

Abstract
Colicins are toxins secreted by Escherichia coli in order to kill their competitors. Colicin D is a 75 kDa protein that consists of a translocation domain, a receptor-binding domain and a cytotoxic domain, which specifically cleaves the anticodon loop of all four tRNAArg isoacceptors, thereby inactivating protein synthesis and leading to cell death. Here we report the 2.0 Å resolution crystal structure of the complex between the toxic domain and its immunity protein ImmD. Neither component shows structural homology to known RNases or their inhibitors. In contrast to other characterized colicin nuclease–Imm complexes, the colicin D active site pocket is completely blocked by ImmD, which, by bringing a negatively charged cluster in opposition to a positively charged cluster on the surface of colicin D, appears to mimic the tRNA substrate backbone. Site-directed mutations affecting either the catalytic domain or the ImmD protein have led to the identification of the residues vital for catalytic activity and for the tight colicin D/ImmD interaction that inhibits colicin D toxicity and tRNase catalytic activity.
Keywords: colicin D–ImmD complex, cytotoxicity, E. coli transfer RNase, protein–protein inhibition
Introduction
The plasmid-encoded antibacterial toxins called colicins are present in many natural strains of Escherichia coli, and secreted into the extracellular medium under a variety of environmental stress conditions. These highly efficient and specific proteins kill competing bacteria that are not able to produce the neutralizing immunity protein (Imm), usually co-synthesized by colicinogenic cells. In the case of nuclease colicins, which target hydrolysable substrates in the cytoplasm, the colicin producer cells are protected against both endogenous and exogenous toxin molecules by forming a tight 1:1-heterodimer complex with the cognate Imm protein (Pugsley, 1984; Braun et al, 1994; James et al, 1996; Kleanthous et al, 1998).
The lethal invasion of the target cell proceeds through three distinct steps: receptor recognition and binding, translocation across both the outer and inner membranes and cytotoxicity. Most colicins display a similar tripartite architecture of functional domains (Lazdunski et al, 1998), each of which is related to a specific step of penetration or killing activity. The central R region is necessary for interacting with the cell surface receptor, whereas the subsequent penetration of the outer cell membrane requires the N-terminal T (translocation) domain. The cytotoxic activity is associated with the 10–15 kDa C-terminal part of the colicin molecule. E-type nuclease colicins bind to the outer membrane BtuB receptor, usually involved in the high-affinity uptake of vitamin B12, and require the outer membrane protein OmpF and the TolA–B–Q–R system for their import (Lazdunski et al, 2000; Bouveret et al, 2002; James et al, 2002; Lazzaroni et al, 2002). The immunity protein is released from the naturally secreted colicin–Imm complex when it is adsorbed to the target cell surface (Krone et al, 1986).
In contrast to E-type colicins, colicins D and B bind to the outer membrane receptor FepA, necessary for the internalization of ferric enterobactin, a siderophore, and penetrate target bacteria by appropriating the multi-protein system of the host cell required for Fe3+ uptake, namely the energy-dependent TonB/ExbB–D pathway (Braun et al, 2002; Ferguson and Deisenhofer, 2002; Postle and Kadner, 2003). The first 313 N-terminal amino acids of colicins B and D, sufficient for their translocation across the outer membrane, are 96% identical (Roos et al, 1989). In contrast, the C-terminal regions carrying the killing function are unrelated, as de facto colicin B has pore-forming lethal activity rather than nucleolytic activity (Pressler et al, 1986). In the colicin B polypeptide, the common T and R domains are immediately followed by the killing pore-forming domain, whereas in colicin D a linker region of 280 residues, with no known function, separates the import and the catalytic domains (de Zamaroczy and Buckingham, 2002). The small C-terminal catalytic domain of colicin D, 91 amino acids long and starting at Lys607, has been shown to be necessary and sufficient for full enzymatic activity (de Zamaroczy et al, 2001). In the case of colicin D, the inner membrane leader peptidase LepB was shown to be an essential and specific requirement for cell killing (de Zamaroczy et al, 2001). Although LepB is necessary for colicin D proteolysis in vitro, its precise role remains unknown.
Target cell death results from either a nonspecific DNase activity (colicins E2, E7, E8 and E9; Toba et al, 1988; Eaton and James, 1989; Chak et al, 1991) or a highly specific RNase activity. Among these latter, colicins E3, E4, E6 and the closely related cloacin DF13 are ribosomal endonucleases, referred to as ribotoxins, whose catalytic activity results in a specific nucleolytic break of the 16S ribosomal RNA (Bowman et al, 1971; de Graaf et al, 1973; Lasater et al, 1989). Colicins E5 and D hydrolyse different classes of transfer RNAs. The first specifically cleaves tRNAs in which the guanine located at the wobble position is modified to queuine (Ogawa et al, 1999), whereas the second hydrolyses the four arginine isoacceptors at a nearby position in the anticodon loop, between nucleotides 38 and 39 (Tomita et al, 2000). RNase colicins thereby provoke cell death by inactivating the protein biosynthetic machinery.
Here, we show the crystal structure of colicin D transfer-RNase domain with its cognate immunity protein at 2 Å resolution. The components of the binary complex are structurally unrelated to previously described colicins, immunity proteins or RNases. Functionally important residues in both colicin D and ImmD have been identified by structure-based site-directed mutagenesis and analysed both in vivo and in vitro. The highly efficient mechanism by which ImmD inhibits the activity of the tRNase domain by binding directly to the active site is discussed in relation to observations concerning other nuclease–immunity protein complexes.
Results
Structure determination
Analysis of the solvent content of crystals obtained from the colicin D–ImmD complex revealed that these could not contain the entire colicin D complex, despite the fact that this was the starting protein material in the crystallization drops. SDS–PAGE and N-terminal peptide sequencing indicated that the crystallized protein material corresponded to a complex between the tRNase domain of colicin D and ImmD. The crystal structure of this complex was solved to 2.0 Å resolution by the Single wavelength Anomalous Dispersion (SAD) phasing method from a selenomethionine-labelled protein crystal containing one copy of the complex in the asymmetric unit. Well-defined electron density was observed for residues Leu591–Leu697 from colicin D, comprising the minimal, 91-amino-acid-long catalytic domain necessary and sufficient for lethal tRNase activity, which starts at Lys607 (de Zamaroczy et al, 2001), and all residues (Met1 to Leu87) from ImmD, as well as for 196 water molecules and a PEG 400 molecule from the crystallization conditions. The molecular masses of these fragments are in accordance with those determined by mass spectrometry. The refinement statistics are gathered in Table I.
Table 1.
Data collection statistics
| Native | SAD | |
|---|---|---|
| Data collection | ||
| Resolution (Å) | 99–1.98 (2.02–1.98) | 25–3.0 (3.09–3.0) |
| Space group | P41212 | P41212 |
| Unit cell parameters | a=b=62 Å; c=147.9 Å | a=b=61.9 Å; c=150 Å |
| Total number of reflections | 273269 | 231794 |
| Total number of unique reflections | 20818 | 11111 |
| Rsym (%)a | 5.8 (27.8) | 11.5 (37.8) |
| Completeness (%) | 98.3 (86.7) | 100 (100) |
| I/σ(I) | 18.9 (1.6) | 20.2 (5.1) |
| Redundancy | 13 | 21 |
| Phasing | ||
| Resolution (Å) | 80–3.0 | |
| FoM (after density modification) | 0.25 (0.55) | |
| Refinement | ||
| Resolution (Å) | 20–2.0 | |
| R/Rfree (%)b | 19.4/23.5 | |
| R.m.s.d. bonds (Å) | 0.005 | |
| R.m.s.d. angles (deg) | 1.07 | |
| Mean B factor (Å2) prot/water | 27/34 | |
| Ramachandran plot | ||
| Most favoured | 89% | |
| Allowed |
11% |
|
| Values in parentheses are for the highest resolution shell. | ||
| aRsym=∑h∑i∣Ihi−〈Ih〉∣/∑h∑iIhi, were Ihi is the ith observation of the reflection h, while 〈Ih〉 is the mean intensity of reflection h. | ||
| b Rfactor=∑∥∣Fo∣−∣Fc∥/∣Fo∣. Rfree was calculated with a small fraction (5%) of randomly selected reflections. | ||
Further biochemical analysis of the crystallization drops showed that colicin D is gradually proteolysed during the nucleation process, resulting in a resistant core defined by the complex between the catalytic domain and ImmD, the fragment that finally crystallized. Once this was understood, crystals could also be obtained by cloning and purifying only the catalytic domain of colicin D in complex with ImmD (data not shown).
Structure of colicin D and ImmD
The colicin D catalytic domain adopts a crescent-shaped globular α/β fold of approximate dimensions 25 × 30 × 40 Å3 and is composed of a central four-stranded antiparallel β sheet (β1, β2, β3 and β4) sandwiched between a long helix (α2) on one side and the C-terminal helix (α3) on the other (Figure 1A). A third kinked helix (α1) lies in the plane of the β sheet, interacting with strand β4, perpendicular to helix α2. Searches for structural homologues of the catalytic domain using the EBI Macromolecular Structure Database (http://www.ebi.ac.uk/msd/) revealed mediocre matches with known structures (best Z score obtained: 3.2).
Figure 1.
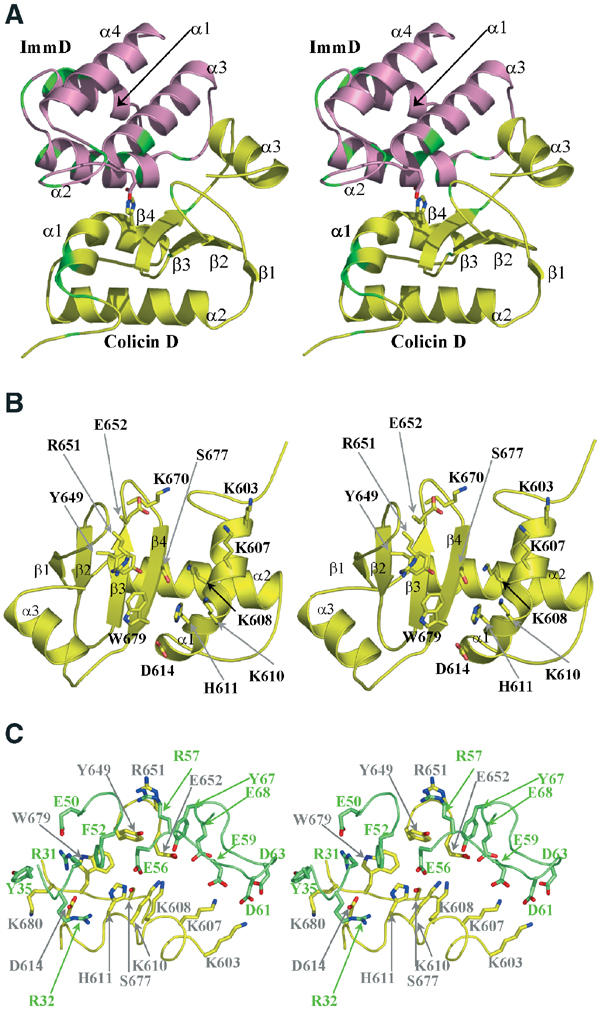
Stereo views. (A) Ribbon representation of the complex between colicin D (yellow) and ImmD (pink). The colicin D His611 and the ImmD Glu56 side chains at the interface are shown as sticks. Residues whose mutation has no effect on the in vivo and in vitro activities are highlighted in green. (B) Close-up of the colicin D putative active site. The colicin D tRNase domain is shown as a ribbon with the potential active site residues represented as sticks. For clarity, residues are identified by their one-letter code in all figures. (C) Close-up of main colicin D (yellow) and ImmD (green) residues involved at the dimer interface.
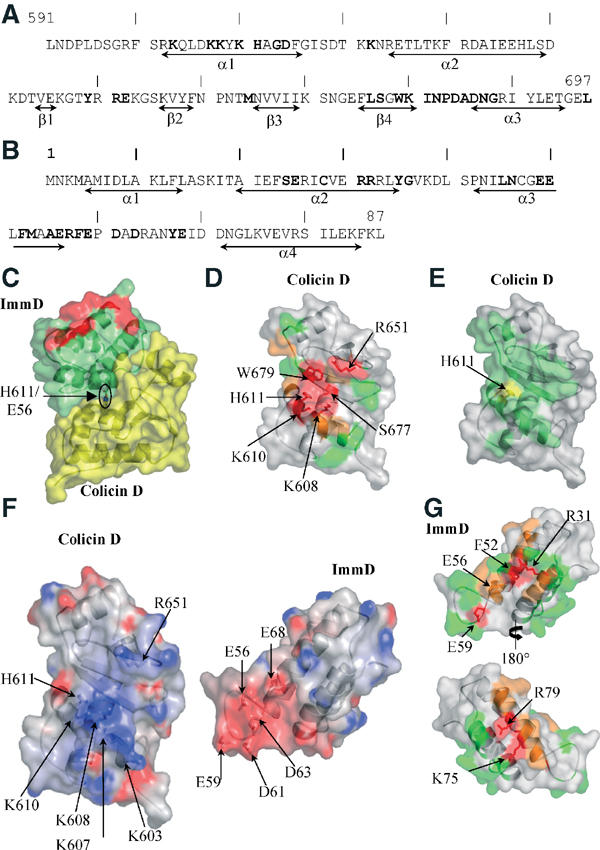
The colicin D immunity protein adopts an antiparallel four helical bundle fold of dimensions 20 × 20 × 40 Å3 (Figure 1A). The helices are tightly packed, forming a compact cylindrical molecule. The first antiparallel helical pair α1, α2 packs against the second pair formed by helices α3 and α4, with an angle of about 30°. No significant structural analogies to other proteins have been found. The location of α helices and β sheets in the peptide sequence of the colicin D and ImmD is depicted in Figure 2A and B.
Figure 2.
Molecular surface representation. (A, B) Secondary structure assignments along the colicin D (A) and ImmD (B) sequences. Residues involved in complex formation are highlighted in bold. (C) The binary colicin D (yellow)–ImmD (green) complex. The orientation is the same as in Figure 1A. The red-coloured region corresponds to ImmD positions located outside the interface, but whose mutation abolishes or reduces the inhibitory capacity. (D) Surface mapping of the colicin D catalytic active site residues. Unmutated positions are coloured grey. Residues whose mutation does not alter tRNase activity are shown in green, those whose substitution results in partially or fully inactivated proteins are depicted in orange and red, respectively. For clarity, only residues whose mutation results in complete loss of activity are labelled. (E) ImmD foot-printing (green) at the colicin D surface. The His611 residue is coloured yellow. The orientation is the same as in (D). (F) Open leaflet representation of the complex illustrating the electrostatic complementarity between colicin D and ImmD. Regions of the surface with negative potential are coloured red and those with positive potential are in blue. The colicin D orientation is similar as in (D). The ImmD is rotated 180° around the vertical axis. (G) Surface mapping of the ImmD residues involved in the inhibitory effect. Colour coding is the same as in (D). The orientations of the ImmD in the top and bottom panels are related by a 180° rotation around the vertical axis.
The colicin D tRNase active site
It has been shown that the smallest colicin D fragment capable of cleaving tRNAArg corresponds to the Lys607–Leu697 fragment (de Zamaroczy et al, 2001). The crystallized colicin D protein in this study contained residues 591–697 and therefore corresponds to a catalytically active fragment. Except for colicin E5, His residues play a crucial role in the catalytic mechanism of RNases. At least one His, consistently present in the active site, is directly involved in proton abstraction of the 2′-OH ribose proton of the RNA substrate (Fersht, 1999). Colicin D specifically cleaves tRNAArg isoacceptors between nucleotides 38 and 39, and mutation of His611 abolishes its cleavage and toxicity activities (Tomita et al, 2000). This His residue is located at the C-terminal end of the α1 helix, which forms the rim of the central groove of the protein (Figure 1B). The bottom of the groove is rather hydrophobic (Leu676 and Trp679 from the β4 sheet). His611 is further surrounded by two polar residues: Ser677 (strand β4) and Asp614 (on helix α1). The opposite rim of the groove carries charged and polar residues (Tyr649, Arg651, Glu652 and Lys670). Helix α1 further carries four Lys residues (Lys603, Lys607, Lys608 and Lys610), creating a positive charge cluster (Figures 1A and B) that could play an important role for substrate binding by neutralizing the negatively charged phosphate groups of the substrate tRNA backbone.
To clarify their functional involvement in tRNA hydrolysis and/or tRNA recognition, most of the residues mentioned above were changed into alanine and both the in vitro ribonuclease activity and the cytotoxicity of the mutants were compared to those of the wild-type protein (Table II). Northern blot analysis of tRNAArgCCG cleavage in vitro revealed that colicin molecules with mutated His611 or lysine residues forming the positive charge cluster (except for Lys603) are either inactive (Lys608, Lys610 and His611) or only partially active (Lys607). Mutant tRNases affecting the main residues forming the central groove (Ser677, Trp679) or its rim (Arg651) are not active, while the Asp614Ala mutant colicin D that had activity in vitro was found to be seriously impaired in its in vivo toxicity (Figure 2D). Finally, mutation of residues located upstream of position 607 (for instance, Met590Ala, Leu591Ala, Asn592Ala (data not shown) and Leu595Pro, Ser601Pro, Arg602Gly, Lys603Glu, Asp606His (Table II)) did not affect the catalytic activity. This is in agreement with previous experiments performed with truncated colicin D proteins constructed to determine the minimal tRNase domain (de Zamaroczy et al, 2001). These structure-based site-directed mutagenesis experiments indicate that the central groove and the positive charge cluster may form the active site of the colicin D catalytic domain.
Table 2.
Site-directed mutagenesisa
| Mutated ColD residue | In vivob |
In vitroc |
Mutated ImmD residue | In vivob | In vitroc | |
|---|---|---|---|---|---|---|
| colD+ImmD | −ImmD | +ImmD | colD+ImmD | colD+ImmD | ||
| wt ColD | + | + | − | wt ImmD | + | − |
| L595P | + | + | − | K11A | + | − |
| S601P | + | + | − | L12A | + | − |
| R602G | + | + | − | S16A | + | − |
| K603E | + | + | − | K17A | + | − |
| D606H | + | + | − | E22A | + | − |
| K607A | +/−− | +/−− | − | R26A | + | − |
| K608A | − | − | ND | E30A | + | − |
| K610A | − | − | ND | R31A | + | + |
| H611A | − | − | ND | R32A | + | − |
| D614A | +/−− | + | +/− | R32Q | + | +/− |
| Y649A | +/− | +/− | − | R33A | + | − |
| R651A | − | − | ND | Y35A | + | − |
| E652A | + | + | − | N46A | + | +/− |
| K670A | + | + | − | E49A | + | +/−− |
| S677A | − | − | ND | F52A | + | + |
| W679A | − | − | ND | M53A | + | − |
| K680A | + | + | − | E56A | + | +/− |
| N682A | +/− | +/− | − | R57A | + | +/−− |
| A685Q | +/− | + | +/− | E59A | + | + |
| D686A | + | +/− | − | D61A | + | − |
| D63A | + | − | ||||
| Y67A | + | − | ||||
| E68A | + | − | ||||
| N72A | + | +/− | ||||
| K75A | + | + | ||||
| V76A | + | +/− | ||||
| R79A | + | + | ||||
| E83A | + | +/− | ||||
| |
|
|
|
K84A |
+ |
+/− |
| Substitutions of functionally important residues are indicated by bold characters. | ||||||
| bThe cytotoxicity of the mutant proteins, spotted directly onto a lawn of sensitive wild-type cells, was determined by halo test. | ||||||
| cThe tRNase catalytic activity was analyzed in vitro by Northern blot: (+) wild-type activity; (+/−)=about 50% of wild-type activity; (+/−−) weak activity; (−) no activity; (ND) not determined. No tRNase activity detected in the presence of ImmD protein, signifies a wild-type level of immunity exhibited by the colicin D–ImmD complex tested. | ||||||
Full-sized wild-type or mutated colicin D molecules, with or without the wild-type or mutated ImmD protein are synthesized in a coupled E. coli S30 transcription/translation system (see Materials and methods).
The colicin D tRNase–ImmD complex
In the complex, ImmD fits smoothly in the central groove described above that potentially harbours the active site of colicin D (Figures 1A and 2C). One face of the ImmD helical bundle, formed by helices α2 and α3, interacts with residues from the bottom and both rims of the colicin D groove, comprising helix α1 and the strand β4–helix 3 segment. The orientation of these helices is parallel to helix α1 of colicin D. Most remarkably, ImmD seems to block completely the active site groove of colicin D (Figure 2D and E). Although the colicin D active site and the colicin D region responsible for ImmD binding seem to overlap, as demonstrated here, three residues were nevertheless identified, the mutation of which specifically affected only either catalytic activity (Tyr649, Asn682 and Asp686) or inhibitory interaction (Ala685) (Table II). This suggests that the regions of the colicin D catalytic domain involved in recognition and interaction of the tRNAArg and ImmD are not strictly identical.
The colicin D–ImmD complex formation buries a total protein surface area of 1920 Å2 with approximately equal contributions from both molecules. This value is in the upper range of the average calculated for other stable protein–protein complexes (Lo Conte et al, 1999). Heterodimer formation involves 25 residues from the colicin D domain and 22 from the immunity protein (Figure 1C). The interface is stabilized by polar and electrostatic interactions: four salt bridges and seven hydrogen bonds were identified (Table III). The most prominent feature is a high degree of charge complementarity between the positively charged patch on colicin D (Lys603, Lys607, Lys608, Lys610) and a corresponding negatively charged patch on ImmD (Glu56, Glu59, Asp61, Asp63, Glu68), resulting in three direct salt bridges (Figure 2F). Another salt bridge is contributed by Asp614 and Arg32 from ImmD. Most of the hydrogen bonds involved in complex formation occur between the main chain atoms from colicin D and side chains from ImmD (Table III).
Table 3.
Salt bridges and hydrogen bonds involved in the ColD–ImmD complex
| Colicin D | Distance (Å) | ImmD |
|---|---|---|
| Lys 607 Nζ | 3.0 | Oɛ1 Glu 59 |
| Lys 608 Nζ | 3.1 | Oɛ2 Glu 56 |
| Lys 610 O | 2.9 | Nη2 Arg 32 |
| His 611 Nɛ2 | 2.6 | Oɛ1 Glu 56 |
| Asp 614 Oδ1 | 2.8 | Nɛ Arg 32 |
| Glu 652 Oɛ1 | 2.6 | Oη Tyr 67 |
| Ser 677 Oγ | 2.7 | Oɛ2 Glu 56 |
| Lys 680 O | 2.7 | Nη1 Arg 31 |
| Lys 680 O | 2.8 | Nη2 Arg 31 |
| Ile 681 O | 2.7 | Oη Tyr 35 |
| Asp 686 N | 2.8 | Oδ1 Asn 46 |
In order to further probe the colicin D–ImmD interaction surface, the ImmD residues involved in complex formation were systematically mutated to alanine and the inhibitory effect of the mutant proteins on the tRNase activity and the toxicity of colicin D was compared to that of the wild-type immunity protein (Table II). This approach revealed that some aromatic and hydrophobic residues buried upon complex formation (Tyr35, Met53 and Tyr67) play a minor role in the interaction. In contrast, the change to alanine of most polar residues contributing to salt bridges and hydrogen bonds (Arg31, Asn46, Glu49 and Glu59), together with the Arg32Gln mutation (but not Arg32Ala), reduced or completely abolished the ImmD inhibitory capacity (Table II and Figure 2G). There are two more residues on the α2 helix whose mutation causes loss of inhibition of ImmD: Arg57 that stacks onto the guanidinium group of colicin Arg651, and Phe52 that is packed against colicin Trp679. Mutation of Glu56, which is in direct interaction with the colicin His611 in the complex structure, led to only 50% inhibition. Taken together, these biochemical and biological results support the importance of the polar interactions identified at the colicin D–ImmD interface and indicate that charge complementarity between the surfaces of both partners is a major factor in recognition.
The tRNase activity of several N-terminally truncated colicin D proteins, starting between positions 455 and 590, was shown to be resistant to inhibition by ImmD. However, the minimal tRNase domain itself was fully inhibited by the co-expressed immunity protein (de Zamaroczy et al, 2001). This suggests that ImmD might recognize more than one region of the whole colicin D molecule. In the case of ImmE3, the interaction with the complete colicin E3 was clearly shown to involve the globular translocation and the catalytic domains (Soelaiman et al, 2001). In order to identify an interaction surface of ImmD other than that in contact with the catalytic domain of colicin D, we have systematically mutated residues that are exposed to solvent and situated at the opposite side of the colicin D–ImmD interface presently observed in the crystal structure. Although substitution by Ala of residues Lys11, Leu12, Ser16, Lys17, Glu22, Arg26 and Glu30, from the α1 and the N terminal part of α2 helices did not affect the inhibitory action of ImmD, substitution of residues belonging to the α4 helix led to a complete (Lys75 and Arg79) or partial (Asn72, Val76, Glu83 and Lys84) loss of wild-type colicin D inhibition (Table II, Figure 2C and G). These observations raise the possibility that in the context of a complex between the intact colicin D–ImmD, the immunity protein is sandwiched between the catalytic and one or more N-terminal domains of colicin D.
Discussion
The studies we report here reveal that the structure of the catalytic tRNase domain of colicin D is unrelated to other RNases. Similarly, ImmD is unlike previously studied RNase inhibitors. Our mutational studies have largely identified the amino-acid residues important both for the catalytic activity of the tRNase domain, and the interaction between this domain and ImmD that efficiently inhibits the enzymatic activity. Unlike other colicin–Imm complexes for which structures have been solved, ImmD completely blocks access of small molecules to the active site, and appears to present to the catalytic domain a pattern of negative charge that mimics the tRNA substrate.
Catalytic site and substrate recognition
The general catalytic mechanism by which ribonucleases hydrolyse RNA has been thoroughly studied by protein engineering and crystallographic analysis over several decades (Gilliland, 1997). RNase A, for instance, has two active site His residues that cooperate during the catalytic cycle (Findlay et al, 1961). One acts as a general base and abstracts a proton from the ribose 2′-OH, thereby catalysing the nucleophilic attack of this hydroxyl group on the 3′-phosphate group, leading to a cyclic intermediate, while the other serves as catalytic acid during the first cyclization step. Their catalytic roles are reversed during the subsequent hydrolysis of the cyclic intermediate. Other ribonucleases, such as barnase (Fersht, 1999) and colicin E3 (Zarivach et al, 2002), proceed probably through a similar mechanism, but with a His and a Glu as catalytic residues.
In the absence of any structural match with already known nuclease structures, it was not possible in the case of colicin D to predict catalytic or substrate binding residues. We therefore probed the potential active site region by systematic site-directed mutagenesis of residues situated in and around a depression on the surface of colicin D. Taken together, the results from the site-directed mutagenesis experiments show a clear picture of the catalytic pocket centred around His611 (Figure 2D). Substitution of this His by Ala or Tyr results in the loss of the catalytic activity, supporting the idea that His611 is the general base of the cyclization step of the reaction (Tomita et al (2000) and this work). From the structure, the only residue susceptible to act as a general acid seems to be Asp614, whose carboxylate group is at 4.5 Å from the His611 side chain. However, as the Asp614Ala mutant retains a significant though low in vivo cytotoxicity and a wild-type tRNase activity (Table II), it is very unlikely that this Asp plays a critical role in the tRNA hydrolysis reaction. Colicin E5, which targets the anticodons of tRNAs for Tyr, His, Asn and Asp, has no sequence analogy with colicin D even in the catalytic domain and lacks His residues altogether. Both tRNases cleave the tRNA anticodon loop, yielding a cyclic 2′, 3′-phosphate and 5′-OH termini. The same is true for a further anticodon nuclease PrrC that provokes depletion of tRNALys in T4 phage-infected E. coli cells (Levitz et al, 1990; Kaufmann, 2000). These observations suggest that there exist several active site solutions to cleave tRNA molecules. As the tRNAArg isoacceptors sensitive to colicin D yield 5′-half fragments with a 2′,3′-cyclic phosphate group (Tomita et al, 2000), this tRNase apparently catalyses only the first cyclization step, releasing the cyclic product from the active site. This may explain why only a single His is present in the putative active site of colicin D and why a second catalytic residue seems to be absent.
The nucleophilic attack of the 2′-OH of the ribose on the 3′-phosphate group creates a negatively charged pentacovalent transition state that can be stabilized by nearby positively charged residues. In colicin D, the amine groups of Lys608 and Lys610 are well positioned to play such a role, as they are located 5.5 and 4.8 Å away, respectively, from His611. Their mutation to Ala abolishes both cytotoxic and RNase activities of colicin D, confirming their role in tRNA substrate recognition or more directly in the catalytic activity. Mutation of Ser677, whose hydroxyl group points towards His611 Nɛ2 (at 3.44 Å), abolishes both cytotoxic and RNase activity, suggesting a role for Ser677 in substrate binding and/or catalysis. Two explanations can be found for this result. First, in the absence of ImmD bound to colicin D, the His611 Nɛ2 atom could be within hydrogen bonding distance of the Ser677 hydroxyl group. As a result, one side of the His611 ring would be accessible to the substrate and oriented to act as a general base. Alternatively, Ser677 could be in direct interaction with the tRNA substrate and play a role in tRNAArg binding. A similar situation occurs in RNase A, where Thr45 is involved in pyrimidine specificity through its OH group (delCardayre and Raines, 1994). Ser677 is involved in both interactions with ImmD and RNase activity. Our assays do not allow the independent evaluation of these activities. Trp679 forms a hydrophobic platform close to the His611 imidazole side chain, exposes one face of its indole ring to the solvent, and could therefore stacks on a tRNA base. The lack of activity for the Trp679Ala mutant is in agreement with this proposal. Site-directed mutagenesis allowed us to map the colicin D active site and to identify residues critical for cytotoxicity (Lys 608, Lys 610, His611, Arg651, Ser677 and Trp679).
The substrate specificity of the tRNase activity of colicin D is remarkably narrow. This activity is conferred by a small domain carrying a modestly sized active site cleft, and subtle recognition mechanisms must be at work to achieve this specificity. In the four tRNAArg isoacceptors, position 38 is occupied by an adenine that is not involved in Watson–Crick base pairing. In general, in the 3D structures of free tRNA, the backbone phosphate moiety of nucleotide 38 is solvent exposed and its base makes a hydrogen bond with the base at position 32. In the structure of the complex between arginyl-tRNA synthetase and yeast tRNAArg, the adenine ring at position 38 flips out of the anticodon loop to interact with the enzyme (Delagoutte et al, 2000). It is therefore tempting to speculate that upon binding to colicin D, the adenine base will do the same and stacks on the exposed face of the Trp679 side chain. How this can result in a highly specific cleavage will have to await structural information on colicine D–tRNAArg complexes.
Interaction between the colicin D catalytic domain and ImmD
The crystal structures of several nuclease domains (colicins E3, E7 and E9) in complex with their bound immunity proteins (Kleanthous et al, 1999a; Ko et al, 1999; Carr et al, 2000), and of the complex between the full-length colicin E3 and its inhibitor Imm3 (Soelaiman et al, 2001), have been reported. In combination with protein engineering experiments, these structures have revealed a common mechanism for nuclease inhibition by the respective Imm proteins. The active site remains exposed, but the Imm protein prevents the access of incoming bulky substrates (ribosome or genomic DNA) to the catalytic groups (Kolade et al, 2002). The predominant interactions involved in complexes between the colicin E7 and E9 DNase domains and their Imm proteins were shown to be charged and hydrophobic, respectively (Kuhlmann et al, 2000). The core of the E-type DNase active site is formed by a H–N–H motif originally found in intron-encoded homing endonucleases (James et al, 2002). Specific ImmE7 binding is conferred by a small number of critical amino-acid residues (Lu et al, 1999). Such a mode of inhibition contrasts with classic enzyme inhibitor complexes such as barnase-barstar (Buckle et al, 1994), uracil-DNA glycosylase–uracil glycosylase inhibitor protein (Savva and Pearl, 1995), where the inhibitors completely block access to the active sites and interact with well-conserved residues.
The crystal structure of the colicin D catalytic domain in complex with ImmD allows a comparison to other immunity proteins and between the different modes of inhibition used to block colicin activity. Comparison of ImmD with other immunity proteins shows that the DNase colicin inhibitors (ImmE7, ImmE8 and ImmE9) are of identical size (87 amino acids) and have a similar four-bundle architecture (Kleanthous et al, 1999b; Ko et al, 1999). However, the lengths and the relative orientations of the helices within the bundle are very different and their structure cannot be superposed on that of ImmD. Similarly, the immunity protein of colicin E3, which inhibits an rRNase-type colicin, adopts a totally different α/β fold (Soelaiman et al, 2001). In the structure of the colicin D–ImmD complex, the ImmD protein associates by inserting two helices into the active site depression, thereby completely blocking the presumed active site pocket. There is therefore a perfect overlap between the colicin D region recognized by ImmD and the catalytic site defined by mutagenesis experiments (Figure 2D and E).
The full-length colicin D protein was previously found to be susceptible to cleavage in cell extracts, in a process dependent on the leader peptidase LepB (de Zamaroczy et al, 2001). The cleavage site has been localized between residues Lys607 and Lys608 (M de Zamaroczy, unpublished data, 2003) and ImmD protects colicin D against this cleavage. The structure we report here explains the protection observed, as these two Lys residues, at the start of the catalytic domain, are completely masked in the colicin D–ImmD complex. Thus, during secretion, ImmD associated with colicin D simultaneously blocks the toxic tRNase activity and the possibility of a proteolytic cleavage.
The present structure is the first example of an Imm protein inhibiting a colicin by completely blocking the active site, rather than by preventing access of the substrate to the active site through binding to an adjacent site. Hence, the mechanism of inhibition of ImmD resembles the more classic enzyme–inhibitor complexes such as barnase-barstar (Buckle et al, 1994) or trypsin-BPTI (Huber et al, 1974). In the complex, the interacting surface is dominated by charge-complementary interactions. Especially noteworthy is the match between the positively charged clamp on helix 1 from colicin D and a negatively charged patch on ImmD (Figure 2F). Mutation of the residues responsible for the positively charged region on colicin D abolishes or reduces colicin D cytotoxicity. This charged surface could then play an important role for the tRNA substrate binding by neutralizing the backbone phosphate groups. The ImmD protein presents a negatively charged surface to this region of colicin D, thus mimicking the phosphate groups of tRNA substrate. A similar mechanism was reported for the inhibition of the uracil-DNA glycosylase by its inhibitor protein (Savva and Pearl, 1995).
Several residues in helix α4 were shown to be essential for the inhibition of colicin D. These residues are distant from the protein surface area located between ImmD and the colicin D active site, and most probably contribute to anchor ImmD to a second domain more towards the N-terminus of colicin D. This may maintain an overall structure of the colicin complex better optimized for its immunity during secretion, and/or for receptor recognition during target cell invasion. Consistent with this proposal, the wild-type colicin E3/ImmE3 complex, comprising a second interaction between ImmE3 and the translocation domain, dissociates two orders of magnitude more slowly than the complex simply formed by the catalytic domain and ImmE3 (Walker et al, 2003). The bipartite interaction of ImmE3 with colicin E3 (Soelaiman et al, 2001) suggested a cascade mechanism for colicin E3 penetration into target cells. The binding of the extended coiled-coil receptor-recognition domain of colicin E3 to BtuB places the N-terminal translocation domain close to a neighbouring OmpF porin (Kurisu et al, 2003). According to the proposed mechanism, the subsequent interaction with OmpF (second receptor) would abolish the contact of the translocation domain with the ImmE3, which in turn could weaken the binding of ImmE3 to colicin and thereby trigger the unfolding necessary for translocation of the C-terminal catalytic domain (Zakharov and Cramer, 2002). However, the N-terminal part of colicin B, highly conserved in colicin D, was recently shown (Hilsenbeck et al, 2004) to adopt a single-domain structure comprising both the receptor recognition and the translocation regions. This structure is completely different from that of the corresponding regions in the E-type colicins. In addition, the immunity proteins of D- and E-type colicins adopt different folds. Altogether, these observations suggest that the ImmD may interact with the central domain of colicin D, of unknown function, and/or with the translocation domain common to colicin B in a manner completely different from what was observed for colicin E3. Further studies will be required to identify the precise target and the functional role of the interaction implicating helix α4 of ImmD, taking into account the fact that no second receptor, involved in the translocation of colicin D across the outer membrane, has so far been identified.
Materials and methods
Expression, purification and activity assay of colicin D
The purification of the native colicin D in complex with its immunity protein was performed from the supernatant of a culture of E. coli strain K12 carrying pColD-CA23 (pJF129) induced with mitomycin C (200 μg/l) (Frey et al, 1986). Labelling with selenomethionine (Se-Met) was achieved by supplementing a culture in minimal medium with L-Se-Met (70 mg/l), and six other L-amino acids (Lys, Phe and Thr at 125 mg/l each; Ile, Leu, Val, each at 62.5 mg/l) to repress the methionine biosynthesis pathway, at an OD600 of 0.7 and after induction by mitomycin C (Doublie, 1997). Expression was continued for 4.5 h. The colicin complex was purified by successive chromatographic steps on Q sepharose FF and Mono Q (Pharmacia) columns (de Zamaroczy et al, 2001), followed by (NH4)2SO4 precipitation (60%), resuspension of the precipitate and gel filtration on a HiLoad Superdex 200 column. The pooled colicin D/ImmD complex was first concentrated on a Mono Q column and finally by ultrafiltration (Amicon Ultra-4 10k NMWL; Millipore). The cytotoxicity of the purified colicin D/ImmD was verified in vivo, by a halo (zone of inhibition) assay. Se-Met incorporation into the purified complex was assayed by mass spectrometry.
Spontaneous fragmentation of colicin D–ImmD complex
Purified colicin D–ImmD complex, usually stored in 20 mM phosphate Na buffer (pH 7.0), 0.15 M NaCl, is stable for several months at 4°C, as judged by the absence of proteolytic degradation and a constant killing activity over time in vivo. Nevertheless, spontaneous fragmentation was regularly observed during colicin D crystallization trials, after storage for 8–12 weeks at 4–20°C, in 20 mM Tris buffer (pH 8.0). The fragments identified from SDS–PAGE consisted mainly of the colicin D C-terminal 12 kDa peptide and the immunity protein. The fragmentation was further analysed by N-terminal peptide sequencing colicin D peptides. In the presence of ‘complete' protease inhibitor complex (Roch), the spontaneous fragmentation over similar periods of storage was eliminated. Historically, some nuclease colicin molecules have been observed to undergo auto-fragmentation on storage, for instance, in the cases of colicin E3 and cloacin DF13 complex (Lau and Richards, 1976; de Graaf et al, 1978). More recently, the proteolytic auto-fragmentation of botulinum A neurotoxin LC was reported and shown to be stimulated by Zn2+ (Ahmed et al, 2001).
Crystallization and resolution of the structure
Native and Se-Met-labelled protein samples were stored in 20 mM Tris–HCl (pH 8). Crystals for the native protein were grown at 18°C from a 1:1 μl mixture of 26 mg/ml protein solution with 30% PEG 400, 0.2 M MgSO4, in the presence of N-octyl β-D-glucoside detergent. Se-Met-labelled protein crystals were grown by mixing 1 μl of a 20 mg/ml protein solution with 1 μl of the reservoir solution composed of 24% PEG 8 K, 0.1 M Na citrate (pH 6.5). Analysis of the crystal content by mass spectrometry, in agreement with the mentioned biochemical analysis, revealed that it contained the intact immunity protein, but only a fragment of colicin D, corresponding to the catalytic domain. For data collection, the crystals were transferred into a cryoprotectant solution composed of the mother liquor and 30% glycerol. Crystals of the native and Se-Met-labelled protein diffracted to 2.0 and 3.0 Å, respectively, on beamline BM30A at the European Synchrotron Radiation Facility (ESRF, Grenoble, France; Roth et al, 2002).
The structure was determined using SAD X-ray diffraction data, collected from the Se-Met derivative crystal at 3 Å resolution. Data were processed using the HKL package (Otwinowski and Minor, 1997). The space group was P41212 (a=b=61.9 Å, c=150.0 Å) with one complex per asymmetric unit. Three Se atom sites out of five were found with the program SOLVE in the 80–3.6 Å resolution range (Terwilliger, 1999). After phase extension to 3 Å and solvent flattening with the program RESOLVE (Terwilliger and Berendzen, 1999), the quality of the electron density map allowed the manual construction of most of the secondary structure elements with the ‘TURBO' molecular modelling program (Roussel and Cambillau, 1991). This partial model was then refined against the 2 Å data set with the Arp/wARP program that allowed automated construction of 98% of the model (Perrakis et al, 1999). After completion of the colicin D–ImmD model, the structure was refined with the CNS program (Brünger et al, 1998). All the residues are well defined in electron density and fall within the allowed regions of the Ramachandran plot, as defined by the program Procheck (Laskowski et al, 1993). Statistics for all the data collections and refinement of the different structures are summarized in Table I. The atomic coordinates and structure factors for the colicin D–ImmD complex have been deposited into the Brookhaven Protein Data Bank under the accession number 1V74.
Cleavage and killing activities of mutated colicin D molecules
Mutations were introduced both at residues of the 591–697 catalytic fragment of colicin D or at residues of ImmD, by two-step PCR amplification. Employing plasmid pColD as a template, the first PCR used a 5′ mutagenic oligonucleotide overlapping the mutated position, together with a 3′ oligonucleotide complementary to a downstream sequence located close to the cda stop codon of the structural gene of colicin D (cda) or ImmD (cdi). After denaturation, the PCR product was used as a 3′ oligonucleotide in a second PCR amplification, coupled with an upstream 113-mer oligonucleotide, containing the tac promoter, known to be efficient both in vitro and in vivo (Huang et al, 1994), and a 14 nt ribosomal binding site from pColD, to generate whole mutated cda and cda-cdi molecules. The resulting PCR products were expressed in vitro in a coupled transcription–translation, Zubay-S30 system from E. coli, optimized for linear DNA templates (Promega) at 37°C for 60 min. tRNA-hydrolysing activity of the mutated colicins D was directly measured from the S30 expression samples, by separation of the intact and cleaved forms of the tRNAArg on PAGE (10% in the presence of 7 M urea), followed by Northern blot analysis (Tomita et al, 2000), using a [32P]-labelled DNA probe specific for tRNAArg(CCG). In parallel, the same test was performed with mutated colicin D–ImmD complexes for studying the inhibitory efficiency of the complex. For semiquantitative determination of cytotoxicity and tRNase activity, the amounts of mutated protein in comparison with that of the wild type, were previously determined from [35S]Met-labelled mutated colicins by phosphorimagery (Molecular Dynamics), and found to be similar (data not shown). In parallel, mutated colicin D/ImmD complexes were directly tested for their in vivo cytotoxic activity on a lawn of sensitive wild-type C600 strain on LB plates.
Acknowledgments
We acknowledge Nancy Diaz for technical assistance during protein purification, P Carpentier (ESRF BM30A beamline) for his help during data collection and Isabelle Sorel for use of the crystallization robot. We thank Professor J Janin for careful reading of the manuscript. This work was supported by the CNRS (UPR 9063, UPR 9073 and UMR 9920), Université Paris 7, the Génopole program and the Association pour la Recherche sur le Cancer (to M Graille).
References
- Ahmed SA, Byrne MP, Jensen M, Hines HB, Brueggemann E, Smith LA (2001) Enzymatic autocatalysis of botulinum A neurotoxin light chain. J Protein Chem 20: 221–231 [DOI] [PubMed] [Google Scholar]
- Bouveret E, Journet L, Walburger A, Cascales E, Benedetti H, Lloubes R (2002) Analysis of the Escherichia coli Tol-Pal and TonB systems by periplasmic production of Tol, TonB, colicin, or phage capsid soluble domains. Biochimie 84: 413–421 [DOI] [PubMed] [Google Scholar]
- Bowman CM, Dahlberg JE, Ikemura T, Konisky J, Nomura M (1971) Specific inactivation of 16S ribosomal RNA induced by colicin E3 in vivo. Proc Natl Acad Sci USA 68: 964–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun V, Patzer SI, Hantke K (2002) Ton-dependent colicins and microcins: modular design and evolution. Biochimie 84: 365–380 [DOI] [PubMed] [Google Scholar]
- Braun V, Pilsl H, Gross P (1994) Colicins: structures, modes of action, transfer through membranes, and evolution. Arch Microbiol 161: 199–206 [DOI] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography+NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Buckle AM, Schreiber G, Fersht AR (1994) Protein–protein recognition: crystal structural analysis of a barnase-barstar complex at 2.0-A resolution. Biochemistry 33: 8878–8889 [DOI] [PubMed] [Google Scholar]
- Carr S, Walker D, James R, Kleanthous C, Hemmings AM (2000) Inhibition of a ribosome-inactivating ribonuclease: the crystal structure of the cytotoxic domain of colicin E3 in complex with its immunity protein. Struct Fold Des 8: 949–960 [DOI] [PubMed] [Google Scholar]
- Chak KF, Kuo WS, Lu FM, James R (1991) Cloning and characterization of the ColE7 plasmid. J Gen Microbiol 137: 91–100 [DOI] [PubMed] [Google Scholar]
- de Graaf FK, Niekus HG, Klootwijk J (1973) Inactivation of bacterial ribosomes in vivo and in vitro by cloacin DF13. FEBS Lett 35: 161–165 [DOI] [PubMed] [Google Scholar]
- de Graaf FK, Stukart MJ, Boogerd FC, Metselaar K (1978) Limited proteolysis of cloacin DF13 and characterization of the cleavage products. Biochemistry 17: 1137–1142 [DOI] [PubMed] [Google Scholar]
- de Zamaroczy M, Buckingham RH (2002) Importation of nuclease colicins into E. coli cells: endoproteolytic cleavage and its prevention by the immunity protein. Biochimie 84: 423–432 [DOI] [PubMed] [Google Scholar]
- de Zamaroczy M, Mora L, Lecuyer A, Geli V, Buckingham RH (2001) Cleavage of colicin D is necessary for cell killing and requires the inner membrane peptidase LepB. Mol Cell 8: 159–168 [DOI] [PubMed] [Google Scholar]
- Delagoutte B, Moras D, Cavarelli J (2000) tRNA aminoacylation by arginyl-tRNA synthetase: induced conformations during substrates binding. EMBO J 19: 5599–5610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- delCardayre SB, Raines RT (1994) Structural determinants of enzymatic processivity. Biochemistry 33: 6031–6037 [DOI] [PubMed] [Google Scholar]
- Doublie S (1997) Preparation of selenomethionyl proteins for phase determination. Methods Enzymol 276: 523–530 [PubMed] [Google Scholar]
- Eaton T, James R (1989) Complete nucleotide sequence of the colicin E9 (cei) gene. Nucleic Acids Res 17: 1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AD, Deisenhofer J (2002) TonB-dependent receptors-structural perspectives. Biochim Biophys Acta 1565: 318–332 [DOI] [PubMed] [Google Scholar]
- Fersht A (1999) Case studies of enzyme structure and mechanism. In Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding, Freeman WH (ed) pp 457–507. Worth Publishers: New York [Google Scholar]
- Findlay D, Herries DG, Mathias AP, Rabin BR, Ross CA (1961) The active site and mechanism of action of bovine pancreatic ribonuclease. Nature 190: 781–784 [DOI] [PubMed] [Google Scholar]
- Frey J, Ghersa P, Palacios PG, Belet M (1986) Physical and genetic analysis of the ColD plasmid. J Bacteriol 166: 15–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliland G (1997) Ribonucleases: Structure and Function. New York: Academic Press [Google Scholar]
- Hilsenbeck JL, Park H, Chen G, Youn B, Postle K, Kang C (2004) Crystal structure of the cytotoxic bacterial protein colicin B at 2.5 A resolution. Mol Microbiol 51: 711–720 [DOI] [PubMed] [Google Scholar]
- Huang B, Shi Z, Tsai MD (1994) A small, high-copy-number vector suitable for both in vitro and in vivo gene expression. Gene 151: 143–145 [DOI] [PubMed] [Google Scholar]
- Huber R, Kukla D, Bode W, Schwager P, Bartels K, Deisenhofer J, Steigemann W (1974) Structure of the complex formed by bovine trypsin and bovine pancreatic trypsin inhibitor. II. Crystallographic refinement at 1.9 A resolution. J Mol Biol 89: 73–101 [DOI] [PubMed] [Google Scholar]
- James R, Kleanthous C, Moore GR (1996) The biology of E colicins: paradigms and paradoxes. Microbiology 142: 1569–1580 [DOI] [PubMed] [Google Scholar]
- James R, Penfold CN, Moore GR, Kleanthous C (2002) Killing of E. coli cells by E group nuclease colicins. Biochimie 84: 381–389 [DOI] [PubMed] [Google Scholar]
- Kaufmann G (2000) Anticodon nucleases. Trends Biochem Sci 25: 70–74 [DOI] [PubMed] [Google Scholar]
- Kleanthous C, Hemmings AM, Moore GR, James R (1998) Immunity proteins and their specificity for endonuclease colicins: telling right from wrong in protein–protein recognition. Mol Microbiol 28: 227–233 [DOI] [PubMed] [Google Scholar]
- Kleanthous C, James R, Hemmings AM, Moore GR (1999a) Protein antibiotics and their inhibitors. Biochem Soc Trans 27: 63–67 [DOI] [PubMed] [Google Scholar]
- Kleanthous C, Kuhlmann UC, Pommer AJ, Ferguson N, Radford SE, Moore GR, James R, Hemmings AM (1999b) Structural and mechanistic basis of immunity toward endonuclease colicins. Nat Struct Biol 6: 243–252 [DOI] [PubMed] [Google Scholar]
- Ko TP, Liao CC, Ku WY, Chak KF, Yuan HS (1999) The crystal structure of the DNase domain of colicin E7 in complex with its inhibitor Im7 protein. Struct Fold Des 7: 91–102 [DOI] [PubMed] [Google Scholar]
- Kolade OO, Carr SB, Kuhlmann UC, Pommer A, Kleanthous C, Bouchcinsky CA, Hemmings AM (2002) Structural aspects of the inhibition of DNase and rRNase colicins by their immunity proteins. Biochimie 84: 439–446 [DOI] [PubMed] [Google Scholar]
- Krone WJ, de Vries P, Koningstein G, de Jonge AJ, de Graaf FK, Oudega B (1986) Uptake of cloacin DF13 by susceptible cells: removal of immunity protein and fragmentation of cloacin molecules. J Bacteriol 166: 260–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann UC, Pommer AJ, Moore GR, James R, Kleanthous C (2000) Specificity in protein–protein interactions: the structural basis for dual recognition in endonuclease colicin–immunity protein complexes. J Mol Biol 301: 1163–1178 [DOI] [PubMed] [Google Scholar]
- Kurisu G, Zakharov SD, Zhalnina MV, Bano S, Eroukova VY, Rokitskaya TI, Antonenko YN, Wiener MC, Cramer WA (2003) The structure of BtuB with bound colicin E3 R-domain implies a translocon. Nat Struct Biol 10: 948–954 [DOI] [PubMed] [Google Scholar]
- Lasater LS, Cann PA, Glitz DG (1989) Localization of the site of cleavage of ribosomal RNA by colicin E3. Placement on the small ribosomal subunit by electron microscopy of antibody-complementary oligodeoxynucleotide complexes. J Biol Chem 264: 21798–21805 [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Lau C, Richards FM (1976) Proteolytic and chemical modification of colicin E3 activity. Biochemistry 15: 3856–3863 [DOI] [PubMed] [Google Scholar]
- Lazdunski CJ, Bouveret E, Rigal A, Journet L, Lloubes R, Benedetti H (1998) Colicin import into Escherichia coli cells. J Bacteriol 180: 4993–5002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazdunski C, Bouveret E, Rigal A, Journet L, Lloubes R, Benedetti H (2000) Colicin import into Escherichia coli cells requires the proximity of the inner and outer membranes and other factors. Int J Med Microbiol 290: 337–344 [DOI] [PubMed] [Google Scholar]
- Lazzaroni JC, Dubuisson JF, Vianney A (2002) The Tol proteins of Escherichia coli and their involvement in the translocation of group A colicins. Biochimie 84: 391–397 [DOI] [PubMed] [Google Scholar]
- Levitz R, Chapman D, Amitsur M, Green R, Snyder L, Kaufmann G (1990) The optional E. coli prr locus encodes a latent form of phage T4-induced anticodon nuclease. EMBO J 9: 1383–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Conte LL, Chothia C, Janin J (1999) The atomic structure of protein–protein recognition sites. J Mol Biol 285: 2177–2198 [DOI] [PubMed] [Google Scholar]
- Lu FM, Yuan HS, Hsu YC, Chang SJ, Chak KF (1999) Hierarchical order of critical residues on the immunity-determining region of the Im7 protein which confer specific immunity to its cognate colicin. Biochem Biophys Res Commun 264: 69–75 [DOI] [PubMed] [Google Scholar]
- Ogawa T, Tomita K, Ueda T, Watanabe K, Uozumi T, Masaki H (1999) A cytotoxic ribonuclease targeting specific transfer RNA anticodons. Science 283: 2097–2100 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS (1999) Automated protein model building combined with iterative structure refinement. Nat Struct Biol 6: 458–463 [DOI] [PubMed] [Google Scholar]
- Postle K, Kadner RJ (2003) Touch and go: tying TonB to transport. Mol Microbiol 49: 869–882 [DOI] [PubMed] [Google Scholar]
- Pressler U, Braun V, Wittmann-Liebold B, Benz R (1986) Structural and functional properties of colicin B. J Biol Chem 261: 2654–2659 [PubMed] [Google Scholar]
- Pugsley AP (1984) The ins and outs of colicins. Part II. Lethal action, immunity and ecological implications. Microbiol Sci 1: 203–205 [PubMed] [Google Scholar]
- Roos U, Harkness RE, Braun V (1989) Assembly of colicin genes from a few DNA fragments. Nucleotide sequence of colicin D. Mol Microbiol 3: 891–902 [DOI] [PubMed] [Google Scholar]
- Roth M, Carpentier P, Kaikati O, Joly J, Charrault P, Pirocchi M, Kahn R, Fanchon E, Jacquamet L, Borel F, Bertoni A, Israel-Gouy P, Ferrer JL (2002) FIP: a highly automated beamline for multiwavelength anomalous diffraction experiments. Acta Crystallogr D Biol Crystallogr 58: 805–814 [DOI] [PubMed] [Google Scholar]
- Roussel A, Cambillau C (1991) Turbo Frodo, Silicon Graphics Geometry Partners Directory. pp 77–78. Silicon Graphics Publisher: Mountain View, CA
- Savva R, Pearl LH (1995) Nucleotide mimicry in the crystal structure of the uracil-DNA glycosylase-uracil glycosylase inhibitor protein complex. Nat Struct Biol 2: 752–757 [DOI] [PubMed] [Google Scholar]
- Soelaiman S, Jakes K, Wu N, Li C, Shoham M (2001) Crystal structure of colicin E3: implications for cell entry and ribosome inactivation. Mol Cell 8: 1053–1062 [DOI] [PubMed] [Google Scholar]
- Terwilliger TC (1999) Reciprocal-space solvent flattening. Acta Crystallogr D Biol Crystallogr 55: 1863–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J (1999) Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr 55: 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toba M, Masaki H, Ohta T (1988) Colicin E8, a DNase which indicates an evolutionary relationship between colicins E2 and E3. J Bacteriol 170: 3237–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita K, Ogawa T, Uozumi T, Watanabe K, Masaki H (2000) A cytotoxic ribonuclease which specifically cleaves four isoaccepting arginine tRNAs at their anticodon loops. Proc Natl Acad Sci USA 97: 8278–8283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker D, Moore GR, James R, Kleanthous C (2003) Thermodynamic consequences of bipartite immunity protein binding to the ribosomal ribonuclease colicin E3. Biochemistry 42: 4161–4171 [DOI] [PubMed] [Google Scholar]
- Zakharov SD, Cramer WA (2002) Colicin crystal structures: pathways and mechanisms for colicin insertion into membranes. Biochim Biophys Acta 1565: 333–346 [DOI] [PubMed] [Google Scholar]
- Zarivach R, Ben-Zeev E, Wu N, Auerbach T, Bashan A, Jakes K, Dickman K, Kosmidis A, Schluenzen F, Yonath A, Eisenstein M, Shoham M (2002) On the interaction of colicin E3 with the ribosome. Biochimie 84: 447–454 [DOI] [PubMed] [Google Scholar]