

Abstract
Terbinafine is increasingly used in combination with other antifungal agents to treat resistant or refractory mycoses due to synergistic in vitro antifungal activity; high doses are commonly used, but limited data are available on systemic exposure, and no assessment of pharmacodynamic target attainment has been made. Using a physiologically based pharmacokinetic (PBPK) model for terbinafine, this study aimed to predict total and unbound terbinafine concentrations in plasma with a range of high-dose regimens and also calculate predicted pharmacodynamic parameters for terbinafine. Predicted terbinafine concentrations accumulated significantly during the first 28 days of treatment; the area under the concentration-time curve (AUC)/MIC ratios and AUC for the free, unbound fraction (fAUC)/MIC ratios increased by 54 to 62% on day 7 of treatment and by 80 to 92% on day 28 compared to day 1, depending on the dose regimen. Of the high-dose regimens investigated, 500 mg of terbinafine taken every 12 h provided the highest systemic exposure; on day 7 of treatment, the predicted AUC, maximum concentration (Cmax), and minimum concentration (Cmin) were approximately 4-fold, 1.9-fold, and 4.4-fold higher than with a standard-dose regimen of 250 mg once daily. Close agreement was seen between the concentrations predicted by the PBPK model and the observed concentrations, indicating good predictive performance. This study provides the first report of predicted terbinafine exposure in plasma with a range of high-dose regimens.
INTRODUCTION
The allylamine antifungal terbinafine is a well-established agent in the treatment of onychomycosis (1). Due to its broad antifungal spectrum, interest in terbinafine has expanded to include its use in a range of cutaneous and subcutaneous mycoses, such as sporotrichosis, eumycetoma, and chromoblastomycosis (2–4), as well as in combination with other antifungal agents to treat resistant or refractory invasive fungal infections (IFIs), as described in numerous case reports (1, 5). In support of the latter indication, synergistic in vitro antifungal activity has been demonstrated with terbinafine in combination with azole antifungals for many important fungal pathogens, including Aspergillus spp., zygomycetes, Fusarium spp., Paecilomyces spp., Candida albicans, dematiaceous molds, and the highly resistant Scedosporium prolificans (6–11).
High dose is a consistent feature of terbinafine use in these indications, with daily doses of up to 1,000 mg daily (1) used to boost the plasma concentrations of terbinafine, which are known to be low following standard dosing, such as the standard dosing regimen of 250 mg daily in onychomycosis, due to extensive accumulation in skin and adipose tissue (12). However, no studies have assessed the systemic exposure of higher-dose terbinafine treatment (>250 mg daily) or divided daily doses (every 12 h [q12h] or q8h), factors that are likely to be crucial to the utility of terbinafine in effectively treating systemic mycoses in combination with other antifungals. This paucity of pharmacokinetic data is particularly important for terbinafine due to its very long terminal elimination half-life (2 to 3 weeks [13]) and substantial accumulation in plasma over time; trough terbinafine concentrations following 250 mg once daily are known to accumulate >10-fold over 12 to 20 weeks (14), with the majority of the accumulation occurring in the first 4 weeks of therapy (13).
In the absence of observed experimental concentration-time data at higher doses, physiologically based pharmacokinetic (PBPK) models are a useful tool to accurately predict drug exposure (15). Using a PBPK model for terbinafine, this study aimed to predict terbinafine concentrations in plasma for a range of high-dose regimens and also assessed pharmacodynamic parameters with terbinafine with each regimen to inform the optimal terbinafine dosing regimen in the treatment of refractory or resistant mycoses.
MATERIALS AND METHODS
The high-dose terbinafine regimens investigated in this study (250 mg q12h, 250 mg q8h, 500 mg q24h, and 500 mg q12h) were selected based on clinical use reported in clinical trials investigating the use of terbinafine for cutaneous and subcutaneous mycoses (2–4) and case reports of resistant or refractory mycoses (1, 5).
The dosing simulations in this study used a modified version of a previously reported PBPK model for terbinafine (12). Briefly, the model is comprised of several tissue compartments to reflect human physiology, with each compartment associated with a terbinafine-specific tissue-to-plasma partition coefficient and experimentally derived values for organ volume (liter) and blood flow (liter/h). The ability of this PBPK model to predict terbinafine pharmacokinetics in humans has been demonstrated (12).
In this study, the oral absorption rate constant was set to 1 h−1 for 250-mg doses and 0.7 h−1 for 500-mg doses to reflect the less-than-proportional increase in peak terbinafine concentrations reported with single doses of ≥500 mg (16). To accurately predict the prolonged accumulation of terbinafine, the previously low estimate of volume of adipose tissue in the model (10 liters) was increased (20 liters [17, 18]). Dosing simulations using the human PBPK model considered five terbinafine dosing regimens over three dosing durations (1, 7, and 28 days) with a focus on three key pharmacokinetic metrics—maximum concentration (Cmax), minimum concentration (Cmin), and area under the concentration-time curve (AUC)—for the bound and unbound terbinafine concentrations. Dosing simulations were performed with the Scientist software program (version 3.0; Micromath Scientific Software, Salt Lake City, UT). Further analysis was performed in Microsoft Excel 2010.
Pharmacodynamic parameters that are predictive of drug efficacy, including AUC/MIC, time above the MIC, and Cmax/MIC, have been established for most antifungal agents (19); however, to our knowledge, no studies have investigated pharmacodynamic parameters for terbinafine. Therefore, the AUC/MIC and Cmax/MIC ratios and time above the MIC were investigated in this study, with each of these parameters being assessed for both total terbinafine plasma concentration (bound to plasma proteins and unbound) and unbound terbinafine plasma concentration (free). The predicted pharmacodynamic parameters were calculated on day 1, day 7, and day 28 of treatment for each terbinafine dosing regimen to investigate the effects of drug accumulation. The AUC/MIC and AUC for the free, unbound fraction (fAUC)/MIC ratios were calculated using the linear trapezoidal rule for the AUC from 0 to 24 h (AUC0–24) corresponding to each day of treatment investigated. Plasma protein binding of 99% was assumed for the calculation of unbound terbinafine concentrations and predicted pharmacodynamic parameters (20).
RESULTS
The predicted total terbinafine exposure in plasma following high-dose regimens compared to the standard dose (250 mg q24h) are shown in Fig. 1, and the predicted AUC, Cmax, and Cmin for total and unbound terbinafine are shown in Table 1. By day 7 of treatment, total terbinafine trough concentrations following 250 mg once daily reached 0.14 mg/liter; the trough terbinafine concentrations were approximately 2-fold, 2.2-fold, 3.8-fold, and 4.4-fold higher for the 500 mg q24h, 250 mg q12h, 250 mg q8h, and 500 mg q12h regimen, respectively. Both the 250 mg q8h and 500 mg q12h regimen resulted in total trough concentrations above 0.5 mg/liter at day 7 of treatment. The highest peak terbinafine concentrations occurred with the 500 mg q12h regimen, reaching 2.8 mg/liter and 3 mg/liter on day 7 and day 28 of treatment, respectively.
FIG 1.

Predicted total terbinafine concentration-time profiles in plasma over the first 7 days of treatment using the PBPK model. The dashed line represents terbinafine dosed at 250 mg once daily (q24h) in each panel, with the solid line representing 250 mg every 12 h (q12h) (A), 250 mg every 8 h (q8h) (B), 500 mg q24h (C), and 500 mg q12h (D).
TABLE 1.
Pharmacokinetic parameters for predicted total and unbound terbinafine plasma concentrations on day 1, day 7, and day 28 for standard and high-dose terbinafine regimens
| Parameter | Day of therapy | Values [total (unbound)] for indicated dose regimena |
||||
|---|---|---|---|---|---|---|
| 250 mg q24h | 250 mg q12h | 500 mg q24h | 250 mg q8h | 500 mg q12h | ||
| AUC0–24 (mg · h/liter) | 1 | 5.40 (0.0540) | 10.4 (0.104) | 10.8 (0.108) | 15.2 (0.152) | 20.6 (0.206) |
| 7 | 8.30 (0.0830) | 16.4 (0.164) | 16.6 (0.166) | 24.6 (0.246) | 32.9 (0.329) | |
| 28 | 9.73 (0.0973) | 19.4 (0.194) | 19.5 (0.195) | 29.1 (0.291) | 38.9 (0.389) | |
| Cmax (mg/liter) | 1 | 1.32 (0.0132) | 1.35 (0.0135) | 2.20 (0.0220) | 1.39 (0.0139) | 2.24 (0.0224) |
| 7 | 1.46 (0.0146) | 1.61 (0.0161) | 2.46 (0.0246) | 1.80 (0.0180) | 2.78 (0.0278) | |
| 28 | 1.52 (0.0152) | 1.74 (0.0174) | 2.59 (0.0259) | 1.99 (0.0199) | 3.04 (0.0304) | |
| Cmin (mg/liter) | 1 | 0.0332 (0.000332) | 0.0659 (0.000659) | 0.0666 (0.000666) | 0.162 (0.00162) | 0.147 (0.00147) |
| 7 | 0.143 (0.00143) | 0.308 (0.00308) | 0.288 (0.00288) | 0.537 (0.00537) | 0.632 (0.00632) | |
| 28 | 0.198 (0.00198) | 0.427 (0.00427) | 0.397 (0.00397) | 0.722 (0.00722) | 0.872 (0.00872) | |
For dose regimens where more than one dose is administered in a 24-h period, Cmax and Cmin values for each day of therapy represent the average of the Cmax and Cmin values for each dosing interval in the corresponding 24-h period.
The predicted AUC/MIC and fAUC/MIC ratios following standard- and high-dose terbinafine regimens are shown in Fig. 2. The AUC/MIC and fAUC/MIC ratios increased by 54 to 62% on day 7 of treatment and by 80 to 92% on day 28 compared to day 1, depending on the dose regimen. On day 7 of therapy, the predicted AUC/MIC and fAUC/MIC ratios were approximately 2-fold higher for the 500 mg q24h and 250 mg q12h regimens and 3-fold and 4-fold higher for the 250 mg q8h and 500 mg q12h regimen, respectively, than for standard terbinafine dosing.
FIG 2.
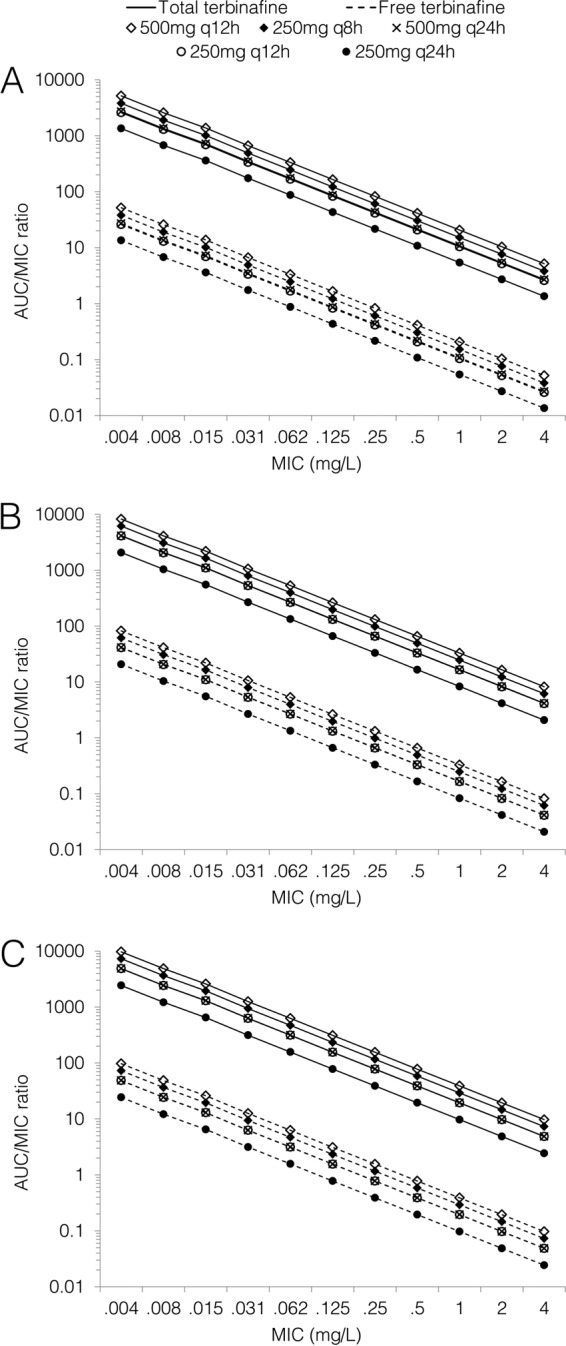
Predicted AUC/MIC ratios for total and free terbinafine on day 1 (A), day 7 (B), and day 28 (C) of treatment for standard and high-dose regimens.
The predicted Cmax/MIC and fCmax/MIC ratios are shown in Fig. 3. The Cmax/MIC and fCmax/MIC ratios increased by 10 to 29% on day 7 of treatment and by 15 to 43% on day 28 compared to day 1, depending on the dose regimen. On day 7 of therapy, the predicted Cmax/MIC and fCmax/MIC ratios were approximately 11%, 24%, 69%, and 91% higher for the 250 mg q12h, 250 mg q8h, 500 mg q24h, and 500 mg q12h regimen, respectively, than for standard terbinafine dosing.
FIG 3.
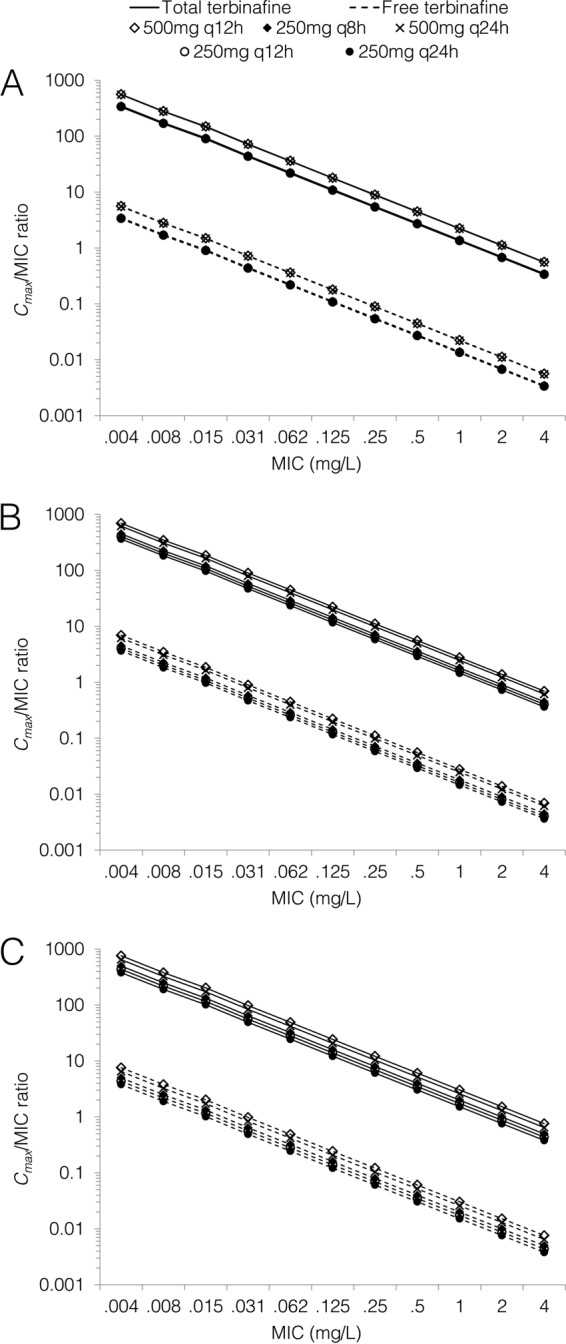
Predicted Cmax/MIC ratios for total and free terbinafine on day 1 (A), day 7 (B), and day 28 (C) of treatment for standard and high-dose regimens.
The predicted times above the MIC on day 1, day 7, and day 28 of treatment for both total and free terbinafine are shown in Fig. 4. On day 1, the 250 mg q8h and 500 mg q12h regimens resulted in times above the MIC for total terbinafine concentrations of close to 100% for MICs of up to 0.125 mg/liter, with times above the MIC of approximately 60% for free terbinafine concentrations at a MIC of 0.004 mg/liter. By day 7, these regimens achieved 100% time above the MIC for total terbinafine concentrations for MICs of up to 0.5 mg/liter, whereas standard dosing resulted in a time above the MIC of 19% at this threshold. For unbound terbinafine concentrations at day 28, the times above the MIC were 100% for MICs of up to 0.008 mg/liter and 44% at a MIC of 0.015 mg/liter with the highest-dose regimen (500 mg q12h); standard dosing resulted in times above the MIC of 14% and 1% at these thresholds.
FIG 4.
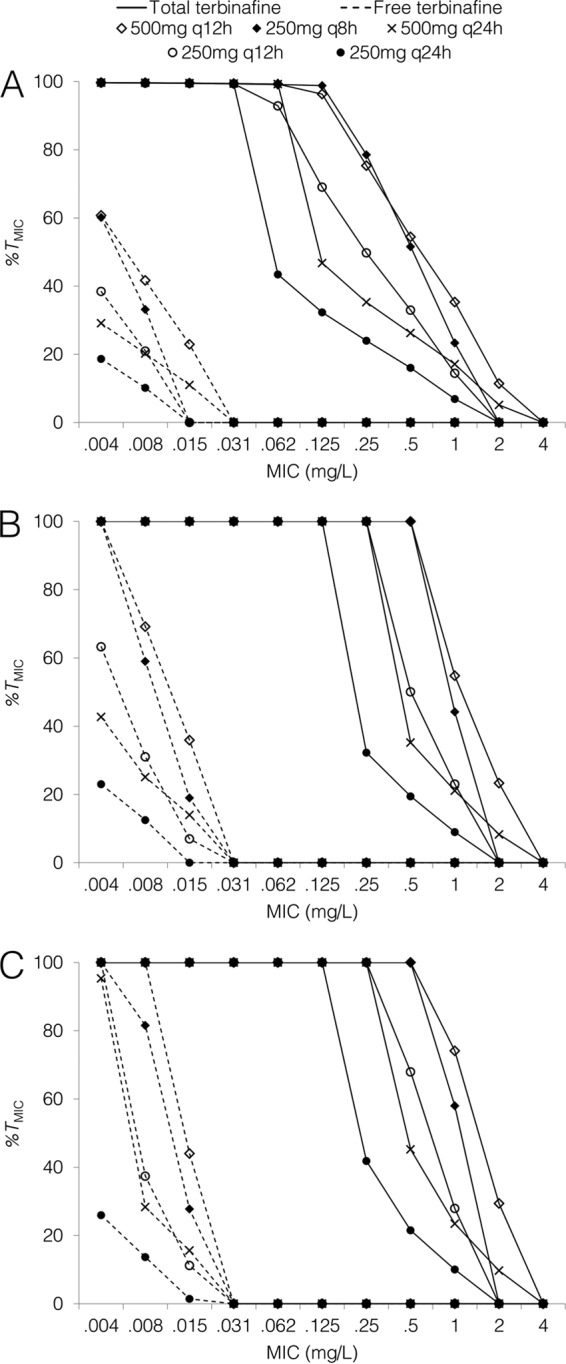
Predicted percentages of time above MIC for total and free terbinafine concentrations on day 1 (A), day 7 (B), and day 28 (C) of treatment for standard and high-dose regimens.
As observed pharmacokinetic data are available for terbinafine at standard doses (250 mg once daily), the model-predicted concentrations following this dose were compared with observed data to assess the predictive performance of the model. There was close agreement between predicted and observed terbinafine concentrations in plasma (Fig. 5) (13, 14, 16, 21, 22).
FIG 5.
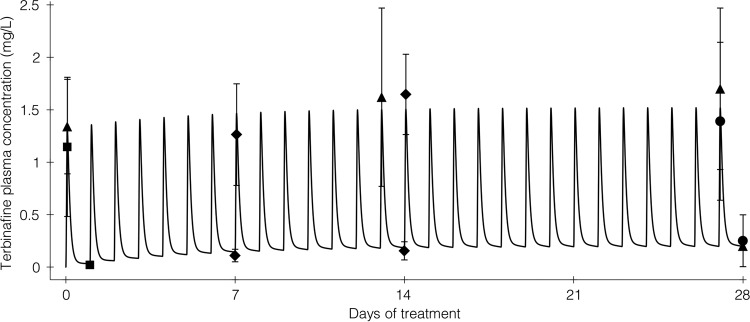
Predicted versus observed terbinafine concentration-time profile following terbinafine 250 mg q24h for 28 days. The solid line represents the PBPK model-predicted terbinafine concentration-time profile, with closed symbols representing mean observed peak and trough terbinafine concentrations during the first 4 weeks of treatment reported in four pharmacokinetic studies. Observed data are from references 13 (closed triangles), 21 (closed diamonds), 16 (closed squares), and 22 (closed circles; reported in reference 14). Symbol error bars represent the standard deviations of the observed concentrations; approximate standard deviations were calculated as range/4 for data from reference 21 because standard deviations were not reported.
DISCUSSION
Using a PBPK model, this study provides the first report of predicted terbinafine exposure in plasma with a range of high-dose regimens. The trough terbinafine concentrations accumulated significantly by day 7 of treatment, with peak concentrations accumulating to a lesser extent (Fig. 1 and Table 1) (14). Pharmacokinetic studies suggest that higher terbinafine exposure would be expected in older patients and those with renal or hepatic impairment and lower exposure in smokers (23, 24).
The ability to provide optimal dosing recommendations for high-dose terbinafine from this work is limited by the absence of studies investigating pharmacodynamic parameters that predict its antifungal efficacy. For antifungals where this parameter is known, dosing regimens may be adapted to maximize antifungal activity (19). Despite this, it is clear that terbinafine is increasingly used at high doses (1), and thus, a practical approach is warranted for the selection of high-dose terbinafine regimens. Of the dosing regimens investigated in this study, terbinafine at 500 mg q12h achieves the highest trough and peak concentrations and AUC values in plasma, suggesting that this terbinafine regimen would achieve the highest antifungal activity irrespective of the pharmacodynamic target (AUC/MIC, time above MIC, or Cmax/MIC).
Terbinafine is known to have an unbound fraction of approximately 1% in serum due to nonsaturable binding to albumin and lipoproteins (20, 25). While there have been conflicting reports on the applicability of the unbound drug hypothesis to some highly bound azole antifungals (26), a marked increase in terbinafine MICs in serum compared to the MICs in protein-free media has been reported (25), suggesting that this assumption is appropriate for terbinafine. Therefore, it is likely that pharmacodynamic measures based on unbound terbinafine concentrations, such as fAUC/MIC, will correlate more closely with antifungal efficacy than will total drug concentrations.
Ryder and colleagues have reported MICs for 5 isolates of Aspergillus spp. for terbinafine in combination with other antifungals; they determined terbinafine MICs of between 0.004 to 0.016 mg/liter in combination with itraconazole and 0.016 to 0.125 mg/liter with voriconazole (27). At a MIC of 0.016 mg/liter on day 7 of treatment, the predicted fAUC/MIC ratio for the highest-dose regimen (terbinafine at 500 mg q12h) is 20.55, compared to 5.19 with the standard dose. Further studies of terbinafine pharmacodynamics and the effect of protein binding on antifungal activity are needed to inform the clinical relevance of these findings.
While concentrations in important sites of infection, such as the brain, remain undefined for terbinafine in humans, lung concentrations of terbinafine have been reported (28). In 11 patients who were administered terbinafine for 3 days prior to undergoing pulmonary lobectomy, the concentrations in lung tissue were approximately 4-fold higher than those in plasma (28), indicating that therapeutic concentrations of terbinafine above pathogen MICs may be achievable in the lung. These findings suggest that terbinafine may be a useful adjunct to azole antifungals in the treatment of refractory or resistant pulmonary mycoses (1).
The potent in vitro synergy observed between terbinafine and azole antifungals for a wide range of fungal pathogens enhances the potential role of terbinafine in the treatment of refractory IFIs (6–11). The complementary mechanisms of action of terbinafine and azoles, which inhibit squalene epoxidase and lanosterol 14α-demethylase, respectively, theoretically result in dual inhibition of fungal ergosterol production (29). These synergistic relationships are particularly important in the treatment of multiresistant Scedosporium prolificans; several case reports have demonstrated in vitro synergy for terbinafine with other agents despite resistance to all available antifungals, leading to successful combination therapy (30–33). However, the choice of antifungal agents should be made cautiously, as in vitro antagonistic interactions have been observed for both terbinafine and azoles when combined with amphotericin B for several isolates of Aspergillus fumigatus (6). An apparent limitation of the present study is the difficulty of linking pharmacokinetic/pharmacodynamic parameters to efficacy when synergism or antagonism is present in combination therapy; where synergistic relationships exist, it is possible that low concentrations of terbinafine would be beneficial when combined with other agents.
An important concern with the use of high-dose terbinafine is the relatively unknown safety profile. At regular doses (≤250 mg daily), terbinafine is generally well tolerated, although cases of hepatotoxicity have been reported (34). At higher doses, similar rates of adverse events have been reported in 401 patients receiving terbinafine at 500 mg daily and in patients receiving 250 mg daily (5); more recently, a trial in 63 patients of terbinafine at 250 mg q12h versus 500 mg q12h in the treatment of sporotrichosis reported a slightly higher rate of adverse events in patients in the 500 mg q12h arm (2). Terbinafine is associated with fewer clinically significant drug interactions than azole antifungals, with the exception of medicines metabolized by CYP2D6, for which it is a known inhibitor (35).
The assessment of antifungal efficacy in animal models is particularly useful in the treatment of infection caused by relatively rare fungal pathogens, due to the poor feasibility of randomized controlled trials in humans (36). The combination of liposomal amphotericin B and terbinafine was recently found to prolong survival and reduce fungal burden in a murine model of disseminated Fusarium verticillioides infection (37). Further studies that include terbinafine are needed in this area, particularly in combination with first-line azole antifungals, such as voriconazole. Furthermore, future studies should address terbinafine pharmacodynamics and the influence of the high protein binding observed with terbinafine on its antifungal efficacy.
In light of the high mortality associated with resistant or refractory IFIs (38), synergistic interactions with other antifungals, and case reports of clinical success, the use of high-dose terbinafine in combination with other antifungal agents for the treatment of resistant or refractory IFIs appears promising and warrants further investigation.
ACKNOWLEDGMENTS
All authors declare that they have no conflicts of interest.
No funding supported the development of this article. M.J.D. is supported by an Australian postgraduate award.
Footnotes
Published ahead of print 14 October 2013
REFERENCES
- 1.Revankar SG, Nailor MD, Sobel JD. 2008. Use of terbinafine in rare and refractory mycoses. Future Microbiol. 3:9–17. 10.2217/17460913.3.1.9 [DOI] [PubMed] [Google Scholar]
- 2.Chapman SW, Pappas P, Kauffmann C, Smith EB, Dietze R, Tiraboschi-Foss N, Restrepo A, Bustamante AB, Opper C, Emady-Azar S, Bakshi R. 2004. Comparative evaluation of the efficacy and safety of two doses of terbinafine (500 and 1000 mg day-1) in the treatment of cutaneous or lymphocutaneous sporotrichosis. Mycoses 47:62–68. 10.1046/j.1439-0507.2003.00953.x [DOI] [PubMed] [Google Scholar]
- 3.N′diaye B, Dieng MT, Perez A, Stockmeyer M, Bakshi R. 2006. Clinical efficacy and safety of oral terbinafine in fungal mycetoma. Int. J. Dermatol. 45:154–157. 10.1111/j.1365-4632.2004.02392.x [DOI] [PubMed] [Google Scholar]
- 4.Esterre P, Inzan CK, Ratsioharana M, Andriantsimahavandy A, Raharisolo C, Randrianiaina E, Roig P. 1998. A multicentre trial of terbinafine in patients with chromoblastomycosis: effect on clinical and biological criteria. J. Dermatolog. Treat. 9(Suppl 1):29–34 [Google Scholar]
- 5.Villars VV, Jones TC. 1992. Special features of the clinical use of oral terbinafine in the treatment of fungal diseases. Br. J. Dermatol. 126(Suppl 39):61–69. 10.1111/j.1365-2133.1992.tb00013.x [DOI] [PubMed] [Google Scholar]
- 6.Vazquez JA. 2008. Clinical practice: combination antifungal therapy for mold infections: much ado about nothing? Clin. Infect. Dis. 46:1889–1901. 10.1086/588475 [DOI] [PubMed] [Google Scholar]
- 7.Gómez-López A, Cuenca-Estrella M, Mellado E, Rodríguez-Tudela JL. 2003. In vitro evaluation of combination of terbinafine with itraconazole or amphotericin B against zygomycota. Diagn. Microbiol. Infect. Dis. 45:199–202. 10.1016/S0732-8893(02)00509-6 [DOI] [PubMed] [Google Scholar]
- 8.Ortoneda M, Capilla J, Javier Pastor F, Pujol I, Guarro J. 2004. In vitro interactions of licensed and novel antifungal drugs against Fusarium spp. Diagn. Microbiol. Infect. Dis. 48:69–71. 10.1016/j.diagmicrobio.2003.09.003 [DOI] [PubMed] [Google Scholar]
- 9.Ortoneda M, Capilla J, Pastor FJ, Pujol I, Yustes C, Serena C, Guarro J. 2004. In vitro interactions of approved and novel drugs against Paecilomyces spp. Antimicrob. Agents Chemother. 48:2727–2729. 10.1128/AAC.48.7.2727-2729.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barchiesi F, Falconi Di Francesco L, Scalise G. 1997. In vitro activities of terbinafine in combination with fluconazole and itraconazole against isolates of Candida albicans with reduced susceptibility to azoles. Antimicrob. Agents Chemother. 41:1812–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biancalana FS, Lyra L, Schreiber AZ. 2011. In vitro evaluation of the type of interaction obtained by the combination of terbinafine and itraconazole, voriconazole, or amphotericin B against dematiaceous molds. Antimicrob. Agents Chemother. 55:4485–4487. 10.1128/AAC.01015-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hosseini-Yeganeh M, Mclachlan AJ. 2002. Physiologically based pharmacokinetic model for terbinafine in rats and humans. Antimicrob. Agents Chemother. 46:2219–2228. 10.1128/AAC.46.7.2219-2228.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kovarik JM, Mueller EA, Zehender H, Denouël J, Caplain H, Millerioux L. 1995. Multiple-dose pharmacokinetics and distribution in tissue of terbinafine and metabolites. Antimicrob. Agents Chemother. 39:2738–2741. 10.1128/AAC.39.12.2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zehender H, Cabiac MD, Denouël J, Faergemann J, Donatsch P, Kutz K, Humbert H. 1994. Elimination kinetics of terbinafine from human plasma and tissues following multiple-dose administration, and comparison with 3 main metabolites. Drug Invest. 8:203–210. 10.1007/BF03258479 [DOI] [Google Scholar]
- 15.Rowland M, Peck C, Tucker G. 2011. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu. Rev. Pharmacol. Toxicol. 51:45–73. 10.1146/annurev-pharmtox-010510-100540 [DOI] [PubMed] [Google Scholar]
- 16.Kovarik JM, Kirkesseli S, Humbert H, Grass P, Kutz K. 1992. Dose-proportional pharmacokinetics of terbinafine and its N-demethylated metabolite in healthy volunteers. Br. J. Dermatol. 126(Suppl 39):8–13. 10.1111/j.1365-2133.1992.tb00002.x [DOI] [PubMed] [Google Scholar]
- 17.Shen W, Punyanitya M, Wang Z, Gallagher D, St Onge M-P, Albu J, Heymsfield SB, Heshka S. 2004. Total body skeletal muscle and adipose tissue volumes: estimation from a single abdominal cross-sectional image. J. Appl. Physiol. 97:2333–2338. 10.1152/japplphysiol.00744.2004 [DOI] [PubMed] [Google Scholar]
- 18.Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP. 1997. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol. Ind. Health 13:407–484. 10.1177/074823379701300401 [DOI] [PubMed] [Google Scholar]
- 19.Sinnollareddy M, Peake SL, Roberts MS, Lipman J, Roberts JA. 2012. Using pharmacokinetics and pharmacodynamics to optimise dosing of antifungal agents in critically ill patients: a systematic review. Int. J. Antimicrob. Agents 39:1–10. 10.1016/j.ijantimicag.2011.07.013 [DOI] [PubMed] [Google Scholar]
- 20.Novartis 2013. Lamisil tablets (terbinafine hydrochloride) prescribing information, revised April. Novartis, Basel, Switzerland: http://www.novartis.com.au/PI_PDF/lam.pdf [Google Scholar]
- 21.Nedelman J, Cramer JA, Robbins B, Gibiansky E, Chang CT, Gareffa S, Cohen A, Meligeni J. 1997. The effect of food on the pharmacokinetics of multiple-dose terbinafine in young and elderly healthy subjects. Biopharm. Drug Dispos. 18:127–138. [DOI] [PubMed] [Google Scholar]
- 22.Faergemann J, Zehender H, Denouël J, Millerioux L. 1993. Levels of terbinafine in plasma, stratum corneum, dermis-epidermis (without stratum corneum), sebum, hair and nails during and after 250 mg terbinafine orally once per day for four weeks. Acta Derm. Venereol. 73:305–309 [DOI] [PubMed] [Google Scholar]
- 23.Nedelman JR, Gibiansky E, Robbins BA, Cramer JA, Riefler JF, Lin T, Meligeni JA. 1996. Pharmacokinetics and pharmacodynamics of multiple-dose terbinafine. J. Clin. Pharmacol. 36:452–461. 10.1002/j.1552-4604.1996.tb05032.x [DOI] [PubMed] [Google Scholar]
- 24.Balfour JA, Faulds D. 1992. Terbinafine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in superficial mycoses. Drugs 43:259–284 [DOI] [PubMed] [Google Scholar]
- 25.Ryder NS, Frank I. 1992. Interaction of terbinafine with human serum and serum proteins. J. Med. Vet. Mycol. 30:451–460. 10.1080/02681219280000611 [DOI] [PubMed] [Google Scholar]
- 26.Lignell A, Löwdin E, Cars O, Chryssanthou E, Sjölin J. 2011. Posaconazole in human serum: a greater pharmacodynamic effect than predicted by the non-protein-bound serum concentration. Antimicrob. Agents Chemother. 55:3099–3104. 10.1128/AAC.01671-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ryder NS, Leitner I. 2001. Synergistic interaction of terbinafine with triazoles or amphotericin B against Aspergillus species. Med. Mycol. 39:91–95. 10.1080/714030977 [DOI] [PubMed] [Google Scholar]
- 28.Schiraldi G, Della Patrona S, Colombo MD, Ravini MC, Bianchi M, Sabolla L. 2006. Plasma and lung concentrations of terbinafine: a pilot study. G. Ital. Mal. Torace 60:235–240 [Google Scholar]
- 29.Ghannoum MA, Rice LB. 1999. Antifungal agents: mode of action, mechanisms of resistance, and correlation of these mechanisms with bacterial resistance. Clin. Microbiol. Rev. 12:501–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howden BP, Slavin MA, Schwarer AP, Mijch AM. 2003. Successful control of disseminated Scedosporium prolificans infection with a combination of voriconazole and terbinafine. Eur. J. Clin. Microbiol. Infect. Dis. 22:111–113. 10.1007/s10096-002-0877-z [DOI] [PubMed] [Google Scholar]
- 31.Gosbell IB, Toumasatos V, Yong J, Kuo RS, Ellis DH, Perrie RC. 2003. Cure of orthopaedic infection with Scedosporium prolificans, using voriconazole plus terbinafine, without the need for radical surgery. Mycoses 46:233–236. 10.1046/j.1439-0507.2003.00878.x [DOI] [PubMed] [Google Scholar]
- 32.Li JYZ, Yong TY, Grove DI, Coates PTH. 2008. Successful control of Scedosporium prolificans septic arthritis and probable osteomyelitis without radical surgery in a long-term renal transplant recipient. Transpl. Infect. Dis. 10:63–65. 10.1111/j.1399-3062.2007.00240.x [DOI] [PubMed] [Google Scholar]
- 33.Bhat SV, Paterson DL, Rinaldi MG, Veldkamp PJ. 2007. Scedosporium prolificans brain abscess in a patient with chronic granulomatous disease: successful combination therapy with voriconazole and terbinafine. Scand. J. Infect. Dis. 39:87–90. 10.1080/00365540600786564 [DOI] [PubMed] [Google Scholar]
- 34.Hall M, Monka C, Krupp P, O'Sullivan D. 1997. Safety of oral terbinafine: results of a postmarketing surveillance study in 25,884 patients. Arch. Dermatol. 133:1213–1219. 10.1001/archderm.1997.03890460029004 [DOI] [PubMed] [Google Scholar]
- 35.Abdel-Rahman SM, Gotschall RR, Kauffman RE, Leeder JS, Kearns GL. 1999. Investigation of terbinafine as a CYP2D6 inhibitor in vivo. Clin. Pharmacol. Ther. 65:465–472. 10.1016/S0009-9236(99)70065-2 [DOI] [PubMed] [Google Scholar]
- 36.Guarro J. 2011. Lessons from animal studies for the treatment of invasive human infections due to uncommon fungi. J. Antimicrob. Chemother. 66:1447–1466. 10.1093/jac/dkr143 [DOI] [PubMed] [Google Scholar]
- 37.Ruíz-Cendoya M, Pastor FJ, Capilla J, Guarro J. 2011. Treatment of murine Fusarium verticillioides infection with liposomal amphotericin B plus terbinafine. Int. J. Antimicrob. Agents 37:58–61. 10.1016/j.ijantimicag.2010.08.008 [DOI] [PubMed] [Google Scholar]
- 38.Husain S, Muñoz P, Forrest G, Alexander BD, Somani J, Brennan K, Wagener MM, Singh N. 2005. Infections due to Scedosporium apiospermum and Scedosporium prolificans in transplant recipients: clinical characteristics and impact of antifungal agent therapy on outcome. Clin. Infect. Dis. 40:89–99. 10.1086/426445 [DOI] [PubMed] [Google Scholar]