

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) is an etiological agent of several AIDS-associated malignancies, including Kaposi's sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman's disease (MCD). Its lytic replication cycle has been proven to be critical for the pathogenesis of KSHV-associated diseases. In KS lesions, lytic viral replication, production of virion particles, and reinfection of endothelial cells are essential to sustain the population of infected cells that otherwise would be quickly lost as spindle cells divide. Thus, antivirals that block KSHV replication could be a strategy in the treatment of KSHV-associated diseases. However, there is no effective anti-KSHV drug currently available. Our previous work showed that human topoisomerase II (Topo II) is indispensable for KSHV lytic replication and is suggested to be an effective target for antiviral drugs. Here, we report the discovery and characterization of a novel catalytic inhibitor of human Topo IIα, namely, (+)-rutamarin. The binding mode of (+)-rutamarin to the ATPase domain of human Topo IIα was established by docking and validated by molecular dynamics (MD) simulations. More importantly, (+)-rutamarin efficiently inhibits KSHV lytic DNA replication in BCBL-1 cells with a half-maximal inhibitory concentration (IC50) of 1.12 μM and blocks virion production with a half-maximal antiviral effective concentration (EC50) of 1.62 μM. It possesses low cytotoxicity, as indicated by the selectivity index (SI) of 84.14. This study demonstrated great potential for (+)-rutamarin to become an effective drug for treatment of human diseases associated with KSHV infection.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus 8 (HHV-8), is a member of the human gammaherpesvirus family. It has been proven to be the etiologic agent of Kaposi's sarcoma (KS), a multicentric malignant neoplasm of endothelial origin (1–5). Although classic KS is a rare disease and in general nonfatal, it often occurs in immunocompromised patients who are under immunosuppression treatment after organ transplantation. In the AIDS epidemic, KS has become the most common AIDS-associated malignancy and has led to significant mortality (6). Approximately 20% of AIDS patients develop KS during the course of their disease, and AIDS-associated KS is estimated to contribute to 10% of the deaths of AIDS patients (7). Besides KS, two B cell-associated lymphoproliferative disorders, namely, primary effusion lymphoma (PEL) and multicentric Castleman's disease (MCD), are also related to KSHV infection and mainly occur in AIDS patients (6, 8). The incidence of KSHV-associated MCD has risen since the advent of highly active antiretroviral therapy (HAART) (9).
The contemporary treatment modalities for KS and other KSHV-associated malignancies include conventional cancer therapies, such as radiation and chemotherapy, or AIDS treatment, such as HAART (6). These cancer therapies in general have serious side effects, and the response of tumors to them is only transient. The success of human papillomavirus (HPV) vaccine in preventing cervical cancer has proven that antiviral-oriented therapies have the potential to be used to treat human malignant diseases that are caused by viruses. Thus, an antiviral targeting KSHV could prove successful at treating KSHV-associated tumors (10). However, currently, there is no therapy for KSHV-associated disease based on targeting of KSHV.
Like all herpesviruses, KSHV has two types of replication cycle, latent and lytic replication. In KS lesions, most KSHV-transformed spindle-shaped cells are in the latent replication phase, but a small percentage of infected cells undergo spontaneous lytic replication (11–13). Increasing evidence suggests that the lytic replication in these cells is necessary for maintaining stable infection and viral pathogenicity. The majority of KSHV genes, including those responsible for malignant cell growth and alteration of the microenvironment, are expressed only in the lytic phase (14). Furthermore, lytic replication is necessary for sustaining the population of latently infected cells that otherwise would be quickly lost by segregation of latent viral episomes as spindle cells divide (15). Thus, KSHV lytic replication and constant primary infection of fresh cells are crucial, not only for viral propagation, but also for viral pathogenesis. Blockade of the lytic replication of KSHV could be a good strategy for the treatment of KS or other KSHV-associated human diseases.
Viral DNA replication is considered an ideal target for antivirals. Our laboratory has been studying the mechanisms that control KSHV lytic DNA replication (16–19). One of our findings was that several host cellular proteins, including topoisomerase I (Topo I) and Topo II, MSH2/6, RecQL, and poly(ADP-ribose) polymerase 1 (PARP-1), are involved in KSHV lytic DNA replication (19). We also demonstrated that both Topo I and Topo II are indispensable for KSHV lytic replication and that specific inhibitors of Topo I and II can effectively inhibit KSHV lytic replication. One category of Topo II inhibitors, namely, catalytic Topo II inhibitor, was found to provide marked inhibition of KSHV replication with minimal cytotoxicity, as indicated by their high selectivity indices (e.g., 31.6 for novobiocin) (20). As such, Topo II can serve as an effective target for antivirals, and catalytic inhibitors of Topo II represent promising antiviral agents for the treatment of malignancies associated with KSHV infection.
In this study, we attempted to search for new compounds with higher Topo II inhibition properties and lower cytotoxicities. With a ligand-based virtual screening approach, we screened more than 7,200 compound structures in our compound repository and identified a set of coumarin derivatives that showed activities in inhibition of KSHV DNA replication equal to or greater than that of novobiocin. In particular, (+)-rutamarin exhibits the highest efficiency in blocking KSHV lytic replication. We demonstrated that (+)-rutamarin has human Topo II inhibition activity and belongs to the category of catalytic Topo II inhibitors.
MATERIALS AND METHODS
Cells and plasmids.
BCBL-1 and JSC-1 are two primary effusion lymphoma cell lines that are latently infected with KSHV (21, 22). The BJAB cell line is a KSHV-negative B cell line isolated from Burkitt's lymphoma. These B cells were grown in RPMI 1640 medium (Gibco-BRL, Gaithersburg, MD) supplemented with 10% fetal bovine serum (Gibco-BRL), penicillin-streptomycin (50 units/ml), and 1.25 g/ml amphotericin B-1.25g/ml sodium deoxycholate (Fungizone). Peripheral blood mononuclear cells (PBMC) were isolated from whole blood of a healthy donor and cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, penicillin-streptomycin, and amphotericin B-sodium deoxycholate.
Plasmid pOri-A contains an EcoRI-PstI fragment (nucleotides 22409 to 26491) of KSHV DNA in pBluescript at the EcoRI/PstI site, as previously described (16). pCR3.1-ORF50 is an RTA expression vector described previously (16).
Chemicals and treatment of cells.
The chemicals used in this study were from the Guangdong Small Molecule Tangible Library (GSMTL) (23). The compounds were dissolved in dimethyl sulfoxide (DMSO) and serially diluted before being added to BCBL-1, JSC-1, or BJAB cell cultures (4 × 105 cells/ml). The final DMSO concentration in the culture medium was maintained at 1%. For lytic replication, BCBL-1 cells were induced by tetradecanoylphorbol acetate (TPA) as previously reported (20). JSC-1 cells were induced by 3 mM sodium butyrate (22). Three hours postinduction, the compounds in dilutions were added to BCBL-1 or JSC-1 cells, and subsequently, the antiviral effects and cytotoxicity were assayed at different time points.
Analysis of intracellular KSHV genomic DNA content and chemical effects.
TPA-induced and uninduced BCBL-1 cells were harvested at 48 h postinduction, and total DNA was purified using a DNeasy kit according to the manufacturer's protocol (Qiagen). The KSHV genomic copy number was quantified by real-time PCR on a Roche LightCycler instrument using the LightCycler FastStart DNA MasterPlus SYBR green kit with primers for the detection of LANA (forward, 5′-CGCGAATACCGCTATGTACTCA-3′; reverse, 5′-GGAACGCGCCTCATACGA-3′). The intracellular viral genomic DNA in each sample was normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) using primers directed to GAPDH (forward, 5′-ACATCATCCCTGCCTCTAC-3′; reverse, 5′-TCAAAGGTGGAGGAGTGG-3′).
The half-maximal inhibitory concentration (IC50) values of compounds were determined from a dose-response curve of KSHV DNA content values from TPA-induced and chemical-treated cells. The viral DNA contents with those of uninduced cells subtracted were divided by those of the control cells with no drug treatment and then represented on the y axes of dose-response curves: y axis value = (TPAX − no TPAX)/(TPA0 − no TPA0), where X is any concentration of the drug and 0 represents nondrug treatment. The IC50 on viral DNA synthesis for each compound was calculated with the aid of GraphPad Prism software.
Analysis of extracellular KSHV virion production and chemical effects.
Five days postinduction with TPA, BCBL-1 culture media were collected and extracellular virions were pelleted from the medium supernatant. To remove contaminating DNA outside viral particles, the concentrated viruses were treated with Turbo DNase I (Ambion) at 37°C for 1 h, followed by proteinase K digestion. Virion DNA was extracted with phenol-chloroform, precipitated with ice-cold ethanol, and then dissolved in Tris-EDTA (TE) buffer. The KSHV genomic copy numbers were determined by real-time PCR, and the values were corrected as described above. The half-maximal antiviral effective concentration (EC50) values were calculated from dose-response curves with GraphPad Prism software.
Cytotoxicity assay.
The viabilities of BCBL-1 cells treated or not with chemicals were assessed by counting Trypan blue-stained cells 2 or 5 days posttreatment using a light microscope. Cell viabilities were defined relative to control cells (non-drug treated). The half-maximal cytotoxic concentration (CC50) was calculated from dose-response curves with GraphPad Prism software.
Cell proliferation assay.
BCBL-1 and BJAB cells (both starting with 2 × 105 cells/ml) were treated with (+)-rutamarin for 5 days at two different concentrations: the IC50 and 5 times the IC50. The cells were stained with Trypan blue and counted every day for 5 days. To maintain the exponential growth, fresh medium (supplemented or not with the drug) was added to the cultures every 2 days.
Cell cycle assay.
BCBL-1 and BJAB cells (both starting with 4 × 105 cells/ml) were treated with (+)-rutamarin and novobiocin at two different concentrations: the IC50 and 5 times the IC50. Two days posttreatment, cells were collected and fixed with cold 70% ethanol for 15 min, permeabilized, and stained with propidium iodide (PI) solution (50 g/ml of PI, 0.1 mg/ml of RNase A, and 0.05% Triton X-100) for 2 h. Cell cycle progression was measured using a FACStar Plus cell sorter flow cytometer (Becton, Dickinson).
ori-Lyt-dependent DNA replication assay.
To assess the effect of (+)-rutamarin on ori-Lyt-dependent DNA replication, BCBL-1 cells were cotransfected with plasmids pOri-A (2.5 μg) and pCR3.1-ORF50 (2.5 μg) by nucleoporation (Amaxa) and cultured with different concentrations of (+)-rutamarin. Seventy-two hours posttransfection, extrachromosomal DNA was prepared from cells using the Hirt DNA extraction method as previously described (20). The extracted extrachromosomal DNA was treated with RNase A at 25°C for 30 min, followed by proteinase K at 50°C for 30 min. Five micrograms of DNA was digested with KpnI/SacI or KpnI/SacI/DpnI (New England Bio-Labs), separated on a 1% agarose gel, and transferred onto GeneScreen membranes (PerkinElmer, Boston, MA). The Southern blots were hybridized with a 32P-labeled pBluescript plasmid in 5× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), 2× Denhardt's solution, 1% SDS, and 50 μg/ml denatured salmon sperm DNA at 68°C.
Kinetoplast DNA decatenation assay.
The decatenation assay is designed to measure type II topoisomerase activity and inhibition of the enzyme by testing compounds. The kinetoplast DNA (kDNA) (200 ng) was incubated in a 20-μl reaction mixture containing 50 mM Tris-HCl (pH 7.5), 85 mM KCl, 10 mM MgCl2, 0.5 mM Na2EDTA, 0.5 mM dithiothreitol, 1 mM ATP, 30 μg/ml bovine serum albumin (BSA), and 2 units of Topo IIα or Topo IIβ for 30 min at 37°C in the absence or the presence of (+)-rutamarin or control compounds (novobiocin and etoposide). The reactions were analyzed by electrophoresis on agarose gels. The decatenated kDNAs were measured with the Gel Dox XR+ imaging system (Bio-Rad).
Malachite green assay for ATPase activity of Topo IIα.
Malachite green reagent was prepared by mixing malachite green (0.0812% [wt/vol]), polyvinyl alcohol (2.32% [wt/vol]), ammonium molybdate (5.72% [wt/vol] in 6 M HCl), and ultrapure water in a ratio of 2:1:1:2. An ATP hydrolysis reaction with a mixture (20 μl) containing 50 mM Tris-HCl, pH 7.4, 85 mM KCl, 10 mM MgCl2, 0.5 mM dithiothreitol (DTT), 0.5 mM Na2EDTA, 30 μg/ml BSA, 200 ng pBR322 DNA, 2 units Topo IIα, 500 μM ATP, and various concentrations of (+)-rutamarin was initiated by adding ATP to the reaction mixture and incubated at 37°C for 2 h. The reaction was stopped by adding 80 μl of the malachite green reagent to the reaction mixture, followed by adding 10 μl of 34% sodium citrate to develop a blue-green color change. The absorbance at 620 nm was measured using a plate reader. The optical density at 620 nm (OD620) was used to represent the ATP hydrolysis level.
Molecular docking study.
The X-ray crystal structure of the ATPase domain of human Topo IIα in complex with ADPNP (Protein Data Bank [PDB] ID, 1ZXM; resolution, 1.87Å) was retrieved from the Protein Data Bank (24). Only chain A was kept for docking study. The missing loop (residues 345 to 350) was added and optimized using Prepare Protein Module encoded in Discovery Studio 3.5 (Accelrys Inc.) (25). The initial structures of (+)-rutamarin and ADPNP were optimized using MMFF force field (26), and the Powell method was used for energy minimization by default parameters in Discovery Studio 3.5.
The docking program FlexX encoded in SYBYL 7.3 (Tripos Inc.) was applied to identify the potential binding of (+)-rutamarin to the Topo IIα ATPase domain. A previous study suggested that two conserved water molecules were not neglected to distinguish the active, moderate, and inactive ligands (27). In the present study, the receptor for docking simulation consisted of protein chain A, two conserved water molecules, and Mg2+. To validate the molecular-docking protocol, ADPNP was redocked into the crystal structure of the ATPase domain of Topo IIα. The docked ADPNP has a binding pose similar to that of the cocrystallized ligand, with a root mean square deviation (RMSD) of about 1.6 Å. The active sites were defined as all residues within a 6.5-Å radius of the bound ADPNP. Other FlexX parameters were set to default values. Thirty poses were retained during the docking process. After running FlexX, all the poses were visually inspected, and the most suitable docking pose was selected on the basis of the score and interactions with key residues of the active site. The complex of Topo IIα and the most suitable docking conformer of (+)-rutamarin was used as the initial coordinates for the subsequent molecular dynamics (MD).
MD simulations.
MD simulations were performed in AMBER 12 (28) with the AMBER ff99SB force field (29, 30). The docked structure of the Topo IIα ATPase domain with (+)-rutamarin was used as the initial coordinates for MD simulations. Parameters for (+)-rutamarin were obtained from the ANTECHAMBER module using the Generalized Amber Force Field (GAFF) (31) with the RESP charge-fitting procedure with input from Hartree-Fock calculations at the 6- to 31-G* level by using the Gaussian03 program (32). All hydrogen atoms of the protein were added using the tleap module, considering ionizable residues set at their default protonation states at a neutral pH. The system was solvated in a truncated octahedron box of TIP3P water molecules with a margin distance of 10 Å. Neutralizing counterions were added to the simulation system. The detailed MD simulations can be found in the supplemental material. The molecular-mechanics Poisson-Boltzmann surface area (MM-PBSA) and molecular-mechanics generalized born surface area (MM-GBSA) methods (33) in the AMBER 12 suite were used to calculate the binding free energies (see the supplemental material).
RESULTS
(+)-Rutamarin inhibits the lytic replication of KSHV.
To search for a novel Topo II inhibitor(s) that effectively blocks KSHV lytic DNA replication, a ligand-based virtual screening using an in-house three-dimensional (3D) molecular superimposing algorithm, WEGA (34), was conducted against the molecular structures in our compound repository, GSMTL (23). Using the novobiocin structure as the template, we screened approximate 7,200 compounds from the GSMTL. The top 33 compounds that are three-dimensionally most similar to novobiocin were selected and subsequently tested for antiviral activities. Exponentially growing BCBL-1 cells were induced into lytic replication with TPA. Three hours after induction, the cells were exposed to each of the compounds, as well as novobiocin, at a concentration of 20 μM. Forty-eight hours postinduction, the KSHV genomic DNA contents of the cells treated with these compounds were analyzed by quantitative real-time PCR. Treatment with novobiocin resulted in 42% inhibition of viral replication compared to the control cells treated with no drug. Seven compounds showed inhibitory effects similar to or better than those of novobiocin on viral DNA replication (Fig. 1A). Four of these, C28, C31, C32, and C33, also displayed strong activities in blocking virion production (Fig. 1B). With the exception of C31, the compounds possess significant cytotoxicity, as demonstrated by low cell viability rates with the treatment (Fig. 1C). C31, which was identified as the natural product (+)-rutamarin (Fig. 2A), exhibited significant inhibition of both viral DNA synthesis and virion production, with relatively low cytotoxicity at a concentration of 20 μM. Therefore, it was chosen for further investigation.
FIG 1.

Compound screening. (A) Thirty-three compounds (see Table S2 in the supplemental material) derived from a virtual screening were assayed for their effects on KSHV lytic DNA replication. BCBL-1 cells were induced with TPA and treated with each of the compounds at a concentration of 20 μM. Forty-eight hours postinduction, the intracellular KSHV genomic copy numbers were determined using quantitative real-time PCR. Seven compounds (marked with asterisks) exhibited considerable inhibitory effects on viral lytic DNA replication. (B) Effects of the compounds on KSHV virion production. Five days postinduction, the KSHV virions in supernatants were collected and measured by quantifying encapsidated viral DNA in the preparations. (C) Effects of the compounds on host cell viability. Uninduced BCBL-1 cells were treated with each of the compounds at 20 μM for 48 h, and cell viability was assessed by trypan blue staining as described in Materials and Methods. The compounds are shown with ascending molecular weights. The error bars indicate standard deviations.
FIG 2.
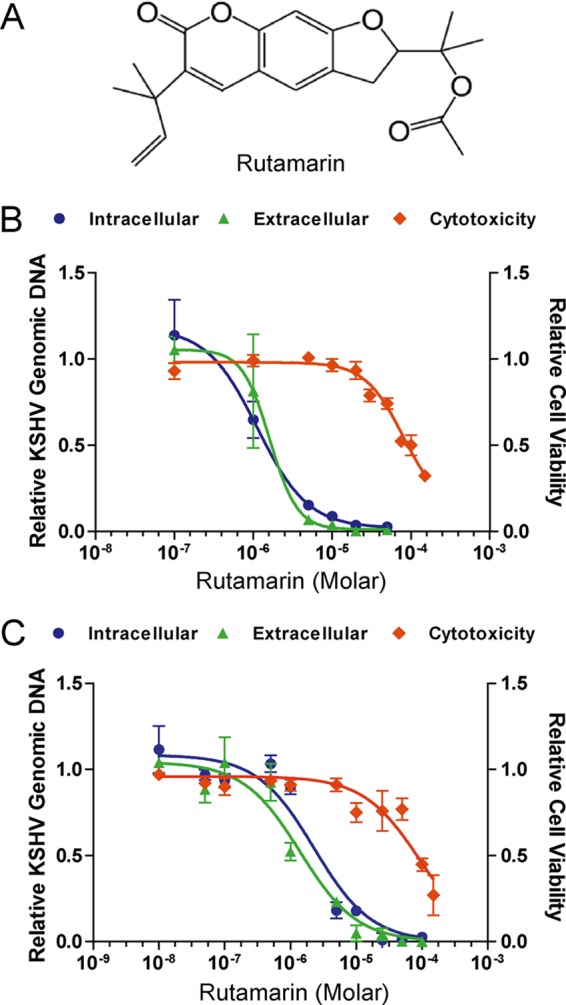
Effect of (+)-rutamarin on KSHV replication and its associated cytotoxicity. (A) Chemical structure of (+)-rutamarin. (B) Effect of (+)-rutamarin on KSHV lytic replication and its associated cytotoxicity in BCBL-1 cells that were treated with a wide range of concentrations of (+)-rutamarin 3 h after lytic replication was induced by TPA. Intracellular KSHV genomic DNA replication (blue), extracellular virion production (green), and cell viability (orange) were determined as described in Materials and Methods. The values were compared to those from the control cells (nondrug treatment). The mean values of results from three independent experiments and standard deviations are presented on the y axis of dose-response curves. (C) Effect of (+)-rutamarin on KSHV lytic replication and its associated cytotoxicity in JSC-1 cells that were induced with 3 mM sodium butyrate. Intracellular KSHV genomic DNA replication (blue), extracellular virion production (green), and cell viability (orange) were determined as described for panel B.
The IC50 values of (+)-rutamarin were determined from the dose-response curve of KSHV DNA content in TPA-induced BCBL-1 cells and were found to be 1.12 μM (Fig. 2B). The effect of (+)-rutamarin on progeny virion production was also determined by quantification of encapsidated viral DNA in BCBL-1 cell culture media. The EC50 calculated from the dose-response curve of extracellular virions is 1.62 μM (Fig. 2B). The cytotoxicity of (+)-rutamarin on BCBL-1 cells was examined in parallel with inhibition of KSHV DNA replication and virion production. Cells treated with (+)-rutamarin at differing concentrations were subjected to the trypan blue exclusion method to determine cell viability in response to (+)-rutamarin treatment. The CC50 was determined to be 94.24 μM (Fig. 2B). Low cytotoxicity of (+)-rutamarin results in an appreciable selectivity index (SI) (SI = CC50/IC50) of 84.14.
To make sure that (+)-rutamarin inhibits KSHV lytic replication regardless of host cells and the means of lytic cycle induction, we tested the effect of (+)-rutamarin on KSHV replication in JSC-1 cells induced by sodium butyrate for lytic replication. The IC50, EC50, and CC50 of (+)-rutamarin on butyrate-induced JSC-1 cells were found to be 2.29 μM, 1.40 μM, and 94.34 μM, respectively, and are very similar to those obtained with BCBL-1. In addition, the cytotoxicity of (+)-rutamarin to primary lymphocytes was also assessed with PBMC. The CC50 for PBMC was calculated to be 60.91 μM (see Fig. S1 in the supplemental material).
Evaluation of (+)-rutamarin for its effects on host cell proliferation and cell cycle progression.
To further investigate the potential of (+)-rutamarin to become an effective and safe antiviral, the effect of the compound on cell proliferation was assessed. KSHV-carrying BCBL-1 and virus-free BJAB cells were cultured in the presence and absence of (+)-rutamarin, and live cells were counted over 5 days. (+)-Rutamarin did not exhibit a cell growth-inhibitory effect in the two cell lines at the IC50. At an excess concentration (5 times the IC50), (+)-rutamarin showed an adverse effect on cell growth, but the extent of inhibition was less than that with novobioicin (Fig. 3). The effect of (+)-rutamarin on cell cycle progression was also examined. BCBL-1 cells treated with (+)-rutamarin were stained with propidium iodide and subjected to flow cytometric analysis. At the IC50, no effect on the cell cycle pattern was observed with either (+)-rutamarin or novobiocin in comparison with the vehicle control. At an excess concentration (5 times the IC50) of (+)-rutamarin, a slight increase in the S phase was observed in comparison to untreated cells. However, the influence of (+)-rutamarin on cell cycle progression was observed to be less than that of novobiocin (Fig. 4). Overall, these results demonstrated that (+)-rutamarin has minimal effects on host cell proliferation and cell cycle progression in its pharmaceutical dose range.
FIG 3.
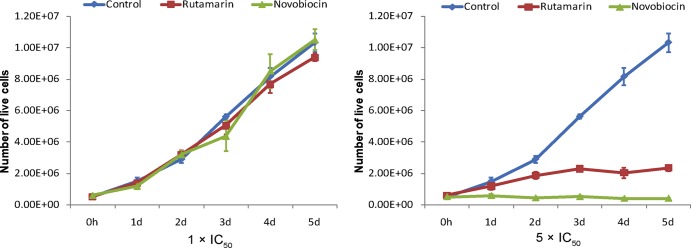
Effects of (+)-rutamarin and novobiocin on BCBL-1 cell proliferation. BCBL-1 cells (starting with 2 × 105 cells/ml) were exposed to (+)-rutamarin and novobiocin at their IC50s and 5 times their IC50s. Data were obtained from three independent determinations and are presented as means and standard deviations.
FIG 4.
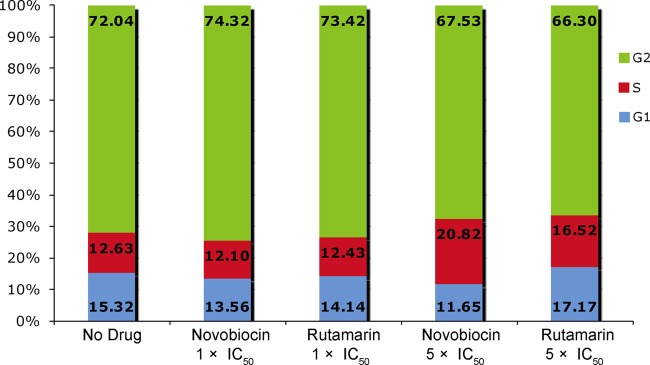
Effects of (+)-rutamarin and novobiocin on BCBL-1 cell cycle progression. BCBL-1 cells (starting with 2 × 105 cells/ml) were exposed to (+)-rutamarin and novobiocin at their IC50s and 5 times their IC50s for 48 h. Cell cycle progressions were measured by PI staining, followed by flow cytometric analysis. The data are presented as means obtained from three independent experiments.
(+)-Rutamarin inhibits KSHV lytic replication by blocking viral ori-Lyt-dependent DNA replication.
We have demonstrated that (+)-rutamarin is capable of inhibiting KSHV DNA synthesis and virion production. To confirm whether the inhibitory effect of (+)-rutamarin on KSHV lytic replication is directly due to the blocking of ori-Lyt-dependent DNA replication, we conducted an ori-Lyt DNA replication assay. BCBL-1 cells were cotransfected with an ori-Lyt-containing plasmid (pOri-A) and an RTA expression vector. Lytic DNA replication was induced by RTA expression (35). The transfected cells were cultured in the presence of (+)-rutamarin at various concentrations, and the effect of (+)-rutamarin on ori-Lyt-dependent DNA replication was measured by a DpnI assay (16, 17). In brief, DNA was isolated from the treated cells 72 h posttransfection and digested with KpnI/SacI and KpnI/SacI/DpnI. Replicated plasmid DNA was distinguished from input plasmids by DpnI restriction digestion, which cleaves input DNA that has been Dam+ methylated in Escherichia coli but leaves intact the DNA that has been replicated in at least one round in eukaryotic cells. Thus, only newly replicated plasmid DNA in BCBL-1 cells, which is resistant to DpnI digestion, can be detected in Southern blot analysis. Replicated pOri-A DNA was detected as a DpnI-resistant DNA band in the cells that were cotransfected with pOri-A and an RTA expression vector. However, the replicated DNA decreased with elevated concentrations of (+)-rutamarin in the cell culture (Fig. 5). This result supports the idea that (+)-rutamarin inhibits KSHV ori-Lyt-dependent DNA replication.
FIG 5.
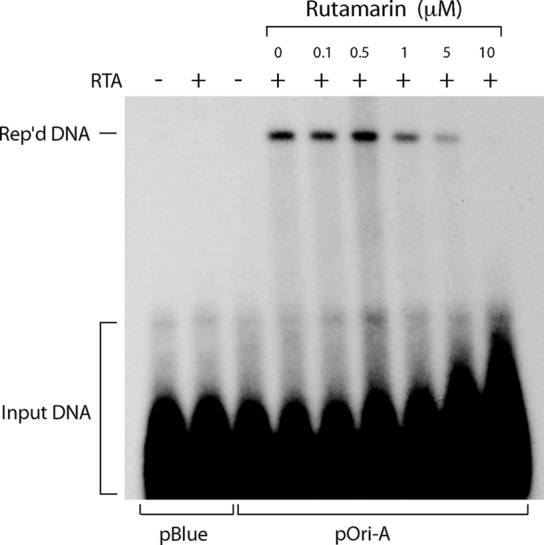
Inhibition of KSHV ori-Lyt-dependent DNA replication with (+)-rutamarin. BCBL-1 cells were transfected with a KSHV ori-Lyt-containing plasmid (pOri-A) and an RTA expression vector (pCR3.1-ORF50). The transfected cells were treated with increasing concentrations of (+)-rutamarin and incubated for 72 h. Hirt DNAs were extracted and digested with KpnI/SacI or KpnI/SacI/DpnI. DpnI-resistant viral replicated DNA (Rep'd DNA) was detected by Southern blotting with 32P-labeled pBluescript plasmid.
To rule out the possibility that (+)-rutamarin blocks KSHV DNA replication by inhibiting RTA expression, the effect of (+)-rutamarin on RTA expression in TPA-induced BCBL-1 cells was examined by Western analysis. The result showed no adverse effect of the compound on RTA expression up to 25 μM (22-fold excess over the IC50) (see Fig. S2 in the supplemental material). The reduced levels of RTA in the treatments with 50 and 100 μM (+)-rutamarin may result from the cytotoxicity of the compound in a concentration near its IC50 (94.24 μM).
(+)-Rutamarin is an inhibitor of human topoisomerase IIα.
The next question was whether (+)-rutamarin is a Topo II inhibitor that blocks KSHV ori-Lyt-dependent DNA replication by inhibiting the catalytic activity of host cell Topo II. Furthermore, there are two Topo II isoforms in mammals, Topo IIα and Topo IIβ. We wanted to know which isoform (+)-rutamarin affects if it is a Topo II inhibitor. To accomplish this, a DNA decatenation-based topoisomerase II assay was established. In this system, kinetoplast DNA consisting of a large network of interlocked DNA minicircles can be decatenated into separate minicircles in the presence of ATP and Topo II in either isoform. (+)-Rutamarin in a wide range of concentrations was added to the reaction mixture, and the effect of the compound on Topo II activity was measured by determining decatenated kDNA levels using agarose gel electrophoresis. The results showed that (+)-rutamarin is able to inhibit the enzymatic activity of Topo IIα in a dose-dependent manner (Fig. 6A and B). The IC50 of (+)-rutamarin in Topo IIα inhibition was calculated as 28.22 ± 6.2 μM (Fig. 6C and D). At a concentration of 100 μM, the inhibition rate of (+)-rutamarin is significantly higher than that of novobiocin and close to that of the Topo II poison etoposide (Fig. 6A). In contrast, (+)-rutamarin did not exhibit any inhibitory effect on Topo IIβ activity (Fig. 6E).
FIG 6.

Identification of (+)-rutamarin as a catalytic inhibitor of Topo IIα. (A) A Topo II-mediated kinetoplast DNA decatenation assay was performed to evaluate the effects of testing compounds for Topo IIα activity at a concentration of 100 μM. The catenated (cat) and decatenated (decat) DNA positions in gels are indicated. (A and B) The Topo IIα activities in the reactions were quantitated (A), and inhibition rates of compounds were calculated (B). (C and E) Inhibition of Topo IIα (C) and Topo IIβ (E) activities by (+)-rutamarin in a wide range of concentrations. (D) The IC50 of (+)-rutamarin on Topo IIα inhibition was calculated as 28.22 ± 6.2 μM. The data are means and standard deviations for three experimental replicates. (E) (+)-Rutamarin exhibits no obvious inhibitory effect on human Topo IIβ. Rut, (+)-rutamarin; Eto, etoposide; Nov, novobiocin.
To confirm if (+)-rutamarin is a catalytic inhibitor of Topo IIα and acts in blocking the ATPase activity of the enzyme, as novobiocin does, we studied the inhibition effect of rutamarin on ATP hydrolysis of Topo IIα using the malachite green assay. As shown in Fig. 7, the ATPase activity of Topo IIα was inhibited by (+)-rutamarin in a dose-dependent manner, suggesting that, like novobiocin, (+)-rutamarin inhibits human Topo IIα by binding to the ATP pocket of the enzyme and blocking its ATP hydrolysis.
FIG 7.
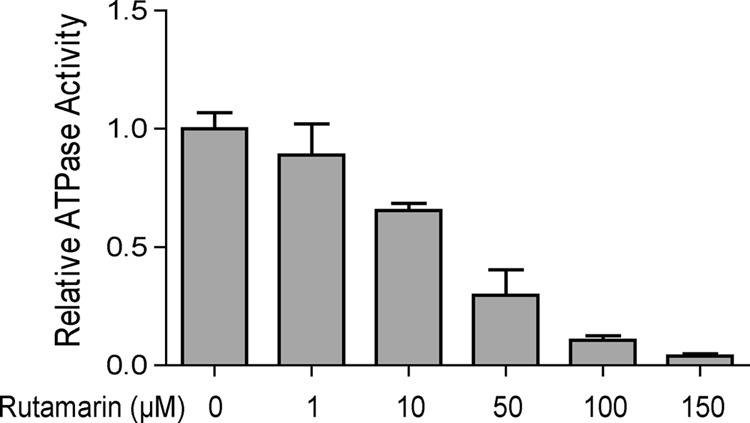
Effect of (+)-rutamarin on ATPase activity of Topo IIα. The inhibitory effect of (+)-rutamarin on Topo IIα ATPase activity was measured using the malachite green assay as described in Materials and Methods. The OD620 represents the ATP hydrolysis level and reflects the inhibition of ATPase activity by (+)-rutamarin. The error bars indicate standard deviations.
A binding model of (+)-rutamarin in the ATP binding pocket of Topo IIα.
The mode of (+)-rutamarin binding in the ATPase domain of Topo IIα was investigated by docking and further defined by MD simulations. Figure 8A illustrates the time dependence of the RMSD values for the X-ray reference enzyme structure of backbone atoms (C, C151, N, and O) over the production phase of simulation. The RMSD values of simulation converged after ∼3 ns, indicating that the system is stable and equilibrated. The RMSD value of ligand compared docking pose is swinging within 1.8 Å, which illustrates that the docking pose is reliable. Previous studies indicated that Mg2+ is necessary for the ATPase domain to hydrolyze the ATP molecule and is stabilized by two conserved water molecules and residues Asn91 and Glu87 (24, 27). The time evolution of oxygen- and nitrogen-Mg2+ distances for these residues and the water molecules is shown in Fig. 8C. The oxygen- and nitrogen-Mg2+ distances for these residues and the water molecules stabilized at 1.9 to 2.0 Å during the simulation, suggesting that Mg2+ is stabilized by these factors. Our simulation results are consistent with previous experimental results, suggesting that our model is reliable and reasonable.
FIG 8.

Stability properties of the simulation system and binding model of (+)-rutamarin with Topo IIα. (A) RMSD plot for the backbone atoms and (+)-rutamarin during MD simulations after equilibration. (B) Distance distributions between Asn95, Ala167, and conserved water molecule 2 and (+)-rutamarin in water at 310 K. The cutoff value for the formation of a hydrogen bond is 3.5 Å. (C) Time dependence of the distance between the carboxyl oxygen of Glu87, the amide nitrogen of Asn91, the two conserved water molecules, and Mg2+ during the 10-ns MD simulations. (D) Detailed binding model between (+)-rutamarin and the residues in the ATPase domain of Topo IIα. Hydrogen bonds are shown by red dashed lines, and magenta spheres represent Mg cations. (E) Polar and hydrophobic surface profile of the ATPase domain of Topo IIα with (+)-rutamarin. Red and gray represent polar and hydrophobic areas, respectively. (F) Superposition of (+)-rutamarin with ADPNP in the ligand binding pocket.
The details of binding of (+)-rutamarin to the ATPase domain of Topo IIα at an atomic level were revealed in a conformation clustering analysis. The results suggest that (+)-rutamarin forms a hydrogen bond with the side chain of Asn95, Ala167, and conserved water molecule 2 and hydrophobically interacts with Ile125, Ile141, Phe142, Ala167, Thr215, Gly166, and Lys123 in the catalytic site of Topo IIα (Fig. 8D). The distance distribution analysis also showed that the hydrogen bonds between conserved water molecule 2- and Ala167–(+)-rutamarin are comparatively stable, whereas the hydrogen bond of Asn95–(+)-rutamarin is unstable (Fig. 8B). Solvent-accessible-surface area (SASA) analysis suggests that the polar surface of (+)-rutamarin is complementary to the polar surface of the binding pocket of Topo IIα (Fig. 8E). Superimposition of (+)-rutamarin and cocrystallized ADPNP suggests that (+)-rutamarin occupies the ATP binding pocket in a fashion similar to that of an ATP molecule (Fig. 8F). The acetyl groups of (+)-rutamarin, like the three phosphate groups of ATP, is involved in interaction with the highly conserved Walker A consensus motif GXXGXG at residues 161 to 166 of Topo IIα (Fig. 8D and F) (36). This conserved motif is important for the binding of ATP, as well as for the catalytic inhibitors acting on the ATPase domain (27). This notion is supported by a site-directed mutagenesis result showing that the mutations in Arg162Gln and Tyr165Ser in the ATP binding site lead to drug resistance (37, 38). These observations, consistent with the results of the DNA decatenation assay and the malachite green assay, support the idea that (+)-rutamarin functions as a Topo IIα catalytic inhibitor.
The MM-PBSA and MM-GBSA methods were employed to calculate the binding free energies and to gain information on the different components of interaction energy that contribute to (+)-rutamarin binding. The results are listed in Table 1. ΔGMM-PBSA and ΔGMM-GBSA for the Topo IIα-(+)-rutamarin complex are −29.90 and −24.66 kcal/mol, respectively. ΔGMM-PBSA, accounting for the total relative binding free energy, equal to ΔGgas plus ΔGsolv, comes mainly from the van der Waals component, whereas the electrostatic contribution is much less. This notion is also reflected in the results of the MM-GBSA method (Table 1), which show that (+)-rutamarin is a comparatively hydrophobic molecule that can form favorable hydrophobic interactions with residues of the Topo IIα catalytic pocket.
TABLE 1.
Binding free energy for Topo IIα–(+)-rutamarin complex and its different energy components based on MM-PBSA and MM-GBSA methods
| Energy term | Binding free energy (kcal/mol) (SEM) |
|
|---|---|---|
| MM-PBSA | MM-GBSA | |
| ΔEvdwa | −40.12 (2.14) | −40.22 (2.26) |
| ΔEeleb | −0.35 (0.22) | −0.42 (0.15) |
| ΔEpol,solvc | 39.99 (1.93) | 22.09 (1.66) |
| ΔEnonpol,solvd | −29.43 (1.37) | −6.11 (0.34) |
| ΔGgase | −40.47 (2.41) | −40.64 (2.28) |
| ΔGsolvf | 10.56 (1.28) | 15.98 (1.14) |
| ΔGbindingg | −29.91 (1.38) | −24.66 (1.69) |
Nonbonded van der Waals.
Nonbonded electrostatics.
Polar component to solvation.
Nonpolar component to solvation.
Total gas phase energy.
Sum of nonpolar and polar contributions to solvation.
Final estimated binding free energy calculated from the terms above.
DISCUSSION
In the current study, we identified (+)-rutamarin as an effective antiviral agent that inhibits KSHV ori-Lyt-dependent DNA replication with low cytotoxicity. It blocks KSHV DNA replication by inhibiting the catalytic activity of human Topo IIα. The salient features and implications of this finding are as follows.
There is a need for effective drugs targeting KSHV.
KSHV has been proven to be the etiological cause of KS and other KSHV-associated malignancies. However, the current treatment modalities for KSHV-associated diseases include only traditional cancer therapies. For classic KS, the chemotherapeutics that have been approved by the FDA include liposomal anthracycline products (liposomal doxorubicin or liposomal daunorubicin), paclitaxel, and alpha interferon (39). However, the majority of these agents have serious side effects, and the tumor responses to the chemotherapeutic regimens are only transient. In the current AIDS epidemic, even though KS has become a major AIDS-associated malignancy and remains an important cause of morbidity and mortality in AIDS patients, little effort has been made to develop more effective drugs for KS, as there was a general belief that AIDS-associated KS would disappear if AIDS were under control with the advent of HAART. However, despite its dramatic decrease in frequency since the advent of HAART, KS remains the most common AIDS-associated cancer (40). In addition, as experience with HAART has grown, a new HAART-associated syndrome has emerged. In a subset of HIV-seropositive individuals, starting HAART in the setting of advanced HIV infection results in a paradoxical clinical worsening of existing infection or the appearance of a new condition, including KS, in a process known as immune reconstitution inflammatory syndrome (IRIS) (41). IRIS is thought to be the direct result of a reconstituted immune system recognizing pathogens or antigens that were previously present but clinically asymptomatic. The current regimen for IRIS-associated KS is a combination therapy with HAART plus chemotherapeutics, such as liposomal doxorubicin. Since IRIS-KS is the result of responses by a recovered immune system to the KS-causing pathogen, i.e., KSHV, treatment of KSHV-seropositive, HIV-positive patients with a combination of antiretroviral (HAART) and anti-KSHV therapeutics is expected to yield positive results. However, at this time, there are no effective drugs targeting KSHV available. Earlier studies on cohorts of HIV-seropositive subjects suggested that treatment with the antiherpesviral drug ganciclovir or foscarnet could reduce the incidence of KS in AIDS patients (42, 43). However, severe side effects (renal impairment and bone marrow suppression), as well as rapid development of drug resistance, have limited the use of these drugs in the treatment of KS. Therefore, there is a need for effective drugs targeting KSHV.
(+)-Rutamarin is an antiviral-drug candidate for KSHV-associated diseases.
Recently, we reported that host cellular Topo II is absolutely required for KSHV DNA replication, and thus, Topo II inhibitors effectively block KSHV DNA synthesis (20). Topo II inhibitors are divided into two categories: Topo II poisons, which target the topoisomerase-DNA intermediate (cleavable complex), and Topo II catalytic inhibitors, which disrupt catalytic turnover of the enzyme. Although both categories possess inhibitory activities against KSHV DNA replication, Topo II poisons in general exhibit very strong cytotoxicity to host cells. In contrast, Topo II catalytic inhibitors show less cytotoxicity (such as novobiocin, with a CC50 value of 871 μM, and merbarone, with a CC50 value of 212.9 μM for BCBL-1 cells). Both novobiocin and merbarone are effective in halting KSHV DNA synthesis, with IC50s of 27.55 μM and 19.54 μM, respectively. The low cytotoxicities and high inhibition rates for viral replication of catalytic Topo II inhibitors suggest the potential for Topo II catalytic inhibitors to become effective anti-KSHV drugs for treatment of KS and other KSHV-associated diseases (20). In addition, given that viral polymerases may introduce mutations which subsequently can give rise to drug resistance, targeting host cellular proteins that viruses rely on for their replication offers the advantage of minimizing drug resistance and thus constitutes a novel therapeutic strategy.
In the current study, we attempted to search for new Topo II catalytic inhibitors that are potent in inhibiting viral replication and that have low cytotoxicity. This effort led to identification of (+)-rutamarin as a promising lead that efficiently inhibits KSHV lytic DNA replication in BCBL-1 cells with an IC50 of 1.12 μM and an EC50 of 1.62 μM, 25- and 17-fold lower than those of novobiocin. In addition, (+)-rutamarin exhibits very low cytotoxicity, and the SI of KSHV lytic replication was calculated to be 84.14, three times better that that of novobiocin. In addition, the molecular weight of (+)-rutamarin (356.42) is smaller than that of novobiocin (634.61), and the mass concentration differences of their IC50s and EC50s would be greater. These data demonstrate great potential for (+)-rutamarin to become an effective antiviral agent with low cytotoxicity.
(+)-Rutamarin is a natural product found in plants, such as Ruta graveolens L (common rue). It has been reported that (+)-rutamarin possesses antiproliferation and anticancer activities against a variety of tumor cell lines (44). It has also been found to induce the expression and translocation of glucose transporter 4 (GLUT4). As impaired translocation or decreased expression of GLUT4 in response to insulin is one of the major pathological features of type 2 diabetes, the function of (+)-rutamarin in ameliorating glucose homeostasis suggests a potential for the compound in anti-type 2 diabetes drug development (45). Our study is the first to demonstrate the capability of the compound in inhibition of viral replication with the potential to be an antiviral.
Anti-proliferation potential of (+)-rutamarin.
At a dose equal to the IC50 for inhibiting KSHV replication, (+)-rutamarin did not show a perceptible effect on cell proliferation and cell cycle progression in BCLB-1 and BJAB cells. At an excess dose (5 times the IC50,), (+)-rutamarin exhibited a degree of suppression of cell proliferation with a delay in the S phase.
The effects of (+)-rutamarin on cell proliferation in a variety of tumor cell lines were estimated, and inhibitory activities against A549, Bel7402, HepG2, and HCT8 cell proliferation were reported (44). The antiproliferation activity of (+)-rutamarin against these tumor cell lines appears to be similar to the effects on B cells observed in our study. Although (+)-rutamarin has an extent of antiproliferative ability that may provide an additional benefit if it becomes a drug to treat KSHV-associated malignancies, the moderate antiproliferation ability may not be potent enough to provide antitumor activity. This may also be true of all Topo II catalytic inhibitors. This notion is supported by studies and observations of some Topo II catalytic inhibitors, including novobiocin and merbarone, which exhibit antiproliferation activities in cell cultures but show no significant effects on tumor development in clinical trials (46, 47).
The moderate effect of (+)-rutamarin on cell proliferation, as well as its low cytotoxicity, may result from redundancy of Topo II activities in mammalian cells. Mammals express two isoforms of Topo II, alpha and beta, which are highly homologous but display differences in expression and subcellular localization at the time of mitosis. The similar functions and domain structures of the two isoforms make it possible that Topo IIβ may be able to partially compensate for the loss of Topo IIα function for cell survival in the presence of an inhibitor, such as (+)-rutamarin. However, KSHV may be dependent on only Topo IIα for its lytic replication, explaining the sensitivity of KSHV replication to Topo IIα catalytic inhibitors.
(+)-Rutamarin is a novel human Topo IIα catalytic inhibitor.
Topo II is an ATPase and uses the energy derived from ATP hydrolysis to resolve the winding problem of double-stranded DNA. The detailed binding mode of (+)-rutamarin and human Topo IIα was identified using molecular docking and MD simulation. The significance of these studies is 2-fold. First, the study provided insight into the binding of (+)-rutamarin to Topo IIα, and the results confirmed that the compound binds to the ATP pocket of Topo IIα. Second, the results of the study will be of great help in our next effort to modify the compound for more potent antiviral candidates. Energy decomposition analysis led to identification of key residues contributing to binding affinity at the active site. As shown in Fig. 9, the major contributing residues can be divided into three clusters, which are consistent with the three binding pocket sites, i.e., (i) the Walker A consensus motif (residues 161 to 167), (ii) the Mg2+ binding area (residues 87, 91, and 94), and (iii) the hydrophobic chamber (key residues, Ile 125, Ile141, Phe142, Lys123, and Thr215). All of these residues except Glu87 positively contribute to the binding affinity for (+)-rutamarin. The incompatibility between the carboxyl group of Glu87 and the acetyl groups of (+)-rutamarin suggests that Glu87 is unfavorable for binding affinity to (+)-rutamarin (Fig. 8D). These results provide a theoretical basis for (+)-rutamarin lead optimization.
FIG 9.
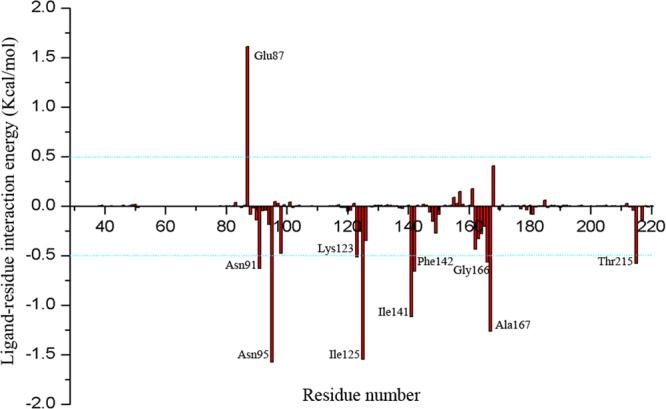
Binding free energy decomposition results based on the MM-GBSA method. The key residues are labeled.
Supplementary Material
ACKNOWLEDGMENTS
We thank Manunya Nuth at the University of Pennsylvania for constructive discussion, suggestions, and critical reading of the manuscript.
This work was supported by Natural Science Foundation of China research grants (no. 81271805 and 81171575), the Guangdong Innovative Research Team program (no. 2009010058), and a research grant from the U.S. National Institutes of Health (R01AI052789).
Footnotes
Published ahead of print 2 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01259-13.
REFERENCES
- 1.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. 10.1126/science.7997879 [DOI] [PubMed] [Google Scholar]
- 2.Moore PS, Chang Y. 1995. Detection of herpesvirus-like DNA sequences in Kaposi's sarcoma in patients with and without HIV infection. N. Engl. J. Med. 332:1181–1185. 10.1056/NEJM199505043321801 [DOI] [PubMed] [Google Scholar]
- 3.Dupin N, Grandadam M, Calvez V, Aubin JT, Agut H, Gorin I, Havard S, Lamy F, Leibowitch M, Huraux JM, Escande JP. 1995. Herpesvirus-like DNA sequences in patients with Mediterranean Kaposi's sarcoma. Lancet 345:761–762. 10.1016/S0140-6736(95)90642-8 [DOI] [PubMed] [Google Scholar]
- 4.Schalling M, Ekman M, Kaaya EE, Linde A, Biberfeld P. 1995. A role for a new herpes virus (KSHV) in different forms of Kaposi's sarcoma. Nat. Med. 1:707–708. 10.1038/nm0795-707 [DOI] [PubMed] [Google Scholar]
- 5.Chuck S, Grant RM, Katongole-Mbidde E, Conant M, Ganem D. 1996. Frequent presence of a novel herpesvirus genome in lesions of human immunodeficiency virus-negative Kaposi's sarcoma. J. Infect. Dis. 173:248–251. 10.1093/infdis/173.1.248 [DOI] [PubMed] [Google Scholar]
- 6.Antman K, Chang Y. 2000. Kaposi's sarcoma. N. Engl. J. Med. 342:1027–1038. 10.1056/NEJM200004063421407 [DOI] [PubMed] [Google Scholar]
- 7.Krentz HB, Kliewer G, Gill MJ. 2005. Changing mortality rates and causes of death for HIV-infected individuals living in Southern Alberta, Canada from 1984 to 2003. HIV Med. 6:99–106. 10.1111/j.1468-1293.2005.00271.x [DOI] [PubMed] [Google Scholar]
- 8.Cesarman E, Knowles DM. 1999. The role of Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in lymphoproliferative diseases. Semin. Cancer Biol. 9:165–174. 10.1006/scbi.1998.0118 [DOI] [PubMed] [Google Scholar]
- 9.Powles T, Stebbing J, Bazeos A, Hatzimichael E, Mandalia S, Nelson M, Gazzard B, Bower M. 2009. The role of immune suppression and HHV-8 in the increasing incidence of HIV-associated multicentric Castleman's disease. Ann. Oncol. 20:775–779. 10.1093/annonc/mdn697 [DOI] [PubMed] [Google Scholar]
- 10.Moore PS, Chang Y. 2011. KSHV: forgotten but not gone. Blood 117:6973–6974. 10.1182/blood-2011-05-350306 [DOI] [PubMed] [Google Scholar]
- 11.Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT. 1997. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 71:715–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun R, Lin SF, Staskus K, Gradoville L, Grogan E, Haase A, Miller G. 1999. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J. Virol. 73:2232–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong W, Wang H, Herndier B, Ganem D. 1996. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. U. S. A. 93:6641–6646. 10.1073/pnas.93.13.6641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cesarman E, Mesri EA, Gershengorn MC. 2000. Viral G protein-coupled receptor and Kaposi's sarcoma: a model of paracrine neoplasia? J. Exp. Med. 191:417–422. 10.1084/jem.191.3.417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grundhoff A, Ganem D. 2004. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J. Clin. Invest. 113:124–136. 10.1172/JCI17803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin CL, Li H, Wang Y, Zhu FX, Kudchodkar S, Yuan Y. 2003. Kaposi's sarcoma-associated herpesvirus lytic origin (ori-Lyt)-dependent DNA replication: identification of the ori-Lyt and association of K8 bZip protein with the origin. J. Virol. 77:5578–5588. 10.1128/JVI.77.10.5578-5588.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Li H, Chan MY, Zhu FX, Lukac DM, Yuan Y. 2004. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: cis-acting requirements for replication and ori-Lyt-associated RNA transcription. J. Virol. 78:8615–8629. 10.1128/JVI.78.16.8615-8629.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Tang Q, Maul GG, Yuan Y. 2006. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: dual role of replication and transcription activator. J. Virol. 80:12171–12186. 10.1128/JVI.00990-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Li H, Tang Q, Maul GG, Yuan Y. 2008. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: involvement of host cellular factors. J. Virol. 82:2867–2882. 10.1128/JVI.01319-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzalez-Molleda L, Wang Y, Yuan Y. 2012. Potent antiviral activity of topoisomerase I and II inhibitors against Kaposi's sarcoma-associated herpesvirus. Antimicrob. Agents Chemother. 56:893–902. 10.1128/AAC.05274-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2:342–346. 10.1038/nm0396-342 [DOI] [PubMed] [Google Scholar]
- 22.Cannon JS, Ciufo D, Hawkins AL, Griffin CA, Borowitz MJ, Hayward GS, Ambinder RF. 2000. A new primary effusion lymphoma-derived cell line yields a highly infectious Kaposi's sarcoma herpesvirus-containing supernatant. J. Virol. 74:10187–10193. 10.1128/JVI.74.21.10187-10193.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gu Q, Xu J, Gu L. 2010. Selecting diversified compounds to build a tangible library for biological and biochemical assays. Molecules 15:5031–5044. 10.3390/molecules15075031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei H, Ruthenburg AJ, Bechis SK, Verdine GL. 2005. Nucleotide-dependent domain movement in the ATPase domain of a human type IIA DNA topoisomerase. J. Biol. Chem. 280:37041–37047. 10.1074/jbc.M506520200 [DOI] [PubMed] [Google Scholar]
- 25.Spassov VZ, Flook PK, Yan L. 2008. LOOPER: a molecular mechanics-based algorithm for protein loop prediction. Protein Eng. Des. Sel. 21:91–100. 10.1093/protein/gzm083 [DOI] [PubMed] [Google Scholar]
- 26.Cheng A, Best SA, Merz KM, Jr, Reynolds CH. 2000. GB/SA water model for the Merck molecular force field (MMFF). J. Mol. Graph. Model. 18:273–282. 10.1016/S1093-3263(00)00038-3 [DOI] [PubMed] [Google Scholar]
- 27.Baviskar AT, Madaan C, Preet R, Mohapatra P, Jain V, Agarwal A, Guchhait SK, Kundu CN, Banerjee UC, Bharatam PV. 2011. N-fused imidazoles as novel anticancer agents that inhibit catalytic activity of topoisomerase II alpha and induce apoptosis in G1/S phase. J. Med. Chem. 54:5013–5030. 10.1021/jm200235u [DOI] [PubMed] [Google Scholar]
- 28.Amaro RE, Swift RV, Votapka L, Li WW, Walker RC, Bush RM. 2011. Mechanism of 150-cavity formation in influenza neuraminidase. Nat. Commun. 2:388. 10.1038/ncomms1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. 2006. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 65:712–725. 10.1002/prot.21123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newhouse EI, Xu D, Markwick PRL, Amaro RE, Pao HC, Wu KJ, Alam M, McCammon JA, Li WW. 2009. Mechanism of glycan receptor recognition and specificity switch for avian, swine, and human adapted influenza virus hemagglutinins: a molecular dynamics perspective. J. Am. Chem. Soc. 131:17430–17442. 10.1021/ja904052q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W, Hou TJ, Qiao XB, Xu XJ. 2003. Parameters for the generalized born model consistent with RESP atomic partial charge assignment protocol. J. Phys. Chem. B 107:9071–9078. 10.1021/jp034613k [DOI] [Google Scholar]
- 32.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. 2004. Gaussian 03, revision E. 01. Gaussian Inc., Pittsburgh, PA [Google Scholar]
- 33.Kollman PA, Massova I, Reyes C, Kuhn B, Huo SH, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE. 2000. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 33:889–897. 10.1021/ar000033j [DOI] [PubMed] [Google Scholar]
- 34.Yan X, Li J, Liu Z, Zheng M, Ge H, Xu J. 2013. Enhancing molecular shape comparison by weighted Gaussian functions. J. Chem. Inf. Model. 53:1967–1978. 10.1021/ci300601q [DOI] [PubMed] [Google Scholar]
- 35.Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A. 95:10866–10871. 10.1073/pnas.95.18.10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walker JE, Saraste M, Runswick MJ, Gay NJ. 1982. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1:945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wessel I, Jensen LH, Jensen PB, Falck J, Rose A, Roerth M, Nitiss JL, Sehested M. 1999. Human small cell lung cancer NYH cells selected for resistance to the bisdioxopiperazine topoisomerase II catalytic inhibitor ICRF-187 demonstrate a functional R162Q mutation in the Walker A consensus ATP binding domain of the alpha isoform. Cancer Res. 59:3442–3450 [PubMed] [Google Scholar]
- 38.Wessel I, Jensen LH, Renodon-Corniere A, Sorensen TK, Nitiss JL, Jensen PB, Sehested M. 2002. Human small cell lung cancer NYH cells resistant to the bisdioxopiperazine ICRF-187 exhibit a functional dominant Tyr165Ser mutation in the Walker A ATP binding site of topoisomerase II alpha. FEBS Lett. 520:161–166. 10.1016/S0014-5793(02)02805-3 [DOI] [PubMed] [Google Scholar]
- 39.Potthoff A, Brockmeyer NH. 2007. HIV-associated Kaposi sarcoma: pathogenesis and therapy. J. Dtsch. Dermatol. Ges. 5:1091–1094. 10.1111/j.1610-0387.2007.06567.x [DOI] [PubMed] [Google Scholar]
- 40.Boshoff C, Weiss R. 2002. AIDS-related malignancies. Nat. Rev. Cancer. 2:373–382. 10.1038/nrc797 [DOI] [PubMed] [Google Scholar]
- 41.Shelburne SA, III, Hamill RJ. 2003. The immune reconstitution inflammatory syndrome. AIDS Rev. 5:67–79 [PubMed] [Google Scholar]
- 42.Glesby MJ, Hoover DR, Weng S, Graham NM, Phair JP, Detels R, Ho M, Saah AJ. 1996. Use of antiherpes drugs and the risk of Kaposi's sarcoma: data from the Multicenter AIDS Cohort Study. J. Infect. Dis. 173:1477–1480. 10.1093/infdis/173.6.1477 [DOI] [PubMed] [Google Scholar]
- 43.Mocroft A, Youle M, Gazzard B, Morcinek J, Halai R, Phillips AN. 1996. Anti-herpesvirus treatment and risk of Kaposi's sarcoma in HIV infection. Royal Free/Chelsea and Westminster Hospitals Collaborative Group. AIDS 10:1101–1105 [PubMed] [Google Scholar]
- 44.Yang QY, Tian XY, Fang WS. 2007. Bioactive coumarins from Boenninghausenia sessilicarpa. J. Asian Nat. Prod. Res. 9:59–65. 10.1080/10286020500382397 [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Zhang H, Yao XG, Shen H, Chen J, Li C, Chen L, Zheng M, Ye J, Hu L, Shen X, Jiang H. 2012. (+)-Rutamarin as a dual inducer of both GLUT4 translocation and expression efficiently ameliorates glucose homeostasis in insulin-resistant mice. PLoS One 7:e31811. 10.1371/journal.pone.0031811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eder JP, Wheeler CA, Teicher BA, Schnipper LE. 1991. A phase I clinical trial of novobiocin, a modulator of alkylating agent cytotoxicity. Cancer Res. 51:510–513 [PubMed] [Google Scholar]
- 47.Chang AY, Kim K, Glick J, Anderson T, Karp D, Johnson D. 1993. Phase II study of taxol, merbarone, and piroxantrone in stage IV non-small-cell lung cancer: The Eastern Cooperative Oncology Group Results. J. Natl. Cancer Inst. 85:388–394. 10.1093/jnci/85.5.388 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.