

Abstract
Motor proteins not actively involved in transporting cargoes should remain inactive at sites of cargo loading to save energy and remain available for loading. KIF1A/Unc104 is a monomeric kinesin known to dimerize into a processive motor at high protein concentrations. However, the molecular mechanisms underlying monomer stabilization and monomer-to-dimer transition are not well understood. Here, we report an intramolecular interaction in KIF1A between the forkhead-associated (FHA) domain and a coiled-coil domain (CC2) immediately following the FHA domain. Disrupting this interaction by point mutations in the FHA or CC2 domains leads to a dramatic accumulation of KIF1A in the periphery of living cultured neurons and an enhancement of the microtubule (MT) binding and self-multimerization of KIF1A. In addition, point mutations causing rigidity in the predicted flexible hinge disrupt the intramolecular FHA–CC2 interaction and increase MT binding and peripheral accumulation of KIF1A. These results suggest that the intramolecular FHA–CC2 interaction negatively regulates KIF1A activity by inhibiting MT binding and dimerization of KIF1A, and point to a novel role of the FHA domain in the regulation of kinesin motors.
Keywords: coiled coil, FHA, kinesin, KIF1A, microtubule binding
Introduction
Within the cell, it stands to reason that activation of motor proteins is tightly regulated to save energy and keep motors at sites of cargo loading (Hackney et al, 1992). Mechanisms of motor activation have been relatively well characterized in conventional kinesin (reviewed in Cross and Scholey, 1999). It is thought that, in the absence of cargo, the kinesin heavy-chain C-terminal domain folds back and inhibits the N-terminal motor domain's microtubule (MT) binding and MT-stimulated ATPase activity. This inhibition is further enhanced by the association of the heavy-chain tail with the light chain. It is hypothesized that this autoinhibition is relieved by cargo binding, upon which processivity is initiated.
Analysis of the human genome predicts a total of 45 kinesin motors (Miki et al, 2001) with a variety of structures (Hirokawa, 1998; Goldstein and Yang, 2000). These motors may also have diverse mechanisms of activation, although little is known about them. One of these kinesins, Unc104/KIF1A, is known to transport synaptic vesicle precursors (Hall and Hedgecock, 1991; Otsuka et al, 1991; Okada et al, 1995) and mediate fast and processive neuronal transport in vivo (Zhou et al, 2001; Lee et al, 2003). Recently, two important molecular mechanisms underlying the activation of KIF1A/Unc104 have been reported; clustering of Unc104 in PtdIns(4,5)P2-containing rafts on the surface of cargo vesicles (Klopfenstein et al, 2002) and conversion of monomeric Unc104/KIF1A into a processive dimer at high motor concentrations (Tomishige et al, 2002). However, there are still a number of unanswered questions. What are the molecular mechanisms underlying the stabilization of KIF1A monomers and the monomer-to-dimer transition of KIF1A? Does the monomer-to-dimer transition of KIF1A previously demonstrated in vitro function as a key regulatory mechanism for KIF1A activation in vivo? And finally, what are the physiological factors that regulate this transition?
In the region following the N-terminal motor domain, KIF1A has two prominent CC domains (CC1 and CC2) that are expected to assist in KIF1A dimerization (Vale, 2003). Between CC1 and CC2, KIF1A contains a forkhead-associated (FHA) domain, which is a phosphoprotein-binding module found in a wide variety of proteins in prokaryotes and higher eukaryotes (Li et al, 2000; Durocher and Jackson, 2002; Tsai, 2002). It is not known, however, whether the FHA domain of KIF1A interacts with phosphoproteins, whether it regulates KIF1A function, or whether it is functionally associated with the flanking CC1 and/or CC2 domains.
Here, we report that an intramolecular interaction between the FHA domain and CC2 negatively regulates KIF1A activity. These results point to a novel role of the FHA domain in the regulation of KIF1A, and suggest the possibility of similar regulation in other FHA domain-containing kinesins.
Results
The CC2 domain negatively regulates KIF1A
KIF1A contains, in addition to the neck CC, two prominent CC domains (CC1 and CC2, aa 429–462 and 625–679, respectively) that flank the FHA domain (aa 488–604; Figure 1A). To determine whether the CC and FHA domains are involved in the regulation of KIF1A, we generated EGFP-tagged deletion variants of KIF1A (Figure 1A) and determined their subcellular distribution in living neurons, as well as their ability to bind MTs. We determined the subcellular distribution of KIF1A deletions by live imaging in living neurons rather than immunostaining of fixed neurons to minimize the potential problems associated with immunostaining signal amplification.
Figure 1.
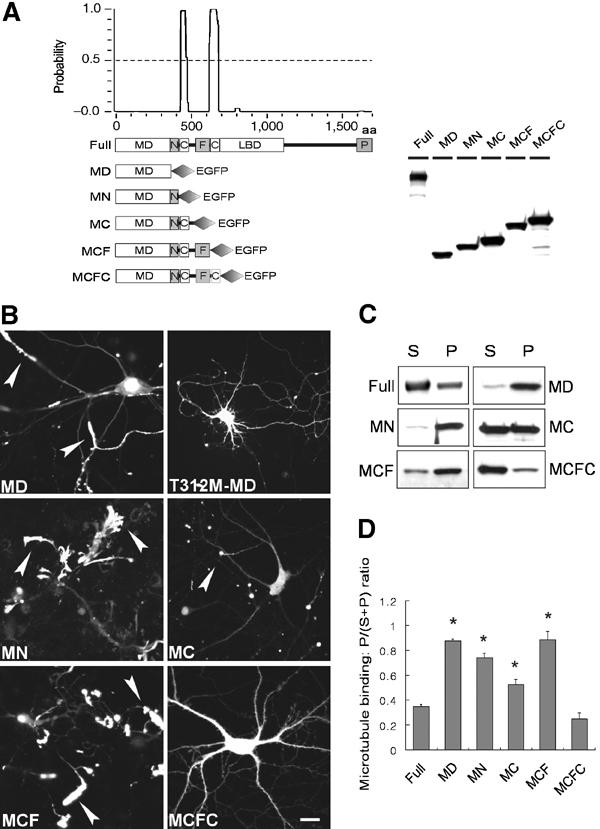
The CC2 domain negatively regulates KIF1A. (A) A schematic showing the coiled-coil probability of KIF1A. EGFP-KIF1A deletion variants are shown along with their expression levels in HEK293T cells. MD, motor domain; N, neck CC; C, CC; F, FHA domain; LBD, liprin-α-binding domain (Shin et al, 2003); P, PH domain. The schematic shows only the two most prominent coiled coils of KIF1A. Note that relatively weak coiled coils, including the neck coiled coil (aa 366–397) and one near aa 800, are invisible and weakly visible, respectively, in this high-stringency prediction. (B) Subcellular distribution pattern of KIF1A deletions in living neurons. Cultured hippocampal neurons at days in vitro (DIV) 14–18 were transfected with EGFP-KIF1A deletions and visualized 48–72 h after transfection. MD, MN, MC, and MCF accumulated in the periphery of neurons (arrowheads), whereas MD-T312M and MCFC were diffusely distributed. Scale bar: 10 μm. (C) MT-binding properties of the KIF1A deletions. Lysates of HEK293T cells transfected with KIF1A deletions were incubated with taxol-stabilized MTs, and the sedimented (P) KIF1A proteins, along with those in the supernatant (S), were analyzed by immunoblotting with EGFP antibodies. (D) Quantitative analysis of the MT binding (mean±s.d.) of KIF1A deletions. Significant increases (*, compared to Full) are indicated.
Interestingly, a fragment of KIF1A containing only the N-terminal motor domain (MD, aa 1–365) showed a dramatic accumulation in distal regions of the neurites in a large proportion of observed neurons (91%, n=32 cells; Figure 1B). In contrast, MD with a point mutation in the motor domain (MD-T312M) known to disrupt KIF1A activity (Lee et al, 2003) showed a greatly reduced peripheral accumulation (16%, n=25) and a mainly diffuse distribution pattern throughout the neurons (Figure 1B), suggesting that peripheral accumulation is driven by the activity of the motor domain. A KIF1A construct containing the motor domain plus the neck CC (MN, aa 1–425; the neck CC spans aa 366–397) showed a high peripheral accumulation (100%, n=24), similar to that of MD (91%). An even broader KIF1A construct containing the motor, neck CC, and CC1 domains (MC, aa 1–489) also exhibited peripheral accumulation (54%, n=28; Figure 1B), although the degree of peripheral accumulation was smaller than that of the MD (91%). A KIF1A construct containing the motor, neck CC, CC1, and FHA domains (MCF, aa 1–615) showed good peripheral accumulation (93%, n=27; Figure 1B). Surprisingly, a KIF1A construct containing the motor, neck CC, CC1, FHA, and CC2 domains (MCFC, aa 1–680), which is identical to KIF1A-MCF except for the addition of the aa 65 residues containing the CC2 domain, showed only minimal peripheral accumulation (10%, n=42) with a mainly diffuse distribution pattern (Figure 1B). In addition, MCFC-FLAG in which the C-terminal EGFP domain was replaced with a much smaller FLAG tag showed a diffuse subcellular distribution similar to that of MCFC-EGFP (data not shown), suggesting that the loss of peripheral accumulation in MCFC is not a result of the EGFP addition to the C-terminus. The expression level of MCFC-EGFP was not different from that of other KIF1A deletion variants in both heterologous cells (Figure 1A) and cultured neurons (data not shown), suggesting that the expression levels of MCFC are unlikely to explain its loss of peripheral accumulation. Taken together, these results suggest that the CC2 domain exerts negative effects on KIF1A activity in vivo.
A previous study demonstrated that the kinesin heavy chain is negatively regulated through inhibition of MT binding (Kuznetsov et al, 1989). To determine whether the CC2-dependent negative regulation of KIF1A involved inhibition of MT binding, we tested MT-binding activity of the KIF1A deletions using the previously described MT cosedimentation assay (Verhey et al, 1998). Among KIF1A deletions expressed in HEK293T cells, MD, MN, MC, and MCF showed cosedimentation with MTs that were significantly better (MD, 0.88±0.01, mean±s.d., n=5, *P<0.0001; MN, 0.74±0.03, n=3, *P<0.0001; MC, 0.52±0.04, n=4, *P<0.0005; 0.89±0.07, n=4, *P<0.0001) than that of full-length KIF1A (Full; 0.35±0.02, n=4; Figure 1C and D). In contrast, MCFC showed limited MT binding (0.25±0.05, n=4) similar to that of Full (Figure 1C and D). These results indicate that the MT-binding characteristics of KIF1A deletion variants correlate with their peripheral accumulation in living neurons, suggesting that MT binding is likely to be one of the key mechanisms mediating CC2-dependent negative regulation of KIF1A.
The CC2 domain is necessary and sufficient for negative regulation of KIF1A
The above results suggest that CC2 is capable of exerting a negative effect on KIF1A activity. To determine whether CC2 is required for the negative regulation of KIF1A, we generated and tested a full-length KIF1A mutant lacking the CC2 domain (Full-ΔCC2; Figure 2A). In transfected neurons, Full-ΔCC2 showed a markedly increased peripheral accumulation (100%, n=30; Figure 2B), which is in sharp contrast to the behavior of the full-length KIF1A (Full-WT; no peripheral accumulation and association with moving particles; Figure 2B) (Lee et al, 2003), but is similar to the peripheral accumulation observed in MD, MN, and MCF (91, 100, and 93%, respectively; Figure 1B). In addition, MT binding of Full-ΔCC2 was significantly increased (0.88±0.03, n=4, *P<0.0001; Figure 2C and D) over that of Full (0.35±0.02, n=4; Figure 2C and D), to levels similar to those of MD, MN, MC, and MF (0.88±0.01, 0.74±0.03, 0.52±0.04, and 0.89±0.07, respectively; Figure 1C and D). Taken together, these results suggest that the CC2 domain is both necessary and sufficient for negative regulation of KIF1A.
Figure 2.

The CC2 and FHA domains are required for the negative regulation of KIF1A. (A) A schematic representation of the deletion variants of KIF1A-Full and MCFC, and their expression levels in heterologous (HEK293T) cells. Full-WT, full-length wild-type KIF1A. (B) Subcellular distribution of KIF1A deletions in living cultured hippocampal neurons. Scale bar: 10 μm. (C) MT binding of the KIF1A deletion variants. (D) Quantitative analysis of the MT binding of KIF1A deletions. Significant increases (*, compared to Full-WT; **, compared to MCFC) are indicated.
The FHA domain is required for CC2-dependent negative regulation of KIF1A
The FHA domain of KIF1A is located immediately upstream of CC2. To test whether the FHA domain is also involved in the regulation of KIF1A, we generated KIF1A deletion variants lacking the FHA domain in the backbone of MCFC and Full (MCFC-ΔFHA and Full-ΔFHA, respectively). Surprisingly, despite the presence of the CC2 domain, MCFC-ΔFHA exhibited enhanced peripheral accumulation (75%, n=32) and MT binding (0.61±0.04, n=4, **P<0.0001) compared to that of MCFC (10% and 0.25±0.05, respectively; Figure 2B–D). Full-ΔFHA also exhibited a significantly increased peripheral accumulation (70%, n=30) and MT binding (0.68±0.08, n=3, *P<0.0005; Figure 2B–D), compared to that of Full (no peripheral accumulation; MT binding, 0.35±0.02). These results, together with the results from Figure 1, suggest that the FHA domain does not exert negative effects on KIF1A by itself, but that it is required for CC2-dependent negative regulation of KIF1A.
The FHA domain of KIF1A interacts with CC2
To test whether the functional association between the FHA domain and CC2 occurs through a physical interaction, we generated a CC2-containing GST fusion protein (GST–CC2) and used a pull-down assay to check for its interaction with EGFP-tagged KIF1A deletion variants expressed in heterologous cells (Figure 3A and B). We found that GST–CC2 was able to pull down MCF, but not MD or MC (Figure 3A), suggesting that CC2 interacts with the FHA domain but not with the motor domain, the neck CC, or CC1. In addition, GST–CC2 did not pull down MCFC, suggesting that the FHA–CC2 intramolecular interaction is favored over an intermolecular interaction. GST alone did not pull down any of the KIF1A deletion variants tested. When tested against the single domains of KIF1A (CC1, FHA, or CC2), GST–CC2 pulled down FHA, but not CC1, as expected (Figure 3B). Intriguingly, GST–CC2 also pulled down CC2, suggesting that the CC2 domains form self-multimers and may participate in the formation of the recently reported KIF1A dimers (Tomishige et al, 2002).
Figure 3.
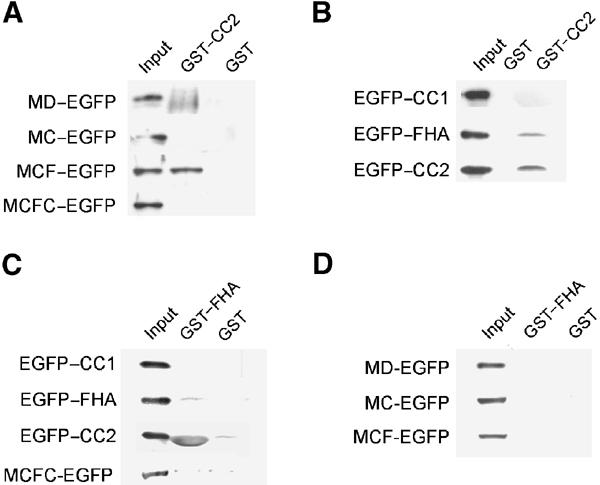
The FHA domain of KIF1A interacts with CC2. (A–D) Pull-down of KIF1A deletions. EGFP-KIF1A deletions expressed in HEK293T cells were incubated with GST–CC2 (A, B), GST–FHA (C, D), or GST alone (A–D), and the precipitates were analyzed by immunoblotting with EGFP antibodies.
In reverse pull-down assays, a GST fusion protein containing the FHA domain (GST–FHA) pulled down CC2, but not CC1 or FHA (Figure 3C), indicating that the FHA domain specifically interacts with CC2, and that it is not involved in the formation of self-multimers. As expected, GST–FHA did not pull down MD, MC, MCF, or MCFC (Figure 3D), further confirming that the FHA domain specifically interacts with CC2 and that the FHA–CC2 intramolecular interaction within MCFC is favored over an intermolecular interaction.
Disrupting the FHA–CC2 interaction by point mutations in the FHA domain increases KIF1A activity
The FHA domain contains several conserved residues, some of which are known to be important for the recognition of target phosphoproteins (Li et al, 2000; Durocher and Jackson, 2002). To determine whether the conserved residues in the KIF1A FHA (G519, R520, H540, and D564) are involved in CC2 recognition, we performed pull-down assays with point mutations of these residues. GST–CC2 failed to pull down mutants G519R and H540A, but was able to bring down mutants R520A and D564A (Figure 4A). This suggests that the two conserved residues (G519 and H540) in the KIF1A FHA are important for CC2 recognition.
Figure 4.
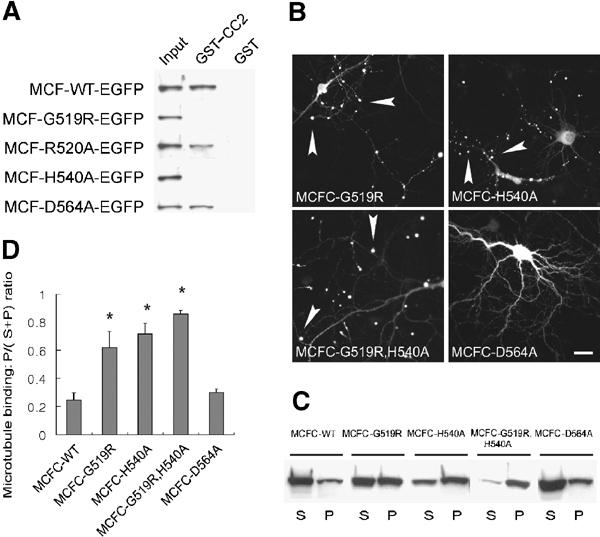
Disrupting the FHA–CC2 interaction by point mutations in the FHA domain increases KIF1A activity. (A) GST–CC2 pull-down of MCF with point mutations in the FHA domain. (B) Subcellular distribution of MCFC-EGFP containing FHA point mutations in living cultured hippocampal neurons. Scale bar: 10 μm. (C) MT binding of MCFC containing FHA point mutations. (D) Quantitative analysis of MT binding of the MCFC mutants. Significant increases (*, compared to MCFC-WT) are indicated.
We further tested the effect of the FHA point mutations on peripheral accumulation of KIF1A in living cultured neurons (Figure 4B). MCFC variants with the FHA point mutations (G519R, H540A, and double mutation of G519R+H540A) showed markedly increased peripheral accumulation (50%, n=22; 57%, n=23; and 87%, n=23, respectively; Figure 4B) that were quite different from the mainly diffuse distribution of wild-type MCFC (10% accumulation; Figure 1B). In contrast, MCFC with the D564A point mutation showed a mainly diffuse distribution (5%, n=20; Figure 4B), similar to that of wild-type MCFC (Figure 1B). The MCFC mutants with enhanced peripheral accumulation (G519R, H540A, and G519R+H540A) also showed a corresponding increase in MT binding (0.62±0.12, n=3, *P<0.005; 0.72±0.07, n=3, *P<0.0001; 0.86±0.03, n=3, *P<0.0001, respectively) to levels that are significantly higher than that of wild-type MCFC (0.25±0.05). In contrast, MCFC with the D564A point mutation showed MT binding (0.30±0.03, n=3) comparable to that of the wild type (Figure 4C and D). A milder mutation at G519 (G519A rather than G519R) showed peripheral accumulation and MT binding similar to that of G519R (data not shown). The G519R point mutation in the full-length context (Full-G519R) also caused enhanced MT binding (0.67±0.10, n=3, *P<0.005) to a level significantly better than that of wild-type KIF1A-Full (0.35±0.02). Taken together, these results suggest that disruption of the FHA–CC2 interaction by point mutations in the FHA domain increases peripheral accumulation and MT binding of KIF1A.
Disrupting the FHA–CC2 interaction by point mutations in CC2 increases KIF1A activity
The importance of some conserved FHA residues in CC2 recognition (Figure 4) suggests that the KIF1A FHA domain may recognize phosphopeptide ligands. To explore this possibility, we first tested whether the phosphorylatable residues of CC2 (Y649, T657, Y658, Y667, or S669) are involved in the interaction with the FHA domain. Intriguingly, GST–FHA failed to pull down EGFP–CC2 with a Y649F mutation, whereas it pulled down all others (wild type, T657A, Y658F, Y667A, and S669A; Figure 5A). In a control pull-down, GST alone did not bring down any of the tested proteins (Figure 5A). Moreover, MCFC with the Y649F point mutation (MCFC-Y649F), but not others (wild-type, T657A, and Y658F), was pulled down by GST–CC2 (Figure 5B), suggesting that this pull-down occurred by an intermolecular interaction between GST–CC2 and the FHA domain that had been released from the intramolecular CC2 interaction by the Y649 point mutation. In control experiments, MCFC-Y649F was not pulled down by GST–FHA (Figure 5B). The circular dichroism pattern of CC2 with the Y649F point mutation was essentially identical to that of wild-type CC2 (data not shown), suggesting that the effects of the Y649F point mutation were not caused by a significant structural change, but rather by a specific loss of the hydroxyl group from the CC2 Y649. Introduction of the same CC2 mutations (Y649F, T657A, Y658F, Y667A, and S669A) into GST–CC2 did not affect their ability to pull down EGFP–CC2 (Figure 5C), suggesting that these mutations, including Y649F, do not affect CC2–CC2 self-multimerization. These results suggest that the Y649 residue in the KIF1A CC2 is required for intramolecular FHA–CC2 interaction, but not for CC2–CC2 multimerization.
Figure 5.

Disrupting the FHA–CC2 interaction by point mutations in CC2 increases KIF1A activity. (A) GST–FHA pull-down of EGFP–CC2 with point mutations. (B) Pull-down of MCFC-EGFP containing point mutations in CC2 with GST–CC2, GST–FHA, or GST alone. (C) Quantitative analysis of the pull-down of mutated EGFP–CC2 with GST–CC2. (D) Subcellular distribution of MCFCs with CC2 point mutations in living cultured hippocampal neurons. Scale bar: 10 μm. (E, F) MT binding of MCFC-EGFP mutants, and quantitative analysis of the MT-binding results. A significant increase (*, compared to MCFC-WT) is indicated. (G) MT binding of MCFC-EGFP (wild type and Y649F) in the presence of AMP-PNP or ATP. (H) MT binding of MCFC-EGFP (wild type and Y649F) at various MT concentrations.
In cultured neurons, MCFC-Y649F showed a dramatic peripheral accumulation (100%, n=30) that sharply contrasts to the mainly diffuse distribution of wild-type MCFC (Figure 5D). Immunofluorescence staining of MCFC Y649F-expressing neurons with antibodies against neuronal markers, including MAP2 and neurofilament-H (markers for dendrites and axons, respectively), revealed that the transfected neurons were morphologically intact, suggesting that the observed peripheral accumulation is not caused by nonspecific morphological disruptions (data not shown). In contrast, MCFC with other point mutations (T657A and Y658F) showed a mainly diffuse distribution similar to that of wild-type MCFC (Figure 5D).
Consistent with these results, MCFC-Y649F, but not MCFC with other mutations (T657A and Y658F), showed a significant increase in MT binding (0.90±0.01, n=3, *P<0.0001; Figure 5E and F) relative to wild-type MCFC (0.25±0.05). MT-binding assays with these KIF1A proteins expressed in cultured hippocampal neurons yielded similar results (data not shown). When AMP-PNP, a nonhydrolyzable ATP analog, was replaced with ATP in the MT-binding assay, the MT binding of MCFC-Y649F (expressed in heterologous cells) was significantly decreased (0.85±0.05 in AMP-PMP versus 0.58±0.06 in ATP, n=4, *P<0.001), whereas that of MCFC-wild type was not significantly changed (0.18±0.05 in AMP-PMP versus 0.15±0.09 in ATP, n=4, P=0.62; Figure 5G), indicating that a portion of the MT binding of MCFC-Y649F is ATP sensitive. The ATP-insensitive portion of the MT binding of MCFC-Y649F may be due to the reported electrostatic interaction between the K loop in the motor domain and the E hook in tubulin (Okada and Hirokawa, 1999, 2000). In support of this, the motor domain only (MD in Figure 1A) also exhibited a significant but partial ATP-dependent reduction in its MT binding (0.91±0.05 in AMP-PMP versus 0.36±0.12 in ATP, n=3, *P<0.005). Further quantitative analysis of the MT binding of MCFC (wild type and Y649F) in the presence of various MT concentrations indicated an ∼30-fold increase in the apparent MT-binding affinity in the mutant, as calculated by comparison of MT concentrations required for half maximal MT binding (6.0 and 0.2 mM for wild type and Y649F, respectively; Figure 5H). In the full-length context, the Y649F point mutation (KIF1A-Full-Y649F) also showed a small but significant increase in MT binding (0.44±0.02, n=3, *P<0.005), when compared to wild-type Full (0.35±0.02, n=4). This suggests that, in the context of full-length KIF1A, the Y649F mutation may not be sufficient to disrupt fully the FHA–CC intramolecular interaction. This possibility is further supported by the fact that KIF1A-Full lacking the whole CC2 domain shows a markedly increased MT binding (0.89±0.03, n=4) (Figure 2C and D). Taken together, our data indicate that disrupting the FHA–CC2 interaction by point mutations in CC2 increases peripheral accumulation and MT binding of KIF1A.
Enhanced MT binding of KIF1A induced by the disruption of the FHA–CC2 interaction occurs through the motor domain
Our results indicate that point mutations disrupting the FHA–CC interaction increase MT binding of KIF1A (Figures 4 and 5). What might be the molecular mechanisms underlying this change? It may occur through the N-terminal motor domain and/or the region outside the motor domain containing the CC and FHA domains. When the Y649F mutation was introduced into an MCFC construct lacking the motor domain (NCFC-Y649F; aa 351–681), its MT binding (0.31±0.07, n=3) was not significantly higher (P=0.139) than that of wild-type NCFC (0.21±0.05, n=3; Figure 6A and B). This is in contrast to the ∼3.6-fold increase in MT binding seen with MCFC-Y649F (0.90±0.01) relative to wild-type MCFC (0.25±0.05; Figures 5E, F and 6A, B). These results suggest that disrupting the FHA–CC2 intramolecular interaction enhances the MT binding of KIF1A through the motor domain.
Figure 6.
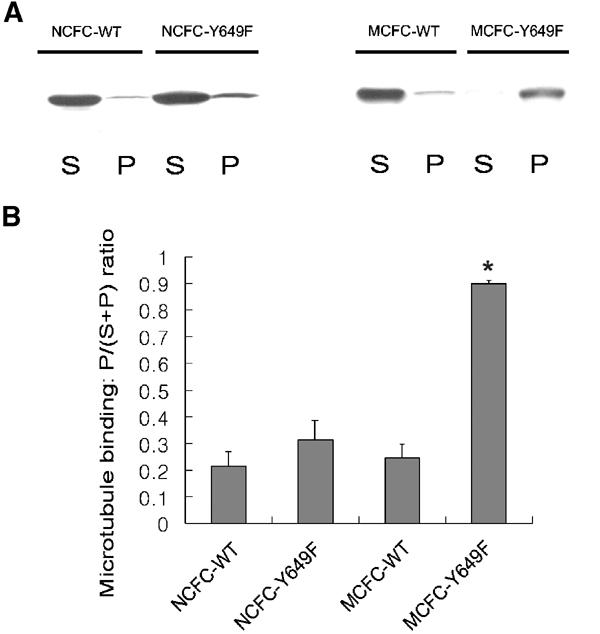
Enhanced MT binding of KIF1A induced by the disruption of the FHA–CC2 interaction occurs through the motor domain. (A) MT binding of EGFP-NCFC (wild type and Y649F) and MCFC-EGFP (wild type and Y649F). (B) Quantitative analysis of the results. A significant increase (*, compared to wild type) is indicated.
Disruption of the FHA–CC2 interaction enhances KIF1A multimerization
Based on the reported dimerization in KIF1A/Unc104 (Tomishige et al, 2002) and our observation that the KIF1A CC2 interacts with both the FHA domain and itself (Figure 3), we hypothesized that, in addition to the increased MT binding, disruption of the FHA–CC2 interaction may lead to enhanced dimerization of KIF1A. To test this hypothesis, we first established a coimmunoprecipitation assay in which KIF1A multimerization could be visualized. When HEK293T cell lysates doubly transfected with differentially tagged MCFC constructs (MCFC-EGFP and MCFC-FLAG) were immunoprecipitated with FLAG antibodies, MCFC-EGFP was co-precipitated with MCFC-FLAG (Figure 7A), whereas singly expressed MCFC-EGFP was not brought down by FLAG antibodies. This indicates that MCFC can form self-multimers, consistent with the reported dimerization of KIF1A/Unc104 (Tomishige et al, 2002). Next, we tested the effect of the Y649F mutation on KIF1A multimerization. When HEK293T cell lysates doubly transfected with MCFC-Y649F-EGFP and MCFC-Y649F-FLAG were incubated with FLAG antibodies, the coimmunoprecipitation of MCFC-Y649F-EGFP was significantly increased by ∼83% (coimmunoprecipitation efficiency of 4.56±0.97%, n=6, *P<0.0005), when compared to that of wild-type MCFC (2.50±0.29%, n=6; Figure 7A and B). These results suggest that disrupting the FHA–CC2 interaction increases KIF1A dimerization.
Figure 7.

Disruption of the FHA–CC2 interaction enhances KIF1A multimerization. (A) Enhanced self-multimerization in MCFC by the introduction of the Y649F mutation. To compare self-multimerization in wild-type and mutant (Y649F) MCFC, HEK293T cells were doubly transfected with MCFC constructs differentially tagged with EGFP and FLAG (MCFC-wild-type-EGFP+MCFC-wild-type-FLAG; lanes 1–4, or MCFC-Y649F-EGFP+MCFC-Y649F-FLAG; lanes 5–8). Cell lysates were immunoprecipitated with FLAG antibodies and characterized by immunoblotting with EGFP and FLAG antibodies. Cells singly transfected with MCFC-wild-type-EGFP or MCFC-Y649F-EGFP were used as controls. FLAG-tagged MCFCs (wild-type and Y649F) are visible in self-immunoprecipitation lanes (lanes 3,7) but not in input lanes (2.5%; lanes 1,5), perhaps due to a relatively low sensitivity of the FLAG antibody. Note that the coimmunoprecipitation level of MCFC-EGFP was increased by the mutation. (B) Quantitative analysis of the coimmunoprecipitation results. A significant increase (*, compared to MCFC-WT) is indicated.
A flexible hinge in CC2 allows the intramolecular FHA–CC2 interaction
We used structural model building to test whether the linker between the FHA domain and CC2 is sufficiently long and flexible to allow intramolecular FHA–CC2 interaction. We first built a model of the FHA domain of KIF1A by comparing it to other FHA domains with known X-ray or NMR structures. This model predicted that the KIF1A FHA domain spans aa 488–604 and contains 11 antiparallel β-strands and a small C-terminal α-helix (Figure 8A). Modeling of the linker connecting the FHA domain and CC2 (aa 605–624; 20 residues) predicted that it contains two distinct regions: a proline-rich region (aa 605–617; 13 residues; the gray region in Figure 8B) and an α-helix (aa 618–624; seven residues; the wheat region in Figure 8B). The proline-rich region is predicted to form a loop structure that has limited flexibility due to its three proline residues (aa 608, 613, and 616). The helix has a weak propensity for coiled-coil formation, but smoothly merges with the α-helical structure of CC2 (aa 625–679), which has a strong propensity for coiled-coil formation. Although the length (20 residues) of the linker is apparently long enough to allow intramolecular interaction, these predicted structures may limit the flexibility that is required for it to function as a hinge.
Figure 8.

A flexible hinge in CC2 allows the intramolecular FHA–CC2 interaction. (A) A ribbon diagram of the FHA domain of KIF1A based upon homology modeling. P488 and R604 indicate the residues at the N- and C-terminal ends of the FHA domain, respectively. G519 and H540 (purple) are two residues critical for the FHA–CC2 interaction. (B) A ribbon diagram of a model for the intramolecular FHA–CC2 interaction and the flexible hinge. The FHA domain (green; aa 488–604) is connected to CC2 (yellow; aa 625–679) via a linker containing a proline-rich region (gray; 13 residues; aa 605–617; prolines at aa 608, 613, and 616) and an α-helix with a weak propensity for coiled-coil formation (wheat; seven residues; aa 618–624). This α-helix merges with the α-helical structure in CC2. The N-terminal region of CC2 contains a small flexible stretch (QGID; aa 630–632) containing a helix-breaking residue (G631). The bidirectional arrow indicates the reversible transition between the bent (yellow) and straight (blue) conformations of CC2. Some side chains are shown by balls and sticks. Our model predicts that the flexible hinge of CC2 allows the intramolecular FHA–CC2 interaction. (C) Increased pull-down of MCFC-G631F. Lysates of HEK293T cells transfected with MCFC-EGFP (wild type, Y649F and G631F) were incubated with GST–CC2 or GST alone, and the precipitates were immunoblotted with EGFP antibodies. Input: 2%. (D, E) Increased MT binding of MCFC-G631F. MT binding of mutant (G631F) MCFC-EGFP was compared to that of MCFC-wild type and MCFC-Y649F (D) and quantified (E). Asterisks in MCFC-G631F (0.60±0.09, n=4) and MCFC-Y649F (0.88±0.11, n=4) indicate significant changes (*P<0.0001 for both) compared to MCFC-WT (0.14±0.06, n=4). (F) Increased peripheral accumulation in MCFC-G631F-expressing neurons. Scale bar: 10 μm.
However, we found a flexible aa stretch in the N-terminal region of CC2 (QGID; aa 630–633) that does not have a predicted α-helical structure and hence may function as a hinge (Figure 8B). To support this hypothesis, we changed Gly631 (a helix breaker) within the QGID stretch to Phe or Ala, which was predicted to change the QGID stretch into an α-helix. We reasoned that these hinge-removing point mutations would change CC2 from bent to straight and make it difficult for Y649 in CC2 to interact with the FHA domain (Figure 8B). As expected, KIF1A constructs containing these point mutations showed various signs of limited intramolecular FHA–CC2 interaction. MCFC-G631F could be significantly pulled down by GST–CC2 (Figure 8C), which contrasts with the lack of pull-down of wild-type MCFC by GST–CC2 (Figure 3A). In addition, MCFC-G631F showed significantly enhanced MT binding (MCFC-G631F, 0.60±0.09, n=4, *P<0.0001 versus wild-type MCFC, 0.14±0.06, n=4; Figure 8D,E) and a dramatic peripheral accumulation in neurons (94%, n=53; Figure 8F). We obtained similar results from the G631A mutation (data not shown). Taken together, these results suggest that the QGID stretch in CC2 may function as a flexible hinge that allows the intramolecular FHA–CC2 interaction.
Discussion
In the present study, we identified an intramolecular interaction between the FHA and CC2 domains of KIF1A that negatively regulate its activity. The FHA domain is generally known as a phosphoprotein-binding module (Li et al, 2000; Durocher and Jackson, 2002; Tsai, 2002). Within KIF1A CC2, we identified Y649 as being critical for FHA binding (Figure 5) and showed that removing the hydroxyl group of Y649 by a point mutation (Y649F) disrupted the FHA–CC2 interaction. These results might seem to suggest that the phosphorylation of Y649 could regulate the FHA–CC2 interaction, thereby regulating KIF1A activity. However, our additional data do not appear to support this possibility. Specifically, treatment of heterologous cells expressing EGFP–CC2 with pervanadate, an inhibitor of protein tyrosine phosphatase, did not increase the phosphotyrosine levels of CC2. In addition, treatment of EGFP–CC2 with various protein tyrosine phosphatases did not affect its pull-down by GST–FHA. More directly, immunoblot analysis with phosphotyrosine antibodies on various KIF1A proteins from the pellet and supernatant fractions of the MT-binding assays did not reveal any differential levels of tyrosine phosphorylation between the two fractions. Furthermore, both wild-type and mutant (Y649F) MCFC constructs showed similar phosphotyrosine levels, suggesting that Y649 is unlikely to be a site of tyrosine phosphorylation. Endogenous KIF1A proteins immunoprecipitated from pervanadate-treated or untreated cultured neurons did not reveal any detectable levels of phosphotyrosine. Taken together, these in vivo and in vitro results suggest that endogenous KIF1A is unlikely to be tyrosine phosphorylated, and that if any tyrosine phosphorylation occurs in KIF1A the site may not be Y649. In addition, these data strongly suggest that the hydroxyl group, but not the phosphate group, of Y649 is a key determinant of the FHA–CC2 interaction. Although the involvement of the hydroxyl group of conserved tyrosine residues in various protein–protein interactions is commonly observed, that is, regulation of the dimerization of a fungal kinesin by a tyrosine in the neck domain (Schafer et al, 2003), the general notion that FHA domain is a phosphoprotein-binding module does not fit with our data.
One possible explanation for this discrepancy is that the KIF1A FHA, unlike other FHA domains, may recognize unphosphorylated peptides. Early studies in the field convincingly demonstrated that the FHA domain recognizes a short stretch of peptides containing either phosphorylated Thr or Tyr (Li et al, 2000; Durocher and Jackson, 2002). NMR and X-ray crystallographic analyses of various FHA domains complexed with peptide ligands indicated that the phosphate group of the peptides appears to be recognized by the same set of FHA residues including the Arg residue that corresponds to Arg 520 of KIF1A (Westerholm-Parvinen et al, 2000; Tsai, 2002). However, our KIF1A R520A point mutation did not disrupt the FHA–CC2 interaction (Figure 4A), suggesting that the FHA–CC2 interaction may not require phosphorylated Y649. Alternatively, a novel surface on the KIF1A FHA that is distant from the conventional phosphopeptide-binding surface may be involved in the recognition of unphosphorylated Y649. It has been shown that both the conventional phosphopeptide-binding surface and a distinct phospho-independent binding surface on the FHA domain of Chk2, a human DNA damage response kinase, are used to recognize target proteins (Li et al, 2002). Of these two possibilities, the first one appears to be supported by our observation that the FHA–CC2 interaction is disrupted by KIF1A FHA point mutations located on the conventional phosphopeptide-binding surface (G519 and H540) (Figure 4). A recent review, however, has pointed out that the highly conserved Gly and His residues, which are not in direct contact with peptide ligands but have been frequently mutated in various studies, may contribute to not only stabilizing the architecture of the peptide-binding surface but also the tertiary structure of the whole FHA domain (Durocher and Jackson, 2002). Thus, our results are compatible with both possibilities, which may not be mutually exclusive.
We demonstrated that the FHA–CC2 interaction negatively regulates the MT binding of KIF1A (Figures 4 and 5) and that disruption of this interaction enhances MT binding of KIF1A mainly through the N-terminal motor domain (Figure 6). A possible explanation for these results is that the FHA and/or CC domains directly interact with the motor domain, sterically hindering the MT binding of the motor domain. However, our results indicate that neither the FHA nor the CC2 domains interact with the KIF1A motor domain (Figure 3A and D). In addition, a protein containing both the FHA and CC2 domains did not interact with the motor domain in a pull-down assay. These results suggest that a physical interaction between the motor domain and the FHA and/or CC2 domains is unlikely to regulate MT binding directly. Alternatively, the FHA or CC2 domain may inhibit the MT binding of the motor domain by an allosteric modulation.
Another important consequence of disrupting the FHA–CC2 intramolecular interaction in KIF1A is enhanced motor multimerization (Figure 7). In addition, our data indicate that the KIF1A CC2 interacts with itself, but not with CC1 and the neck CC (Figure 3A and B). These results, along with the reported KIF1A dimerization (Tomishige et al, 2002), strongly supports a model in which a ‘parallel' dimer of two full-length KIF1A molecules is stabilized by small self-dimerizations of the neck CC, CC1, and CC2 domains (Vale, 2003). Disrupting the FHA–CC2 intramolecular interaction may induce the dimerization of CC2 first, followed by the dimerization of CC1 and finally that of the neck CC, in a ‘zippering' fashion. Conversely, factors favoring the intramolecular interaction would destabilize the CC2–CC2 dimer first and then the others, in an ‘unzipping' fashion.
A flexible hinge located in the N-terminal region of CC2 appears to allow the intramolecular FHA–CC2 interaction (Figure 8). In support of this hypothesis, point mutation of G631 in the hinge to one of two α-helix-forming residues (Phe or Ala) caused disruption of the intramolecular FHA–CC2 interaction, as evidenced by the increased pull-down of MCFC G631F by GST–CC2 (Figure 8C), enhancement in KIF1A MT binding (Figure 8D and E), and marked peripheral accumulation of KIF1A in neurons (Figure 8F). These results suggest that the α-helix-forming mutations make the hinge more rigid and inhibit the intramolecular FHA–CC2 interaction, hence activating KIF1A. A possible argument against these results is that the G631F or G631A mutations in CC2 might simply weaken the affinity between the FHA and CC2 domains. However, this is unlikely because MCF was readily pulled down by GST–CC2 G631F and G631A, to levels comparable to that of wild-type GST–CC2. We propose that the flexible hinge may allow reversible transition between the bent and straight conformations of CC2, which facilitate the intramolecular FHA–CC2 interaction and CC2–CC2 dimerization, respectively. In addition, these mechanisms may contribute to the reported reversible monomer-to-dimer transition of KIF1A (Tomishige et al, 2002).
Recently, a cryo-electron microscopy study revealed that two neck helices (H1 and H2) of Caenorhabditis elegans Unc104, which correspond to the neck CC and CC1 domains of KIF1A, respectively, form a parallel dimer in the self-folded monomeric conformation and an intermolecular dimer in the dimeric state (Al-Bassam et al, 2003). This suggests that the self-folded conformation may inhibit KIF1A monomers, whereas the dimeric conformation likely enhances KIF1A activity. This report and our results are highly complementary in many aspects. The self-folded, repressed conformation of Unc104 may explain our finding that the addition of CC1 to MN (motor+neck CC) slightly inhibits KIF1A in MT binding and neuronal accumulation assays (Figure 1). The decrease in MT-binding affinity of Unc104 in the ATPase assay following addition of the region containing the FHA domain (aa 447–653; corresponding to the FHA+CC2 domains in KIF1A) to Unc104 (aa 1–446; corresponding to the motor+neck CC+CC1 in KIF1A) is consistent with our observation that the addition of the FHA+CC2 domains to MC (motor+neck CC+CC1) in MCFC markedly lowers its MT binding and peripheral accumulation (Figure 1). Our results extend the Unc104 study by further dissecting the FHA-containing region of Unc104 (aa 447–653) into two independent domains (FHA and CC2) and identifying a novel intramolecular interaction between them required for KIF1A inhibition. Conceptually, the notion that an intramolecular interaction between coiled coils (H1/neck CC and H2/CC1) can regulate motor function is similar to our conclusion that a coiled-coil (CC2)-mediated intramolecular interaction can regulate motor function. Finally, the fact that the same outcome (enhanced KIF1A activity) was induced by the disruption of two independent intramolecular interactions (H1/neck CC-H2/CC1 and FHA–CC2) suggests that the two interactions are functionally interconnected. Exploration of these mechanisms would be an interesting direction for future studies.
What are the physiological factors that could induce disruption of the intramolecular FHA–CC2 interaction? It has been shown that high motor concentrations in solution or on membranes can induce the transition of KIF1A/Unc104 monomers to processive dimers (Klopfenstein et al, 2002; Tomishige et al, 2002). Thus, an obvious candidate for disruption of the FHA–CC2 interaction would be high local concentrations of KIF1A, which may weaken the FHA–CC2 interaction, shift the balance toward the CC2–CC2 interaction and favor KIF1A dimerization. Alternatively, the trigger could be the interaction of the KIF1A FHA with other proteins, that is, KIF1A cargoes. It should be noted that the KIF1A FHA contains the conserved Arg520 and thus can possibly interact with phosphate groups in target proteins. Although the R520A point mutation does not disrupt the FHA–CC2 intramolecular interaction (Figure 4A), this does not necessarily exclude the possibility that the KIF1A FHA may bind to phosphoproteins in an intermolecular fashion.
Finally, the domain structure of KIF1A, in particular the position of the FHA domain between CC1 and CC2, is conserved in other members of the KIF1/Unc104 family, including C. elegans Unc104, Drosophila CG8566-PD, and mammalian KIF1 isoforms (KIF1B, KIF1C and KIF1D) (Otsuka et al, 1991; Nangaku et al, 1994; Rogers et al, 1997; Dorner et al, 1998). Moreover, multiple sequence alignment analysis of these kinesins reveals conservation of the critical residues required for the FHA–CC2 intramolecular interaction and the flexible hinge; the FHA G519 and H540, CC2 Y649, and hinge G631 residues are strictly conserved. This suggests that the FHA–CC2 intramolecular interaction may be a common regulatory feature in the KIF1/Unc104 family. Notably, a domain structure similar to that of KIF1/Unc104 is also found in KIF13, another family of FHA-containing kinesins, including Caenorhabditis elegans KLP-4, Drosophila kinesin-73, mouse KIF13B, and human GAKIN (Li et al, 1997; Nakagawa et al, 1997; Hanada et al, 2000). However, the critical residues for the FHA–CC2 interaction are not conserved in these kinesins. Thus, it remains to be determined whether the KIF13 family kinesins are similarly regulated by the FHA–CC2 intramolecular interaction.
In summary, the present study identifies the FHA domain-mediated intramolecular interaction as a novel autoinhibitory switch for the regulation of KIF1A activity. Furthermore, this novel intramolecular mode of FHA action may be applicable to the regulation of other FHA-containing proteins involved in numerous processes including DNA repair, transcription, cell cycle arrest, signal transduction, and protein degradation.
Materials and methods
Constructs
EGFP-full-length KIF1A in GW1 has been described (Lee et al, 2003). MD, MN, MC, and MCF (aa 1–365, 1–425, 1–489, and 1–615) of KIF1A were subcloned into pEGFP-N1 (Clontech). MCFC-EGFP was made by subcloning aa 1–680 of KIF1A+EGFP from Full-EGFP into GW1. MCFC-FLAG was generated by inserting aa 1–681 into pBK(Δ)-FLAG. Full-EGFP-ΔFHA and Full-EGFP-ΔCC2 were made by subcloning aa 1–489+616–1695 and 1–615+680–1695 (from Full-EGFP) into GW1, respectively. For MCFC-ΔFHA, aa 616–681 was added to MC-EGFP. EGFP–CC1, FHA, CC2, and NCFC were made by subcloning aa 396–489, 460–615, 585–681, and 351–681 into pEGFP-C1. Constructs with point mutations were generated by QuickChange kit (Stratagene). For GST fusions, FHA (462–615) and CC2 (574–684) were subcloned into pGEX-4T1 (Amersham Biosciences).
Cell culture and transfection
HEK293T cells, maintained in DMEM containing 10% fetal bovine serum, were transfected by the calcium phosphate method (Invitrogen) at 50–60% confluence. Cell extracts were prepared 2 days after transfection. Primary hippocampal neuronal cultures were prepared as described previously (Lee et al, 2003) and transfected by the calcium phosphate method.
Image acquisition from living neurons and image analysis
Transfected neurons on coverslips were set on a perfusion chamber with a thermostat (37°C) and maintained in MEM supplemented with 1 mM pyruvate and 60 μM N-acetyl L-cystein during the observation. Images were captured by a confocal microscope (LSM510, Zeiss) and analyzed using the MetaMorph software (Universal Imaging). To measure peripheral accumulation of KIF1A, we compared the immunofluorescence intensity of KIF1A at neurite tips (1–3 neurites with the most prominent accumulation per neuron) and an adjacent region in the same neurite (∼10–30 μm away). Cells with fluorescence ratios (tip/adjacent region) greater than a factor of 4 were counted as representing cases of positive peripheral accumulation. Statistical significance was assessed using Student's t-test.
MT-binding assay
The MT-binding assay was performed as described previously (Verhey et al, 1998). Briefly, transfected HEK293T cells were solubilized in ice-cold lysis buffer (LB; 25 mM HEPES/KOH, 115 mM K+CH3COO−, 5 mM Na+CH3COO−, 5 mM MgCl2, 0.5 mM EGTA, 1% Triton X-100, pH 7.2) containing protease inhibitors. After removing insoluble materials by centrifugation of the lysates at 50 000 rpm (TLA 100.2 rotor; Beckman) at 4°C for 30 min, the supernatant was added with AMP-PMP (5′-adenylylimidodiphosphate) or ATP (2.5 mM final), taxol (20 μM), and taxol-stabilized MTs (1 mg/ml). After 30 min incubation at room temperature, the mixture was overlaid on top of an LB bed containing 10% sucrose and 20 μM taxol, centrifuged at 30 000 rpm (SW55Ti rotor) at 22°C for 30 min, and washed with LB+taxol. The samples were immunoblotted with EGFP antibodies.
GST pull-down assay
GST fusion proteins were purified using glutathione sepharose 4B resin (Amersham Biosciences). HEK293 cells transfected with various KIF1A constructs were solubilized with phosphate-buffered saline containing 1% Triton X-100. After centrifugation at 22 000 g at 4°C for 30 min, the supernatant was incubated with glutathione sepharose 4B resin pre-bound with 10 μg of GST fusion proteins for 1 h at 4°C. Precipitated proteins were analyzed by immunoblotting with EGFP antibodies.
Coimmunoprecipitation
Transfected HEK293 cells were solubilized with phosphate-buffered saline containing 1% Triton X-100 and protease inhibitors. After centrifugation at 22 000 g at 4°C for 30 min, the supernatant was incubated with anti-FLAG M2-agarose (Sigma) for 2 h at 4°C. Precipitated proteins were immunoblotted with EGFP (Clontech) and FLAG (Sigma) antibodies.
Structural model building
Homology modeling of the FHA domain of KIF1A was performed using MODELLER v6 (Sali and Blundell, 1993). The homologous structures were detected using the threading servers GenTHREADER (Jones, 1999) and 3D-PSSM (Kelley et al, 2000). The atomic coordinates of the FHA domains from Chk2, Rad53 FHA1, Rad53 FHA2, and KAPP (PDB #1GXC, 1G6G, 1J4K, and 1MZK, respectively) were selected as templates for model building. Before submission to MODELLER, the sequence–structure alignment obtained from the threading servers was manually edited to adjust the boundary of FHA domain (aa G488–A604) in accordance with that of the closely related Xenopus Unc104 FHA domain (Xklp4) (Westerholm-Parvinen et al, 2000). The best model with the lowest MODELLER restraint energy was kept and further refined by energy minimization of the side chains. The backbone of CC2 model was constructed from an α-helix template, and assignment of secondary structures was performed using the NPS server (Combet et al, 2000). The bent helix model of CC2 and the final FHA–CC2 interaction model were manually constructed. The figures were prepared using PyMOL (http://www.pymol.org).
Acknowledgments
This work was supported by the Creative Research Initiatives Program of the Korean Ministry of Science and Technology.
References
- Al-Bassam J, Cui Y, Klopfenstein D, Carragher BO, Vale RD, Milligan RA (2003) Distinct conformations of the kinesin Unc104 neck regulate a monomer to dimer motor transition. J Cell Biol 163: 743–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combet C, Blanchet C, Geourjon C, Deleage G (2000) NPS@: network protein sequence analysis. Trends Biochem Sci 25: 147–150 [DOI] [PubMed] [Google Scholar]
- Cross R, Scholey JM (1999) Kinesin: the tail unfolds. Nat Cell Biol 1: E119–E121 [DOI] [PubMed] [Google Scholar]
- Dorner C, Ciossek T, Muller S, Moller PH, Ullrich A, Lammers R (1998) Characterization of KIF1C, a new kinesin-like protein involved in vesicle transport from the Golgi apparatus to the endoplasmic reticulum. J Biol Chem 273: 20267–20275 [DOI] [PubMed] [Google Scholar]
- Durocher D, Jackson SP (2002) The FHA domain. FEBS Lett 513: 58–66 [DOI] [PubMed] [Google Scholar]
- Goldstein LSB, Yang Z (2000) Microtubule-based transport systems in neurons: the roles of kinesins and dyneins. Ann Rev Neurosci 23: 39–72 [DOI] [PubMed] [Google Scholar]
- Hackney DD, Levitt JD, Suhan J (1992) Kinesin undergoes a 9 S to 6 S conformational transition. J Biol Chem 267: 8696–8701 [PubMed] [Google Scholar]
- Hall DH, Hedgecock EM (1991) Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans. Cell 65: 837–847 [DOI] [PubMed] [Google Scholar]
- Hanada T, Lin L, Tibaldi EV, Reinherz EL, Chishti AH (2000) GAKIN, a novel kinesin-like protein associates with the human homologue of the Drosophila discs large tumor suppressor in T lymphocytes. J Biol Chem 275: 28774–28784 [DOI] [PubMed] [Google Scholar]
- Hirokawa N (1998) Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science 279: 519–526 [DOI] [PubMed] [Google Scholar]
- Jones DT (1999) GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences. J Mol Biol 287: 797–815 [DOI] [PubMed] [Google Scholar]
- Kelley LA, MacCallum RM, Sternberg MJ (2000) Enhanced genome annotation using structural profiles in the program 3D-PSSM. J Mol Biol 299: 499–520 [DOI] [PubMed] [Google Scholar]
- Klopfenstein DR, Tomishige M, Stuurman N, Vale RD (2002) Role of phosphatidylinositol(4,5)bisphosphate organization in membrane transport by the Unc104 kinesin motor. Cell 109: 347–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov SA, Vaisberg YA, Rothwell SW, Murphy DB, Gelfand VI (1989) Isolation of a 45-kDa fragment from the kinesin heavy chain with enhanced ATPase and microtubule-binding activities. J Biol Chem 264: 589–595 [PubMed] [Google Scholar]
- Lee J, Shin H, Ko J, Choi J, Lee H, Kim E (2003) Characterization of the movement of the kinesin motor KIF1A in living cultured neurons. J Biol Chem 278: 2624–2629 [DOI] [PubMed] [Google Scholar]
- Li HP, Liu ZM, Nirenberg M (1997) Kinesin-73 in the nervous system of Drosophila embryos. Proc Natl Acad Sci USA 94: 1086–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Lee GI, Van Doren SR, Walker JC (2000) The FHA domain mediates phosphoprotein interactions. J Cell Sci 113 (Part 23): 4143–4149 [DOI] [PubMed] [Google Scholar]
- Li J, Williams BL, Haire LF, Goldberg M, Wilker E, Durocher D, Yaffe MB, Jackson SP, Smerdon SJ (2002) Structural and functional versatility of the FHA domain in DNA-damage signaling by the tumor suppressor kinase Chk2. Mol Cell 9: 1045–1054 [DOI] [PubMed] [Google Scholar]
- Miki H, Setou M, Kaneshiro K, Hirokawa N (2001) All kinesin superfamily protein, KIF, genes in mouse and human. Proc Natl Acad Sci USA 98: 7004–7011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Tanaka Y, Matsuoka E, Kondo S, Okada Y, Noda Y, Kanai Y, Hirokawa N (1997) Identification and classification of 16 new kinesin superfamily (KIF) proteins in mouse genome. Proc Natl Acad Sci USA 94: 9654–9659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nangaku M, Sato-Yoshitake R, Okada Y, Noda Y, Takemura R, Yamazaki H, Hirokawa N (1994) KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell 79: 1209–1220 [DOI] [PubMed] [Google Scholar]
- Okada Y, Hirokawa N (1999) A processive single-headed motor: kinesin superfamily protein KIF1A. Science 283: 1152–1157 [DOI] [PubMed] [Google Scholar]
- Okada Y, Hirokawa N (2000) Mechanism of the single-headed processivity: diffusional anchoring between the K-loop of kinesin and the C terminus of tubulin. Proc Natl Acad Sci USA 97: 640–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Yamazaki H, Sekine-Aizawa Y, Hirokawa N (1995) The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell 81: 769–780 [DOI] [PubMed] [Google Scholar]
- Otsuka AJ, Jeyaprakash A, Garcia-Anoveros J, Tang LZ, Fisk G, Hartshorne T, Franco R, Born T (1991) The C. elegans unc-104 gene encodes a putative kinesin heavy chain-like protein. Neuron 6: 113–122 [DOI] [PubMed] [Google Scholar]
- Rogers KR, Griffin M, Brophy PJ (1997) The secretory epithelial cells of the choroid plexus employ a novel kinesin-related protein. Brain Res Mol Brain Res 51: 161–169 [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234: 779–815 [DOI] [PubMed] [Google Scholar]
- Schafer F, Deluca D, Majdic U, Kirchner J, Schliwa M, Moroder L, Woehlke G (2003) A conserved tyrosine in the neck of a fungal kinesin regulates the catalytic motor core. EMBO J 22: 450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Wyszynski M, Huh KH, Valtschanoff JG, Lee JR, Ko J, Streuli M, Weinberg RJ, Sheng M, Kim E (2003) Association of the kinesin motor KIF1A with the multimodular protein liprin-alpha. J Biol Chem 278: 11393–11401 [DOI] [PubMed] [Google Scholar]
- Tomishige M, Klopfenstein DR, Vale RD (2002) Conversion of Unc104/KIF1A kinesin into a processive motor after dimerization. Science 297: 2263–2267 [DOI] [PubMed] [Google Scholar]
- Tsai MD (2002) FHA: a signal transduction domain with diverse specificity and function. Structure (Camb) 10: 887–888 [DOI] [PubMed] [Google Scholar]
- Vale RD (2003) The molecular motor toolbox for intracellular transport. Cell 112: 467–480 [DOI] [PubMed] [Google Scholar]
- Verhey KJ, Lizotte DL, Abramson T, Barenboim L, Schnapp BJ, Rapoport TA (1998) Light chain-dependent regulation of kinesin's interaction with microtubules. J Cell Biol 143: 1053–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerholm-Parvinen A, Vernos I, Serrano L (2000) Kinesin subfamily UNC104 contains a FHA domain: boundaries and physicochemical characterization. FEBS Lett 486: 285–290 [DOI] [PubMed] [Google Scholar]
- Zhou HM, Brust-Mascher I, Scholey JM (2001) Direct visualization of the movement of the monomeric axonal transport motor UNC-104 along neuronal processes in living Caenorhabditis elegans. J Neurosci 21: 3749–3755 [DOI] [PMC free article] [PubMed] [Google Scholar]