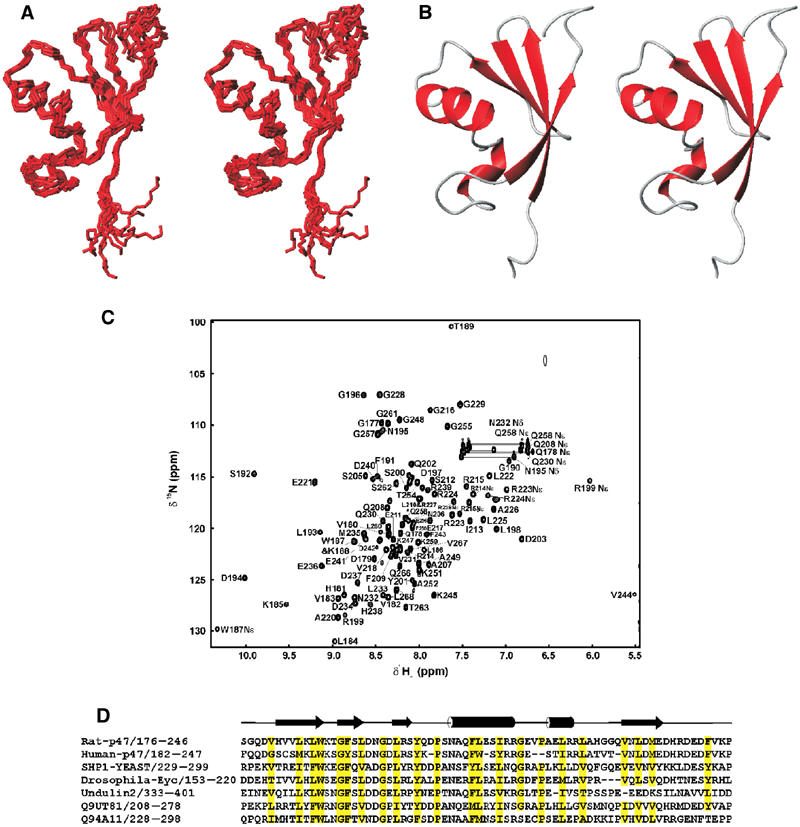
Solution structures of p47 SEP domain. (A) Cα traces representing the superimposition of the 10 refined p47 SEP domain structures. (B) Ribbon representation of the structure of p47 SEP domain for the orientation displayed in (A). (C) 1H–15N HSQC spectrum of the p47(171–270) fragment with SEP domain assignments indicated. (D) Sequence alignment and topology for p47 SEP domain. The approximate location of secondary structure elements is indicated; helices are delineated as tubes and strands as arrows. Numbering is consistent with full-length rat p47. Conserved hydrophobic residues are highlighted in yellow.
For the SEP domain, a total of 1038 structurally significant NOE-derived interproton distance constraints, 40 distance restraints for backbone hydrogen bonds and 76 φ/ϕ dihedral angle restraints were used for the structure determination. The 10 lowest energy structures were chosen from a total of 100 calculated, based on agreement with experimental data and structural quality. In the final family of structures, shown in Figure 5A, no NOE violation greater than 0.4 Å and no torsion angle violation greater than 5° were allowed. The statistics for the structure determination are summarised in Table I. The secondary structure arrangement of the p47 SEP domain is βββααβ, where strand β4 pairs parallel to β1 (Figure 5B). The longer helix α1 packs against the four-stranded β-sheet, where as the shorter helix α2 is located at one edge of the globular structure formed by α1 and the four-stranded β sheet. A number of highly conserved hydrophobic residues are present in the SEP domain (see the sequence alignment in Figure 5D), which are predominantly buried and form the hydrophobic core. A search in the DALI database (Holm and Sander, 1995) revealed no similar structure to the p47 SEP domain, suggesting a novel fold.
Self-association of p47 and interaction with p97 N
Initial NMR structural studies on p47 were carried out with the p47(171–370) construct that contains both SEP and UBX domains. UBX assignments were already completed in our earlier report (Yuan et al, 2001) and it was therefore straightforward to re-assign all the UBX signals in the 1H–15N HSQC spectrum of p47(171–370). There are no changes observed in the UBX resonances when present in the longer fragment, implying that no significant interaction exists between the two domains. In order to quantify the relaxation properties of p47(171–370), 15N relaxation data sets (T1, T2 and heteronuclear 1H–15N NOE) were measured and analysed using the model-free approach (Lipari and Szabo, 1982a, 1982b); these are shown in Figure 6.
Figure 6.
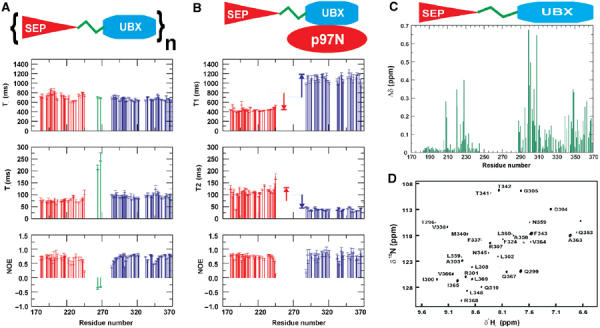
Sequence dependence of the 15N-relaxation data. (A) Sequence dependence of T1, T2 and NOE values for free p47(171–370). (B) Sequence dependence of T1, T2 and NOE values for p47(171–370) in complex with p97 N. The average T1 and T2 values for each domain in free p47(171–370) are also indicated as dotted lines. (C) Chemical shift mapping for the p47(171–370) fragment. Combined chemical shift differences (Δδ) of the backbone 15N and 1HN resonances of the p47(171–370) fragment in free and p97 N-bound state. (D) Difference spectrum between cross-saturation experiment and control irradiated for deuterated p47(171–370) in the presence of protonated p97 N. Only residues at the interface appear in the 15N–1H HSQC spectra, and are predominantly observed in the UBX domain.
Although there is chemical shift evidence (BRMB accession number 5874) for helical propensity between residues 276 and 289, the T1, T2 and NOE values measured for resolved resonances indicate that parts of the linker region exhibit flexibility in solution. Surprisingly, the small SEP domain (67 residues—∼7.5 kDa) has very high overall tumbling time, τm=11.4 ns, which is appreciably larger than expected for an independently tumbling 7.5 kDa domain (τm∼4–6 ns). One explanation for this would be the multimerisation or self-association of the SEP domain or proximal regions within p47(171–370). Since NOE contacts between SEP and UBX domains are not observed in the free p47(171–370) construct, a direct interaction between SEP and UBX is unlikely. The overall tumbling time for the UBX is also somewhat longer than expected for an independent 10 kDa domain (9.6 ns compared to 5–7 ns). This systematic increase could either be due to the UBX domain being tethered to a severely self-associated SEP domain or a second self-association determinant within UBX.
In our previous report, we identified the p97 N-binding site within the p47 UBX domain, which comprises part of the five-stranded β-sheet of the ubiquitin-like fold. Furthermore, previous studies have identified a second p97-binding site within either the SEP domain or the SEP–UBX linker (Uchiyama et al, 2002). Several striking observations are observed in the relaxation data when the p97 N titration is performed with the longer p47(171–370) fragment. First, the overall tumbling of UBX slows dramatically from 9.6 to 20.6 ns upon p97 N binding, which is highly consistent with the correlation time expected for an independent 35 kDa UBX–p97 N complex (Yuan et al, 2001). Quite the reverse is true for the SEP domain since the relaxation parameters remarkably transform to values consistent with a single, independently tumbling 7.5 kDa moiety. The overall correlation time of the SEP domain drops to 6.4 ns, from 11.4 ns, indicating that the observed self-association is abolished in the presence of p97 N (Figure 6). Finally, p97 N binding induces severe broadening of resonances within the linker between UBX and SEP, indicating that it exhibits conformational exchange on an intermediate timescale possibly by forming a direct interaction with p97 N, consistent with recent observations of a second p97-binding site (Uchiyama et al, 2002).
The interaction of p47(171–370) with p97 N was further explored by comparing the backbone amide chemical shifts in free p47(171–370) and p97 N-bound p47(171–370) (Figure 6C). A 1H–15N HSQC spectrum was recorded for each system under identical experimental conditions. Chemical shift perturbations within the UBX domain are primarily the result of the well-characterised interaction with p97 N (Yuan et al, 2001), but there are also significant chemical shift differences observed within the SEP domain. Most notably, residues N206, Q208, F209, A220, E221, R224, A226, H227, G228, G229, Q230, L233 and D237 show significant chemical shift changes upon p97 N–UBX binding (Figure 6C). These residues form part of a region formed by helix α1, α2 and edge of β4, but all lie on one face of the molecule. In order to assess whether a direct interaction existed between the SEP domain and p97 N, a cross-saturation experiment was performed with deuterated p47(171–370) in the presence of unlabelled p97 N. Selective irradiation of the aliphatic region in p97 N while observing the magnetisation transfer to the amides of 2H-p47(171–370) reveals unambiguously the interaction interface (Lane et al, 2001). As expected, the difference spectrum reveals amides from the UBX domain (Figure 6D), which concur with our published binding-site identification (Yuan et al, 2001). Additional resonances that are not attributable to the UBX or SEP domains are also observed and point towards an interaction of p97 N with the SEP–UBX linker. The complete absence of SEP resonances in this spectrum indicates that direct interaction between p47 SEP and p97 N is unlikely and the chemical shift differences observed in Figure 6C are expected to be a result of dissociation of the fragment to monodispersed species or the proximity of a p97 N/SEP–UBX linker interaction.
The self-association properties of the p47(171–370) fragment were further investigated by performing a series of crosslinking experiments with disuccinimidyl suberate (DSS). Figure 7A shows the SDS–PAGE of the crosslinked reactions for full-length p47 and the various fragments shown in Figure 1. In line with our NMR data, the fragment containing the UBA domain does not form significant amounts of crosslinked product, suggesting that there is no tendency to self-associate. The fragment containing both SEP and UBX domains shows a similar profile to full-length p47 in that populations of crosslinked dimers and trimers are detected, signifying that this fragment has a significant propensity to self-associate. This is in agreement with NMR relaxation data and early reports that full-length p47 can exist as a trimer (Kondo et al, 1997). In order to provide further evidence of a specific interaction within self-associated p47 species, we quantified their molecular weight by nano-electrospray mass spectrometry (Nettleton et al, 1998). Figure 7B clearly indicates the presence of multimeric species of p47(171–370) up to tetramer. The SEP–UBX fragment was then dissected into p47(171–285) and p47(286–370) constructs encompassing the SEP and UBX domains, respectively. Crosslinked products could also be identified in both p47(171–285) and p47(286–370) fragments, indicating the presence of two self-association determinants. Two-site contact between p47 molecules provides an explanation for the observation of dimers, trimers and tetramers in the mass spectrum of p47(171–370) (Figures 7A and B). Analysis of NMR relaxation data for the p47(171–270) fragment gives an overall correlation time of ∼11 ns for the SEP domain, which indicates self-associating properties identical to the free SEP–UBX construct. Interestingly, a monomeric status can be generated by removal of the 24-amino-acid residues C-terminal to the SEP domain, which implicates this region of the SEP–UBX linker in self-association. In the context of p47(171–370), the removal of oligomerisation upon p97 N binding can be explained by direct interactions with both the SEP–UBX linker and UBX domain, thereby occluding the self-association interfaces.
Figure 7.
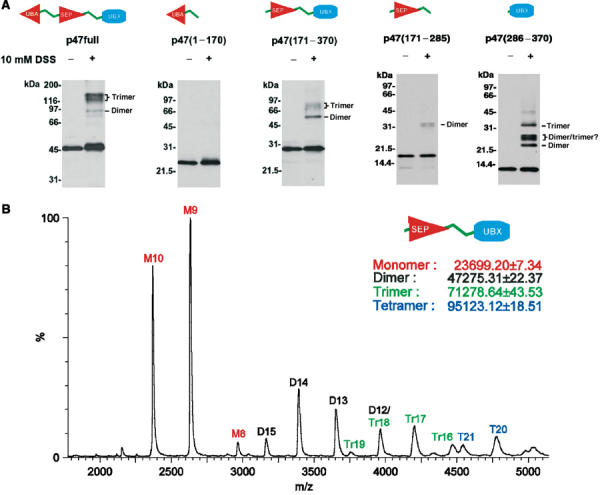
Self-association properties of p47. (A) SDS–PAGE of crosslinked experiments for full-length rat p47 containing UBA, SEP and UBX domains, p47(1–170) containing the UBA domain, p47(171–370) containing the SEP and UBX domains, p47(171–285) containing SEP domain and p47(286–370) containing the UBX domain. (B) Electrospray mass spectrum of p47(171–370). Peaks corresponding in mass to monomer (M), dimer (D), trimer (Tr) and tetramer (T) can be observed.
Conclusions
p47 contains three structured domains (UBA, SEP and UBX) and displays a high degree of flexibility within the linker regions (residues 46–178 and 247–281). The linker between UBA and SEP (residues 46–178) is of great interest since at least one important functional site has been identified in this region. Specifically, the phosphorylation of Ser140 by Cdc2 at mitosis is essential for Golgi disassembly, since the phosphorylated p47 does not bind to Golgi membranes; thus, the p97-mediated Golgi membrane fusion is inhibited (Uchiyama et al, 2003). This in turn implies that the unstructured region around Ser140 is likely to be involved in p47 recognition of the membrane surface receptor complex. It is also plausible that this region adopts a specific structure upon binding to syntaxin 5 and that phosphorylation of Ser140 modulates this interaction by either conformational or electrostatic changes.
The UBA domain from p47 adopts a classic UBA fold and forms a specific complex with ubiquitin. A structural model of the p47 UBA–ubiquitin complex places two critical motifs (TVGA and FY) at the interface in an analogous fashion to the CUE–ubiquitin interaction and verifies that UBA and CUE domains share similar modes of ubiquitin recognition (Kang et al, 2003; Prag et al, 2003; Shih et al, 2003). The inhibitory effect of p47 UBA on substrate poly-ubiquitination can be explained by straightforward occlusion of the intramolecular ubiquitin–ubiqutin interface and restriction of the access to K48.
The unstructured region between the SEP and UBX domain is particularly fascinating. It is highly flexible in free p47 but appears to become more restricted upon p97 N binding, possibly by adopting a defined structure in association with p97 N. A second p97 N-binding site in addition to the UBX domain is consistent with data showing that a construct encompassing the SEP domain and part of the linker interacts with p97 (Uchiyama et al, 2002). In the context of the full-length AAA ATPase p97, it is plausible that structuring of this linker upon binding p97 and subsequent removal of p47 self-association are brought about by changes in the p97 N domain accessibility therefore revealing new interactional surfaces. Cryo-EM studies have revealed that the flexibility of the p97 N domains is modulated upon ATP hydrolysis (Rouiller et al, 2000; Beuron et al, 2003). The more rigid state of the p97 N domain may hinder a complete multipoint p47–p97 N interaction, whereas release of p97 N domains to a flexible state could enable the full interaction. This could unveil potential interaction surfaces within p47, which could be available for association with other p97 domains and cofactors and perhaps amplify the mechanical force exerted on the substrate.
Materials and methods
Protein preparation
p47 has been dissected into several constructs comprising amino acids 1–174, 171–370, 171–270 and 286–370. p47(1–174), p47(171–370), p47(171–270) and p47(286–370) constructs were cloned and expressed as a recombinant protein within Escherichia coli. The expressed fragments contain an N-terminal hexahistidine tag (MRGSHHHHHHGS) for purification and were readily concentrated to mM concentrations. For NMR 15N, 13C double-labelled samples of p47(1–174), p47(171–370) and p47(171–270) were produced in minimal media, containing 0.07% 15NH4Cl and 0.2% 13C-glucose, supplemented with 50 μg/ml ampicillin. Protein expression was induced by the addition of 1 mM isopropyl-β-D-thiogalactopyroside. Clarified cell lysate was purified essentially to homogeneity in a single step on Ni2+-chelating HiTrap Column (Pharmacia). p47(171–246) was prepared by limited proteolysis of the p47(171–270) fragment. Purified proteins was dialysed into 20 mM sodium acetate buffer at pH 5.3 and concentrated to approximately 0.5 mM for NMR.
NMR spectroscopy
The majority of NMR spectra were recorded at 298 K on a 500 MHz four-channel Bruker DRX500 spectrometer equipped with a z-shielded gradient triple resonance cryoprobe. Sequence-specific backbone 1HN, 15N, 13Cα and 13Cβ were determined using standard triple resonance methods (Grzesiek and Bax, 1992a, 1992b; Kay et al, 1994; Muhandiram and Kay, 1994). Secondary structure and side-chain assignments were obtained from HCCH-TOCSY experiments (Bax et al, 1990). 3D 1H–15N/13C NOESY-HSQC (mixing time 100 ms) spectra provided distance restraints used in the final structure calculations. A further 3D 1H–13C NOESY-HSQC spectrum (mixing time 80 ms) was recorded at 800 MHz in order to resolve overlap. Structural restraints for the SEP domain were acquired from NMR data recorded on the p47(171–246) fragment. All spectra were processed with NMRPipe (Delaglio et al, 1995) and analysed with NMRView (Johnson and Blevins, 1994). Amide protons involved in hydrogen bonds were identified by the presence of NH resonances in HSQC spectra recorded 12 h after dissolving in D2O. The hydrogen bond acceptors were identified according to distinctive neighbouring NOEs. Backbone dihedral angle restraints (ϕ and φ angles) were predicted using the program TALOS (Cornilescu et al, 1999).
NMR chemical shift mapping and cross-saturation experiments
For NMR mapping experiments, 15N-labelled p47(1–174) containing the UBA domain was prepared in 20 mM sodium acetate buffer at pH 5.3 at approximately 30 μM in 0.5 ml. Unlabelled ubiquitin (Sigma Inc.) in the same buffer was introduced in four steps, at the molar ratios (ubiquitin:15N-p47(1–174)) of 0, 0.5, 1.0 and 2.7:1, and 15N-1H HSQC spectra were recorded at each stage under identical experimental conditions. The experiment was repeated with 15N-labelled ubiquitin (Silantes Gbm) and unlabelled p47(1–174) at the molar ratios (p47(1–174):15N-ubiquitin) of 0, 0.5, 1.0 and 2.7:1. A suitable cutoff value of the average weighted chemical shift perturbation was defined as the minimum value indicating significant conformational change (Wishart et al, 1991; Williamson et al, 1997). For the cross-saturation experiment on perdeuterated 15N-p47(171–370) in the presence of p97 N, 15N–1H correlation spectra were performed with ∼1 s pre-irradiation over a selective frequency band (Lane et al, 2001). Two experiments were performed with adiabatic pulses centred outside and inside the p97 N aliphatic chemical shift range, respectively. The resulting difference spectrum reveals residues within 15N-p47(171–370) at the p97 N interface.
15N relaxation measurements
15N T1, 15N T2 and 1H–15N heteronuclear NOE data were measured using methods described elsewhere (Kay et al, 1989; Farrow et al, 1994). For free p47(171–370), relaxation delays of 50, 250, 400, 650, 950 and 1400 ms were employed for T1 measurements, and delays of 16.6, 33.2, 49.8, 66.4, 83.0, 99.6, 149.4 and 199.2 ms for T2. For p47(171–370) in complex with p97 N, relaxation delays of 50, 150, 300, 500, 750, 1100 and 1500 ms were employed for T1 measurements, and delays of 16.6, 33.2, 49.8, 66.4, 83.0, 99.6 and 149.4 ms for T2. A single repeat was performed for two delays in each experiment and these were used in evaluating uncertainties in peak intensity. Overlapped resonances were not included in the analysis.
Structure calculation for p47 UBA and SEP domains
Structure calculations were started from extended structures and using a hybrid algorithm including torsion angle dynamics (TAD) and Cartesian dynamics executed within the program CNS/Xplor (Brunger, 1993; Brunger et al, 1998). All parameters were scaled using default CNS values. In all, 100 structures were calculated and were ranked in overall energy terms; the best 10% were selected as representative and are presented in Figures 2 and 5 for p47 UBA and SEP, respectively. No distance violation greater than 0.4 Å and no dihedral violation greater than 5° were tolerated in the final family of structures. The structural coordinates of p47 UBA and SEP have been deposited in the PDB under codes IV92 and IVAZ.
Structure calculation for p47 UBA–ubiquitin complex
The chemical shift perturbation studies delineated clear interaction surface areas in the p47 UBA–ubiquitin complex, making this highly suitable for structure calculation using the HADDOCK approach (Dominguez et al, 2003). For the calculation, the crystal structure of human ubiquitin (Vijaykumar et al, 1985, 1987) and the lowest energy NMR structure from the family of p47 UBA structures were used. In all, 11 amino-acid residues in p47 UBA and 12 in ubiquitin were identified to have weighted chemical shift changes significantly greater than the average; 0.07 ppm in ubiquitin and 0.09 ppm in p47 UBA were chosen as suitable cutoff values. After filtering for a relative solvent accessibility greater than 50%, as calculated using the program Naccess (Hubbard and Thornton, 1993), eight residues in p47 UBA (Q6, V15, G17, Q35, A39, S40, Y42 and D44) and eight for ubiquitin (K6, K11, A46, K48, Q49, H68, R72 and R74) were identified as active residues. Residues juxtaposed to these, which have a relative solvent accessibility greater than 50%, were termed passive residues. These included 15 further residues in p47 UBA (E3, E4, R5, D7, R10, E11, A14, E20, D21, R22, D33, I36, F41, E43 and G45) and 10 in ubiquitin (L8, T9, D39, G47, E51, L67, L69, V70, L73 and G75). An ambiguous distance restraint of 3.0 Å was invoked between all active residues in one partner molecule to any atom within the active and passive residues of the other protein partner. The interfacial residues that were allowed to move during the simulated annealing and water refinement were F12–D21 and G31–E43 for p47 UBA and F44–D52 and S65–G76 for ubiquitin. Residues H68–G76 in ubiquitin were allowed full flexibility during the whole simulated annealing process, as this C-terminal region is flexible in monomeric solution structures (Cornilescu et al, 1998). In all, 1000 initially complex structures were generated by rigid body energy minimisation, and the best 200 by total energy were selected for torsion angle dynamics and subsequent Cartesian dynamics in an explicit water solvent. Default scaling for energy terms is as described previously (Dominguez et al, 2003).
Crosslinking experiments on p47 fragments
His-tagged p47 was prepared as described previously (Kondo et al, 1997). To obtain His-tagged p47 fragments, the corresponding cDNAs with stop codons were subcloned into pQE30 using BamHI and KpnI sites. The fragments were expressed in E. coli and purified with Ni beads, followed by further purification with gel filtration. The protein sample (1 μg) was incubated together with 10 mM DSS in 20 μl of buffer (20 mM HEPES, 0.15 M KCl, 1 mM DTT, 5% glycerol, pH 7.4). After 30 min incubation at room temperature, 1 μl of 1 M Tris (pH 7.4) was added to quench excess DSS. The resulting samples were fractionated by SDS–PAGE. Blots were probed with anti-His antibodies.
Nano-electrospray mass spectrometry
The nano-electrospray mass spectrum was recorded on an LCT MS (Micromass, UK) equipped with a z-spray source. This instrument is modified for high mass operation, with an argon inlet in the first RF hexapole. Analyses were carried out under conditions of increased pressure in the source and intermediate pressure regions of the mass spectrometer (Pirani gauge readback 6.2 mbar and Penning gauge readback 1 × 10−6 mbar) in order to maintain noncovalent interactions. Nano-flow needles were made in-house as described previously (Nettleton et al, 1998). The p47(171–370) protein (concentration 18 mg/ml) was buffer exchanged using Micro Bio-spin 6 columns (Bio-Rad Laboratories, Hercules, CA) into 200 mM ammonium acetate (pH 7.0) immediately prior to analysis.
Acknowledgments
ID, CM, PSF, PS, RW, SM, XZ and XY thank the support from the Wellcome Trust. HK is grateful to the support from the MRC and the Wellcome Trust. CR and CK thank the BBSRC and The Royal Society. We also thank Geoff Kelly and Tom Frenkiel of the 800 MHz NMR service at NIMR.
References
- Acharya U, Jacobs R, Peters JM, Watson N, Farquhar MG, Malhotra V (1995) The formation of Golgi stacks from vesiculated Golgi membranes requires 2 distinct fusion events. Cell 82: 895–904 [DOI] [PubMed] [Google Scholar]
- Bax A, Clore GM, Gronenborn AM (1990) H-1–H-1 correlation via isotropic mixing of C-13 magnetization, a new 3-dimensional approach for assigning H-1 and C-13 spectra of C-13-enriched proteins. J Magn Reson 88: 425–431 [Google Scholar]
- Beal RE, Toscano-Cantaffa D, Young P, Rechsteiner M, Pickart CM (1998) The hydrophobic effect contributes to polyubiquitin chain recognition. Biochemistry 37: 2925–2934 [DOI] [PubMed] [Google Scholar]
- Bertolaet BL, Clarke DJ, Wolff M, Watson MH, Henze M, Divita G, Reed SI (2001b) UBA domains of DNA damage-inducible proteins interact with ubiquitin. Nat Struct Biol 8: 417–422 [DOI] [PubMed] [Google Scholar]
- Beuron F, Flynn TC, Ma JP, Kondo H, Zhang XD, Freemont PS (2003) Motions and negative cooperativity between p97 domains revealed by cryo-electron microscopy and quantised elastic deformational model. J Mol Biol 327: 619–629 [DOI] [PubMed] [Google Scholar]
- Brunger AT (1993) XPLOR Manual Ver 3.1. New Haven: Yale University Press [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr Section D—Biol Crystallogr 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Buchberger A, Howard MJ, Proctor M, Bycroft M (2001) The UBX domain: a widespread ubiquitin-like module. J Mol Biol 307: 17–24 [DOI] [PubMed] [Google Scholar]
- Chen L, Shinde U, Ortolan TG, Madura K (2001) Ubiquitin-associated (UBA) domains in Rad23 bind ubiquitin and promote inhibition of multi-ubiquitin chain assembly. EMBO Rep 2: 933–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook WJ, Jeffrey LC, Carson M, Chen ZJ, Pickart CM (1992) Structure of a diubiquitin conjugate and a model for interaction with ubiquitin conjugating enzyme (E2). J Biol Chem 267: 16467–16471 [DOI] [PubMed] [Google Scholar]
- Cook WJ, Jeffrey LC, Kasperek E, Pickart CM (1994) Structure of tetraubiquitin shows how multiubiquitin chains can be formed. J Mol Biol 236: 601–609 [DOI] [PubMed] [Google Scholar]
- Cornilescu G, Delaglio F, Bax A (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol Nmr 13: 289–302 [DOI] [PubMed] [Google Scholar]
- Cornilescu G, Marquardt JL, Ottiger M, Bax A (1998) Valigation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J Am Chem Soc 120: 6836–6837 [Google Scholar]
- Cummings MD, Hart TN, Read RJ (1995) Monte-Carlo docking with ubiquitin. Prot Sci 4: 885–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe—a multidimensional spectral processing system based on Unix pipes. J Biomol Nmr 6: 277–293 [DOI] [PubMed] [Google Scholar]
- Dominguez C, Boelens R, Bonvin AMJJ (2003) HADDOCK: a protein–protein docking approach based on biochemical and/or biophysical information. J Am Chem Soc 125: 1731–1737 [DOI] [PubMed] [Google Scholar]
- Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Formankay JD, Kay LE (1994) Backbone dynamics of a free and a phosphopeptide-complexed Src homology-2 domain studied by N-15 Nmr relaxation. Biochemistry 33: 5984–6003 [DOI] [PubMed] [Google Scholar]
- Grant RP, Neuhaus D, Stewart M (2003) Structural basis for the interaction between the Tap/NXF1 UBA domain and FG nucleoporins at 1 angstrom resolution. J Mol Biol 326: 849–858 [DOI] [PubMed] [Google Scholar]
- Grzesiek S, Bax A (1992a) An efficient experiment for sequential backbone assignment of medium-sized isotopically enriched proteins. J Magn Reson 99: 201–207 [Google Scholar]
- Grzesiek S, Bax A (1992b) Improved 3d triple-resonance NMR techniques applied to a 31-kDa protein. J Magn Reson 96: 432–440 [Google Scholar]
- Hetzer MW, Meyer HH, Walther TC, Warren G, Mattaj IW (2001) The AAA-ATPase p97 functions in two discrete steps of nuclear envelope formation. Mol Biol Cell 12: 13. [DOI] [PubMed] [Google Scholar]
- Hofmann K, Bucher P (1996) The UBA domain: a sequence motif present in multiple enzyme classes of the ubiquitination pathway. Trends Biochem Sci 21: 172–173 [PubMed] [Google Scholar]
- Holm L, Sander C (1995) Dali—a network tool for protein-structure comparison. Trends Biochem Sci 20: 478–480 [DOI] [PubMed] [Google Scholar]
- Hubbard SJ, Thornton JM (1993) ‘NACCESS', Computer Program. University College London: Department of Biochemistry and Molecular Biology [Google Scholar]
- Johnson BA, Blevins RA (1994) NMRView—a computer-program for the visualization and analysis of NMR data. J Biomol Nmr 4: 603–614 [DOI] [PubMed] [Google Scholar]
- Just M, Herbst H, Hummel M, Durkop H, Tripier D, Stein H, Schuppan D (1991) Undulin is a novel member of the fibronectin–tenascin family of extracellular-matrix glycoproteins. J Biol Chem 266: 17326–17332 [PubMed] [Google Scholar]
- Kang RS, Daniels CM, Francis SA, Shih SC, Salerno WJ, Hicke L, Radhakrishnan I (2003) Solution structure of a CUE–ubiquitin complex reveals a conserved mode of ubiquitin binding. Cell 113: 621–630 [DOI] [PubMed] [Google Scholar]
- Kawashima T, BerthetColominas C, Wulff M, Cusack S, Leberman R (1996) The structure of the Escherichia coli EF-Tu center dot EF-Ts complex at 2.5 angstrom resolution. Nature 379: 511–518 [DOI] [PubMed] [Google Scholar]
- Kay LE, Torchia DA, Bax A (1989) Backbone dynamics of proteins as studied by N-15 inverse detected heteronuclear Nmr-spectroscopy—application to staphylococcal nuclease. Biochemistry 28: 8972–8979 [DOI] [PubMed] [Google Scholar]
- Kay LE, Xu GY, Yamazaki T (1994) Enhanced-sensitivity triple-resonance spectroscopy with minimal H2O saturation. J Magn Reson Ser A 109: 129–133 [Google Scholar]
- Kondo H, Rabouille C, Newman R, Levine TP, Pappin D, Freemont P, Warren G (1997) p47 is a cofactor for p97-mediated membrane fusion. Nature 388: 75–78 [DOI] [PubMed] [Google Scholar]
- Lane AN, Kelly G, Ramos A, Frenkiel TA (2001) Determining binding sites in protein-nucleic acid complexes by cross-saturation. J Biomol Nmr 21: 127–139 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol Nmr 8: 477–486 [DOI] [PubMed] [Google Scholar]
- Latterich M, Frohlich KU, Schekman R (1995) Membrane-fusion and the cell-cycle—Cdc48p participates in the fusion of ER membranes. Cell 82: 885–893 [DOI] [PubMed] [Google Scholar]
- Lipari G, Szabo A (1982a) Model-free approach to the interpretation of nuclear magnetic-resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc 104: 4546–4559 [Google Scholar]
- Lipari G, Szabo A (1982b) Model-free approach to the interpretation of nuclear magnetic-resonance relaxation in macromolecules. 2. Analysis of experimental results. J Am Chem Soc 104: 4559–4570 [Google Scholar]
- Meyer HH, Shorter JG, Seemann J, Pappin D, Warren G (2000) A complex of mammalian Ufd1 and Npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J 19: 2181–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer HH, Wang YZ, Warren G (2002) Direct binding of ubiquitin conjugates by the mammalian p97 adaptor complexes, p47 and Ufd1-Npl4. EMBO J 21: 5645–5652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller TD, Feigon J (2002) Solution structures of UBA domains reveal a conserved hydrophobic surface for protein–protein interactions. J Mol Biol 319: 1243–1255 [DOI] [PubMed] [Google Scholar]
- Muhandiram DR, Kay LE (1994) Gradient-enhanced triple-resonance 3-dimensional NMR experiments with improved sensitivity. J Magn Reson Ser B 103: 203–216 [Google Scholar]
- Nagahama M, Suzuki M, Hamada Y, Hatsuzawa K, Tani K, Yamamoto A, Tagaya M (2003) SVIP is a novel VCP/p97-interacting protein whose expression causes cell vacuolation. Mol Biol Cell 14: 262–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nettleton EJ, Sunde M, Lai ZH, Kelly JW, Dobson CM, Robinson CV (1998) Protein subunit interactions and structural integrity of amyloidogenic transthyretins: evidence from electrospray mass spectrometry. J Mol Biol 281: 553–564 [DOI] [PubMed] [Google Scholar]
- Patel S, Latterich M (1998) The AAA team: related ATPases with diverse functions. Trends Cell Biol 8: 65–71 [PubMed] [Google Scholar]
- Patel SK, Indig FE, Olivieri N, Levine ND, Latterich M (1998) Organelle membrane fusion: a novel function for the syntaxin homolog Ufe1p in ER membrane fusion. Cell 92: 611–620 [DOI] [PubMed] [Google Scholar]
- Phillips CL, Thrower J, Pickart CM, Hill CP (2001) Structure of a new crystal form of tetraubiquitin. Acta Crystallogr Section D—Biol Crystallogr 57: 341–344 [DOI] [PubMed] [Google Scholar]
- Prag G, Misra S, Jones EA, Ghirlando R, Davies BA, Horazdovsky BF, Hurley JH (2003) Mechanism of ubiquitin recognition by the CUE domain of Vps9p. Cell 113: 609–620 [DOI] [PubMed] [Google Scholar]
- Raasi S, Pickart CM (2003) Rad23 ubiquitin-associated domains (UBA) inhibit 26 S proteasome-catalyzed proteolysis by sequestering lysine 48-linked polyubiquitin chains. J Biol Chem 278: 8951–8959 [DOI] [PubMed] [Google Scholar]
- Rabouille C, Kondo H, Newman R, Hui N, Freemont P, Warren G (1998) Syntaxin 5 is a common component of the NSF- and p97-mediated reassembly pathways of Golgi cisternae from mitotic Golgi fragments in vitro. Cell 92: 603–610 [DOI] [PubMed] [Google Scholar]
- Rabouille C, Levine TP, Peters JM, Warren G (1995) An NSF-like ATPase, p97, and NSF mediate cisternal regrowth from mitotic Golgi fragments. Cell 82: 905–914 [DOI] [PubMed] [Google Scholar]
- Rockel B, Walz J, Hegerl R, Peters J, Typke D, Baumeister W (1999) Structure of VAT, a CDC48/p97 ATPase homologue from the archaeon Thermoplasma acidophilum as studied by electron tomography. FEBS Lett 451: 27–32 [DOI] [PubMed] [Google Scholar]
- Rouiller I, Butel VM, Latterich M, Milligan RA, Wilson-Kubalek EM (2000) A major conformational change in p97 AAA ATPase upon ATP binding. Mol Cell 6: 1485–1490 [DOI] [PubMed] [Google Scholar]
- Roy L, Bergeron JJM, Lavoie C, Hendriks R, Gushue J, Fazel A, Pelletier A, Morre DJ, Subramaniam VN, Hong WJ, Paiement J (2000) Role of p97 and syntaxin 5 in the assembly of transitional endoplasmic reticulum. Mol Biol Cell 11: 2529–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang TK, Ready DF (2002) Eyes closed, a Drosophila p47 homolog, is essential for photoreceptor morphogenesis. Development 129: 143–154 [DOI] [PubMed] [Google Scholar]
- Schultz J, Copley RR, Doerks T, Ponting CP, Bork P (2000) SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res 28: 231–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih SC, Prag G, Francis SA, Sutanto MA, Hurley JH, Hicke L (2003) A ubiquitin-binding motif required for intramolecular monoubiquitination, the CUE domain. EMBO J 22: 1273–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama K, Jokitalo E, Kano F, Murata M, Zhang XD, Canas B, Newman R, Rabouille C, Pappin D, Freemont P, Kondo H (2002) VCIP135, a novel essential factor for p97/p47-mediated membrane fusion, is required for Golgi and ER assembly in vivo. J Cell Biol 159: 855–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama K, Jokitalo E, Lindman M, Jackman M, Kano F, Murata M, Zhang XD, Kondo H (2003) The localization and phosphorylation of p47 are important for Golgi disassembly–assembly during the cell cycle. J Cell Biol 161: 1067–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadlamudi RK, Joung I, Strominger JL, Shin J (1996) p62, a phosphotyrosine-independent ligand of the SH2 domain of p56(lck), belongs to a new class of ubiquitin-binding proteins. J Biol Chem 271: 20235–20237 [DOI] [PubMed] [Google Scholar]
- Varadan R, Walker O, Pickart C, Fushman D (2002) Structural properties of polyubiquitin chains in solution. J Mol Biol 324: 637–647 [DOI] [PubMed] [Google Scholar]
- Vijaykumar S, Bugg CE, Cook WJ (1987) Structure of ubiquitin refined at 1.8 A resolution. J Mol Biol 194: 531–544 [DOI] [PubMed] [Google Scholar]
- Vijaykumar S, Bugg CE, Wilkinson KD, Cook WJ (1985) 3-dimensional structure of ubiquitin at 2.8 A resolution. Proc Natl Acad Sci USA 82: 3582–3585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson CRM, Seeger M, Hartmann-Petersen R, Stone M, Wallace M, Semple C, Gordon C (2001) Proteins containing the UBA domain are able to bind to multi-ubiquitin chains. Nat Cell Biol 3: 939–943 [DOI] [PubMed] [Google Scholar]
- Williamson RA, Carr MD, Frenkiel TA, Feeney J, Freedman RB (1997) Mapping the binding site for matrix metalloproteinase on the N-terminal domain of the tissue inhibitor of metalloproteinases-2 by NMR chemical shift perturbation. Biochemistry 36: 13882–13889 [DOI] [PubMed] [Google Scholar]
- Wishart DS, Sykes BD, Richards FM (1991) Relationship between nuclear-magnetic-resonance chemical-shift and protein secondary structure. J Mol Biol 222: 311–333 [DOI] [PubMed] [Google Scholar]
- Withers-Ward ES, Mueller TD, Chen ISY, Feigon J (2000) Biochemical and structural analysis of the interaction between the UBA(2) domain of the DNA repair protein HHR23A and HIV-1 Vpr. Biochemistry 39: 14103–14112 [DOI] [PubMed] [Google Scholar]
- Yuan XM, Shaw A, Zhang XD, Kondo H, Lally J, Freemont PS, Matthews S (2001) Solution structure and interaction surface of the C-terminal domain from p47: a major p97-cofactor involved in SNARE disassembly. J Mol Biol 311: 255–263 [DOI] [PubMed] [Google Scholar]
- Zhang SR, Guha S, Volkert FC (1995) The saccharomyces Shp1 gene, which encodes a regulator of phosphoprotein phosphatase-1 with differential-effects on glycogen-metabolism, meiotic differentiation, and mitotic cell-cycle progression. Mol Cell Biol 15: 2037–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XD, Shaw A, Bates PA, Newman RH, Gowen B, Orlova E, Gorman MA, Kondo H, Dokurmo P, Lally J, Leonard G, Meyer HE, van Heel M, Freemont PS (2000) Structure of the AAA ATPase p97. Mol Cell 6: 1473–1484 [DOI] [PubMed] [Google Scholar]