

Abstract
Griffithsin (GRFT) is a red-alga-derived lectin that binds the terminal mannose residues of N-linked glycans found on the surface of human immunodeficiency virus type 1 (HIV-1), HIV-2, and other enveloped viruses, including hepatitis C virus (HCV), severe acute respiratory syndrome coronavirus (SARS-CoV), and Ebola virus. GRFT displays no human T-cell mitogenic activity and does not induce production of proinflammatory cytokines in treated human cell lines. However, despite the growing evidence showing the broad-spectrum nanomolar or better antiviral activity of GRFT, no study has reported a comprehensive assessment of GRFT safety as a potential systemic antiviral treatment. The results presented in this work show that minimal toxicity was induced by a range of single and repeated daily subcutaneous doses of GRFT in two rodent species, although we noted treatment-associated increases in spleen and liver mass suggestive of an antidrug immune response. The drug is systemically distributed, accumulating to high levels in the serum and plasma after subcutaneous delivery. Further, we showed that serum from GRFT-treated animals retained antiviral activity against HIV-1-enveloped pseudoviruses in a cell-based neutralization assay. Overall, our data presented here show that GRFT accumulates to relevant therapeutic concentrations which are tolerated with minimal toxicity. These studies support further development of GRFT as a systemic antiviral therapeutic agent against enveloped viruses, although deimmunizing the molecule may be necessary if it is to be used in long-term treatment of chronic viral infections.
INTRODUCTION
The glycan structures displayed on envelope glycoproteins frequently play important roles in virus transmission and entry into target cells (1). Viruses that establish chronic infections, such as human immunodeficiency virus (HIV) and hepatitis C virus (HCV), display a dense shield of oligomannose glycans that also assist the pathogen in immune evasion, both through the display of “self”-like epitopes and by induction of immunosuppressive innate immune responses (2–6). Antiviral compounds that target envelope glycoproteins are classified as “carbohydrate binding agents” (CBA) and generally encompass lectins and nonpeptidic antibiotics such as pradimicin A and S and benanomicin A (reviewed by Balzarini [7]). Several different lectins from natural sources show significant antiviral activity in vitro and have been proposed as antiviral prophylactic and therapeutic compounds. While there are a plethora of publications in the scientific literature showing in vitro antiviral activity of CBA against a broad array of enveloped viruses, most in vivo studies on the safety and efficacy of this class of compounds have been performed in pre-exposure prophylaxis models. However, two important studies demonstrated the potential of antiviral therapy with lectins. Smee et al. (8) demonstrated that postexposure treatment with the antiviral lectin cyanovirin-N (CV-N), which targets an α-(1–2)-linked mannobiose substructure on oligomannose glycans, showed significant survival benefit 6 h after infection in a murine influenza model. A recent study by Michelow and colleagues (9) demonstrated that high-dose therapy with human mannose binding lectin (MBL), an endogenous C-type lectin that recognizes glycan structures, including mannose, glucose, and fucose, on the surface of pathogens, could ameliorate Ebola virus infection in a murine model. Despite their demonstrated antiviral activities, the fact that many natural product lectins have significant in vitro and in vivo toxicity, acting as nonspecific T-cell stimulants and red blood cell-agglutinating agents, has limited their development as antiviral therapeutics. However, not all antiviral lectins are toxins and not all antiviral lectins have cell-agglutinating activity.
Griffithsin (GRFT) is a 12.77-kDa red-alga-derived lectin that binds the terminal mannose residues on the asparagine (N)-linked Man5-9GlcNAc2 structures that comprise the vast majority of N-linked glycans in the HIV type 1 (HIV-1) glycan shield (10–13). GRFT displays no human T-cell mitogenic activity and, unlike many other lectins, does not induce production of proinflammatory cytokines in treated human peripheral blood mononuclear cells (14, 15). In collaborative studies, we have shown that GRFT has broad-spectrum antiviral activity against HIV-1 (10, 15, 16), HIV-2 (16), HCV (17, 18), and an array of pathogenic coronaviruses, including severe acute respiratory syndrome coronavirus (SARS-CoV) (19), in addition to influenza A virus and several other enveloped viral pathogens (B. R. O'Keefe, unpublished data). The in vitro inhibitory activity (50% effective concentration [EC50]) of GRFT against HIV-1 is in the mid-picomolar to low-nanomolar range for most isolates (10, 15, 20). The GRFT EC50 against HCV is 13.9 nM (17) and against SARS-CoV is 48 nM (12, 19). There is a growing body of published evidence indicating that GRFT also has antiviral activity ex vivo as well as in vivo: topical application of GRFT prevents HIV-1 infection of human cervical explants (15); intranasal treatments with GRFT prevents disease in mice challenged with SARS-CoV (19); intraperitoneal (i.p.) treatment with GRFT prevents Japanese encephalitis virus (JEV) infection in mice (21); and subcutaneous (s.c.) treatment with GRFT shows some efficacy against HCV challenge in a mouse-human chimeric liver models (17, 18). The JEV and HCV studies demonstrated that GRFT is relatively well tolerated by mice exposed to the drug systemically at doses of 5 mg/kg of body weight but did not report a comprehensive assessment of GRFT safety as a potential systemic antiviral treatment. Here, we report that minimal toxicity is induced by a range of subcutaneous doses of GRFT in two rodent species. The drug was systemically distributed and accumulated to high levels in the serum and plasma after subcutaneous delivery. Furthermore, we demonstrated that serum from GRFT-treated animals retained antiviral activity against HIV-1-enveloped pseudoviruses in a cell-based neutralization assay. Overall, these findings support further investigation into GRFT's potential as a systemic antiviral therapeutic agent against enveloped viruses, including HIV-1.
MATERIALS AND METHODS
Lectin reagents.
Recombinant GRFT was produced in Nicotiana benthamiana plants as described previously, purified to >99% purity, and formulated in phosphate-buffered saline (PBS), pH 7.4 (15). Phytohemagglutinin A (PHA) was purchased from Sigma.
Animal housing and care.
For the study, 6-to-8-week-old female BALB/c mice (Jackson Laboratory) and Hartley guinea pigs (Cavia porcellus; Charles River Laboratories) were housed in a temperature- and humidity-controlled room with an alternating light/dark cycle of 12 h, with standard diet and water ad libitum. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Louisville.
Mouse treatments and sample collection.
To evaluate effects of a single high dose of GRFT, mice were injected subcutaneously with 50 mg/kg GRFT (n = 30) or PBS (n = 15). At 1, 7, and 14 days posttreatment, 10 mice treated with GRFT and 5 control animals were sacrificed and blood was collected by cardiac puncture. Kidneys, livers, and spleens were excised. For chronic administration, mice were treated with 10 mg/kg GRFT (n = 15) or PBS (n = 15) daily for 14 days. Each group was further subdivided into 3 groups of 5 mice each. Blood was collected from the submandibular vein every other day, alternating between subgroups. Animals were sacrificed on day 14 (9 mice per treatment group), day 16, and day 21 (3 mice per treatment group at each time point) and the samples collected as outlined above.
Guinea pig treatment and sample collection.
For chronic administration in guinea pigs, two studies were conducted at different times. In the first experiment, 10 mg/kg GRFT (n = 12) or 1 ml/kg PBS (n = 6) was subcutaneously administered daily for 10 days. The second experiment was similar to the first, except for the number of animals (GRFT, n = 10; PBS, n = 6). Half of the animals in each group were sacrificed on day 11 after cardiac puncture exsanguination under conditions of isoflurane anesthesia, and the remaining animals were euthanized on day 15. Blood and organs were collected at sacrifice.
Extraction of GRFT from mouse organs.
Pooled organ tissues (100 to 500 mg) were homogenized in 1 ml of PBS supplemented with complete, EDTA-free Protease Inhibitor Cocktail (Roche) and the samples cleared by a series of two centrifugation steps performed at 10,000 and 15,000 × g, respectively, for 10 min each time. The supernatants were stored at −20°C until use.
GRFT capture immunoassay using the HIV-1 gp120 envelope glycoprotein.
To detect trace amounts of GRFT present in serum and plasma and in homogenized organ tissues, we used an HIV-1 gp120 binding enzyme-linked immunosorbent assay (ELISA) as previously described (14) with a few modifications. Briefly, Maxisorp plates (Nunc) were coated with 25 ng purified gp120 (Protein Sciences) and incubated overnight at 4°C. Plates were blocked with 3% (wt/vol) bovine serum albumin (BSA) in PBS containing 0.05% Tween 20 (PBS-T). Samples were diluted 1:10 in blocking buffer and were incubated at room temperature (RT) for 1 h. Serial dilutions of purified GRFT were run in parallel to generate a standard curve. The gp120-bound GRFT was detected by rabbit anti-GRFT antiserum (1:25,000) followed by horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (1:10,000). Plates were developed with SureBlue TMB Microwell peroxidase substrate, and reactions were stopped with 1 N H2SO4. Absorbance at 450 nm and 570 nm was measured using a BioTek Synergy HT plate reader.
Evaluation of anti-HIV activity.
HIV-1 neutralization activity of heat-inactivated serum or plasma was measured using pseudovirus neutralization assays as previously described (22). Briefly, molecularly cloned DU156 env-pseudotyped virus particles were generated by transfection of 293T cells and titrated in TZM-bl cells. Antiviral activity was measured as a function of luciferase reporter gene activity. The 50% infective dose (ID50) values were defined as the sample dilution required to reduce luminescence by 50% in comparison to wells with no sample added.
Hematology parameters and serum chemistry.
A complete blood count (CBC) was run for guinea pig samples using a Hemavet 950 system (Drew Scientific) standardized for guinea pig blood. The following parameters were quantified in potassium-EDTA anticoagulated whole blood: counts of red blood cells (RBC; 104/μl), total and differential leukocyte counts (neutrophils, lymphocytes, monocytes, eosinophils, and basophils quantitated as 103/μl or %), hemoglobin concentration (HGB; g/dl), hematocrit (HCT; %), mean corpuscular volume (MCV; fl), mean cell hemoglobin (MCH; pg), mean cell hemoglobin concentration (MCHC; g/dl), red cell distribution width (RDW; %), platelets (PLT; 104/μl), and mean platelet volume (MPV; fl).
In the first guinea pig experiment, levels of the following serum chemistries were assessed and the differences analyzed by two-way analysis of variance (ANOVA): serum albumin (Alb), alkaline phosphatase (ALKP), amylase (Amy), alanine aminotransferase (ALT), blood urea nitrogen (BUN), calcium (Ca), cholesterol (Chol), creatinine (Creat), globulin (Glob), glucose (Glu), phosphorus (Phos), total bilirubin (TBil), and total protein (TP). Based on the results obtained, we decided to further measure the effect of GRFT on selected markers, including serum albumin, alkaline phosphatase, and amylase, in the second experiment using a VetTest Chemistry Analyzer (IDEXX Laboratories).
Hemagglutination assays.
Specimens of guinea pig, sheep, and human blood (Innovative Research) and blood collected from untreated mice were washed and resuspended at a final concentration of 1% to 2% (vol/vol) in 1× PBS containing 3 g/liter BSA and 1 g/liter sodium azide. PBS, phytohemagglutinin (PHA), or GRFT was mixed with an equal volume of erythrocytes in a 96-well round-bottom plate. The plate was incubated for 1 h at RT, followed by overnight incubation at 4°C. Finally, wells were dried and hemagglutination activity was determined by visual examination.
Statistical analysis.
Statistical analysis was conducted using Graph Pad Prism 5 and SAS software version 9.3. Because of the stratification by guinea pig study, day of sacrifice, and treatment group, it was decided to increase statistical power by using all the data from both studies. Three-way ANOVA (23) was utilized, which allowed testing for treatment effect while at the same time adjusting for differences between the two studies and the days of sacrifice. For the initial univariable analysis, the two-sample t test (24) or Wilcoxon rank sum test (25) was used to test for differences between studies, days of sacrifice, and treatments. For the sets of data such as weight change and nonselected serum chemistries collected in only one guinea pig study, a two-way ANOVA was utilized. A P value < 0.05 was deemed significant.
RESULTS
Griffithsin serum concentrations.
Plasma samples collected from mice injected with a single high dose of 50 mg/kg GRFT showed up to 4 nM GRFT (4 nM equals 51.08 ng/ml) in animals sacrificed 1 day posttreatment (Fig. 1A). These levels decreased considerably to less than 0.5 nM by day 7 and persisted through day 14.
FIG 1.
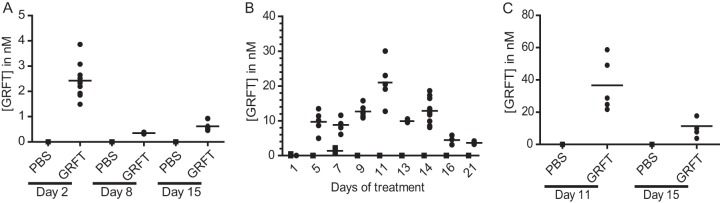
Griffithsin concentrations in plasma and serum samples. Data were obtained from mouse plasma after a single high-dose administration of 50 mg/kg GRFT (A) and a chronic daily administration of 10 mg/kg GRFT (B) and from guinea pig serum after 10 daily administrations of 10 mg/kg GRFT (C). PBS was administered to control animals, and bars indicate mean group concentrations.
In chronically dosed mice, plasma concentrations of GRFT peaked at 25 nM (Fig. 1B) by day 11, followed by a gradual decrease in detectable GRFT even with subsequent treatments. This trend continued throughout the recovery time. Notably, concentrations of GRFT persisted in the plasma at levels of approximately 4 nM after a week of recovery (Fig. 1B). GRFT concentrations in sera from chronically treated guinea pigs were similar to those in sera from chronically treated mice, with mean concentrations of 36 nM at day 11 and 11.36 nM at day 15 (Fig. 1C).
Anti-HIV activity of plasma and serum.
Plasma samples collected from mice treated with a single dose of 50 mg/kg GRFT neutralized HIV-1 pseudoviruses (clade C primary sexually transmitted isolate Du156) with a mean ID50 of 1,500 on experimental day 2 (Fig. 2A). This neutralization activity decreased in samples obtained at days 8 (ID50 of 300) and 15 (ID50 of 200). Plasma from chronically treated mice neutralized the HIV-1 Du156 pseudovirus with an ID50 value of approximately 800 (Fig. 2B) at day 14. After the 7-day recovery period, the ID50 values decreased to approximately 200. Serum samples collected from guinea pigs after chronic treatment with 10 mg/kg GRFT displayed a mean ID50 of approximately 3,277 at day 11 and 576 at day 15. The HIV-1 Du156 neutralization activity of guinea pig sera correlated well with the concentration of GRFT detected in the serum; however, the GRFT serum concentrations detected in mouse samples predicted only about 14% to 25% of the actual serum HIV-1 neutralization activity we observed, confirming that additional nonspecific HIV-1 neutralizing activities in mouse serum contributed to the higher-than-expected overall HIV-1 inhibitory activity in the mouse sera.
FIG 2.
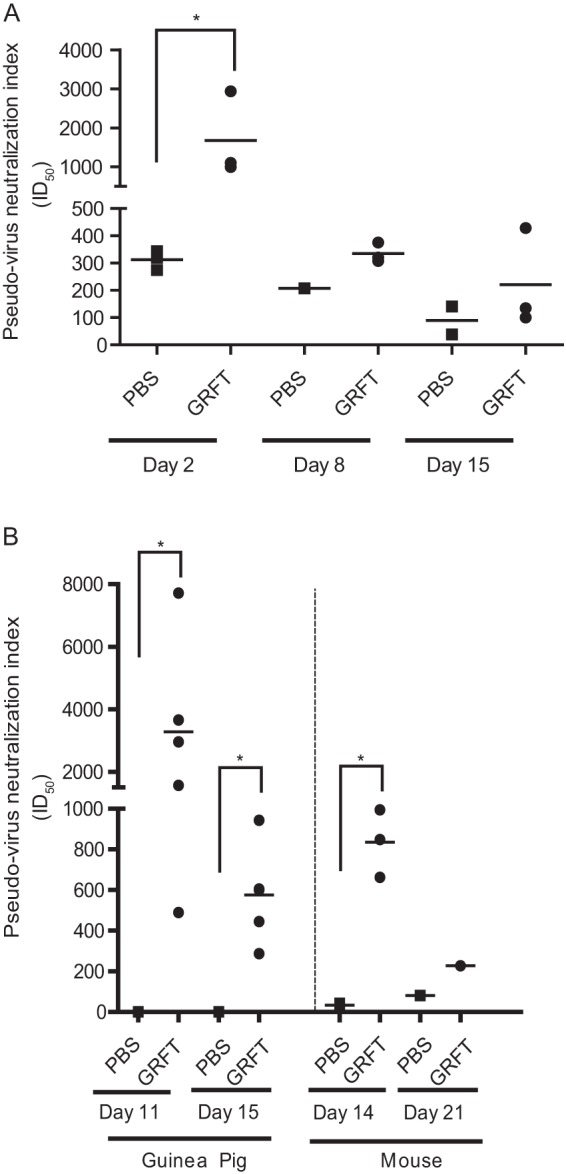
Antiviral activity of samples collected from GRFT-treated animals. (A) For HIV-1 env, pseudovirus neutralization activity was assessed for mouse plasma after a single high-dose administration of 50 mg/kg GRFT and is expressed as ID50. (B) The ID50 values were obtained for samples from guinea pigs and mice chronically treated with 10 mg/kg GRFT. Control animals were treated with PBS bars indicate mean group concentrations. Statistical significance (P < 0.05; 1-way ANOVA) is indicated by asterisks (*).
GRFT distribution into tissues.
Organs from mice chronically treated with GRFT were harvested, and total protein was extracted. We measured the total amounts of GRFT that accumulated in these tissues by gp120 binding ELISA. Figure 3 shows that GRFT accumulated in all three organs assayed, with most GRFT accumulating in the spleen. Nonquantitative immunofluorescence studies detected GRFT in the same organs (liver, kidney, and spleen) harvested from treated guinea pigs (data not shown).
FIG 3.
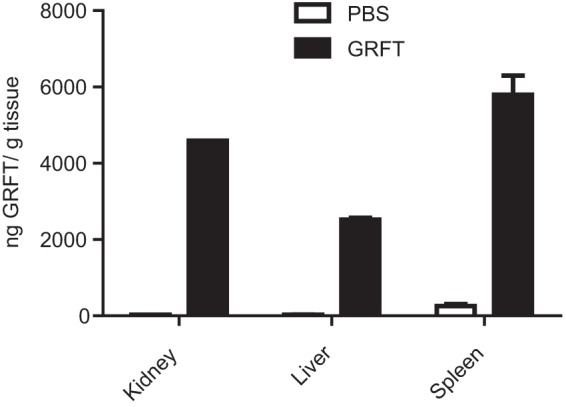
Quantitation of GRFT in mouse organs. Pooled protein samples were extracted from kidneys, livers, and spleens after a chronic subcutaneous administration of GRFT or PBS, and GRFT was detected using gp120 binding ELISA.
GRFT is tolerated after subcutaneous administration.
We studied the toxicity of GRFT in guinea pigs using several parameters, including mortality, behavior, animal body and organ weight changes, tissue pathology, and changes in blood properties.
All animals survived, and no change in behavior was observed. Since the guinea pigs used in this work were juvenile, we evaluated their overall fitness after GRFT treatment using body weight as a surrogate marker. Animals were weighed at day 1 and at the time of sacrifice. Using a 2-way ANOVA to measure the impact of treatment and time on body weight gains, we found that GRFT treatment resulted in significantly less body weight gain in comparison with that seen with PBS-treated controls (P = 0.0011; Fig. 4).
FIG 4.

Guinea pig body weight gain as an indicator of overall health. Body weights were measured on experimental day 1 and on the termination day (day 11 or day 15). Statistical significance (P < 0.05) is indicated by asterisks (*).
Liver, kidney, and spleen weights measured at termination were normalized to total body weights and compared to time-matched controls. A statistically significant increase of the normalized weights of guinea pig livers and spleens was observed for GRFT-treated animals (P = 0.008 and 0.005, respectively) (Fig. 5; see also Fig. S1 in the supplemental material). Tissue sections from guinea pigs in the second experiment were stained with hematoxylin and eosin and evaluated in a blind fashion by a veterinary pathologist (O. Foreman). No distinct pathologies were observed as a result of GRFT treatment. Of all CBC parameters tested, a statistically significant difference was observed only in red blood cell width (RDW) (P = 0.016) (Table 1; see also Fig. S3 to S6 in the supplemental material). Of note, the RDW values obtained for GRFT-treated animals were still within the normal physiological range described for guinea pigs (26).
FIG 5.
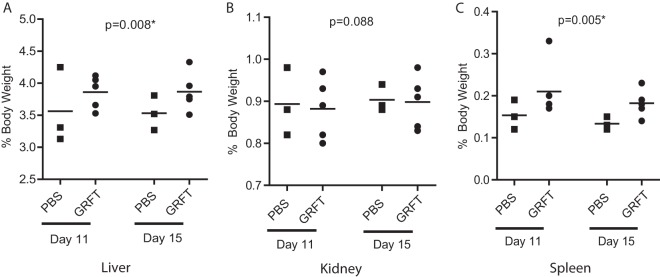
Effect of GRFT on guinea pig organ weights. Liver (A), kidney (B), and spleen (C) weights were measured relative to body weight at sacrifice.
TABLE 1.
Hematological profile for guinea pigs after chronic treatment with GRFTa
| Cell type | Parameter | Unit | Values |
|||
|---|---|---|---|---|---|---|
| Day 11 |
Day 15 |
|||||
| PBS (n = 3) | GRFT (n = 5) | PBS (n = 3) | GRFT (n = 5) | |||
| Leukocyte | WBC | k/μl | 4.92 ± 2.43 | 2.15 ± 0.23 | 4.35 ± 1.28 | 2.90 ± 1.36 |
| NE | k/μl | 2.30 ± 1.46 | 0.96 ± 0.13 | 1.67 ± 0.45 | 1.01 ± 0.42 | |
| LY | k/μl | 2.49 ± 1.11 | 1.14 ± 0.28 | 2.60 ± 0.89 | 1.82 ± 0.99 | |
| MO | k/μl | 0.09 ± 0.10 | 0.03 ± 0.03 | 0.07 ± 0.03 | 0.06 ± 0.04 | |
| EO | k/μl | 0.03 ± 0.02 | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.01 ± 0.02 | |
| BA | k/μl | 0.00 ± 0.01 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | |
| Erythrocyte | RBC | M/μl | 4.30 ± 0.06 | 4.12 ± 0.18 | 4.75 ± 0.21 | 4.37 ± 0.22 |
| Hb | g/dl | 12.23 ± 0.64 | 11.64 ± 0.46 | 13.10 ± 0.46 | 12.22 ± 0.61 | |
| HCT | % | 38.30 ± 2.31 | 36.12 ± 1.47 | 41.57 ± 1.76 | 39.52 ± 1.94 | |
| MCV | fl | 88.97 ± 4.73 | 87.58 ± 1.29 | 87.53 ± 1.70 | 90.40 ± 0.52* | |
| MCH | pg | 28.43 ± 1.33 | 28.26 ± 1.52 | 27.63 ± 0.61 | 27.96 ± 1.04 | |
| MCHC | g/dl | 31.97 ± 0.25 | 32.26 ± 1.33 | 31.50 ± 0.26 | 30.94 ± 1.11 | |
| RDW | % | 13.83 ± 1.43 | 12.92 ± 0.73 | 14.13 ± 0.29 | 12.66 ± 0.70* | |
| Thrombocyte | PLT | k/μl | 595.33 ± 54.31 | 628.60 ± 44.77 | 677.33 ± 11.93 | 659.00 ± 34.26 |
| MPV | fl | 4.40 ± 1.14 | 3.92 ± 0.23 | 3.57 ± 0.12 | 3.84 ± 0.24 | |
Data represent the mean values ± standard deviations for white blood cells (WBC), neutrophils (NE), lymphocytes (LY), monocytes (MO), eosinophils (EO), basophils (BA), red blood cells (RBC), hemoglobin (Hb), hematocrit (HCT), mean corpuscular volume (MCV), mean cell hemoglobin (MCH), mean cell hemoglobin concentration (MCHC), red cell distribution width (RDW), platelets (PLT), and mean platelet volume (MPV). Statistical significance (P < 0.05) is indicated by asterisks (*).
In addition to CBCs, serum chemistries were examined. When data collected from both experiments were combined for analysis, a statistically significant increase was observed for alkaline phosphatase after GRFT treatment compared with control results (P = 0.001) (Fig. 6; see also Fig. S2 in the supplemental material). There were no statistically significant differences between the GRFT-treated group and the PBS control group in the remaining serum chemistry when examined by 2-way ANOVA (Fig. 6; see also Fig. S2).
FIG 6.
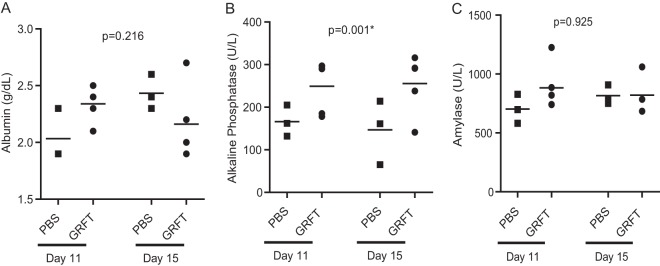
Serum chemistry data for guinea pigs after chronic treatment with GRFT. Levels of serum albumin (A), alkaline phosphatase (B), and amylase (C) were obtained from GRFT- or PBS-treated animals at sacrifice. Bars indicate mean group concentrations.
Since many natural product lectins cause hemagglutination, we investigated GRFT's hemagglutination activity in several species. Blood samples from guinea pig, mouse, sheep, and human were tested. Interestingly, only red blood cells from guinea pigs were affected by GRFT—at concentrations over 5 μg/ml (Fig. 7). As expected, the vehicle (PBS) did not show any hemagglutination activity on erythrocytes, and the known hemagglutinating agent PHA demonstrated activity at concentrations of 5 μg/ml and above for all species tested.
FIG 7.
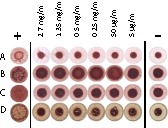
Hemagglutination activity of GRFT. A wide range of GRFT concentrations (5 μg/ml equals 391 nM) was used to examine GRFT's potential hemagglutination activity on erythrocytes from multiple species, including sheep (A), guinea pig (B), human (C), and mouse (D). PHA (5 μg/ml) and PBS were used as positive and negative controls, respectively.
DISCUSSION
In this study, we demonstrated that GRFT persists in serum and plasma of laboratory rodents at concentrations well above the EC50 described for several known enveloped viruses after subcutaneous administration (10, 17, 19, 27). Although decreasing in concentrations after final administration, functionally active concentrations of GRFT, as determined by gp120 binding ELISA, remain in circulation for many days after treatment cessation in regimens of both single and chronic dosing. These findings support further investigations of the utility of GRFT in treatment of both acute and chronic viral infections.
Every drug candidate must show a favorable safety profile to advocate for its further development. Previously, we demonstrated that GRFT was devoid of any mitogenic and cytotoxic activity, was unable to induce cell mediators of inflammation, and had only minimal off-target effects on human cells (14), corroborating the results of other works (10, 15) and unlike other anti-HIV lectins, including CV-N and ConA (28, 29). In the present in vivo studies, GRFT did not alter experimental animal behavior and no animal died as a result of treatment. Using juvenile animals, we did notice that guinea pigs treated with GRFT gained weight significantly more slowly than those injected with PBS. This is not the case when adult mice are treated s.c. with GRFT (unpublished data). Organ toxicity was also assessed both by measuring weights and by histopathology. While liver and spleen percentages displayed a statistically significant increase in comparison with those of the controls, histopathological examination of the organs from GRFT-treated animals did not show any pathology.
When a complete blood count was performed and serum chemistries were analyzed, we observed that most of the parameters were not significantly changed as a result of GRFT treatment. The only exception was RDW. Although significantly different from those of the PBS controls, RDW values obtained from GRFT-treated animals remained within the normal range described for guinea pigs. In serum chemistries, alkaline phosphatase levels were significantly elevated in GRFT-treated guinea pigs. Whether this elevation is related to the increased liver-mass-to-body-weight ratio seen with GRFT-treated animals is yet to be determined since the alkaline phosphatase isotype was not determined.
As a xenogeneic protein, a key concern is a possible immune response to GRFT which could lead to anaphylaxis (30). Our data showing treatment-associated increases in spleen- and liver-mass-to-body-weight ratios is suggestive of a nascent immune response to GRFT treatment. However, we were unable to detect anti-GRFT antibodies in sera from these treated animals, probably reflective of the short duration of these studies. Although GRFT is a relatively weak immunogen, we are able to raise high-titer antibodies in animals immunized with GRFT in the presence of adjuvant; these hyperimmune sera were binding but nonneutralizing (data not shown). Immunogenicity of biologic drugs is a widely acknowledged issue, and our data suggest that future efforts to deimmunize GRFT by structure-guided elimination of T-cell epitopes (31, 32) may be necessary before the product can be used for chronic treatment of viral infection in humans. Lectins are well known for their mitogenic and agglutinating properties (28, 29, 33–36) which prevent their use as therapeutics. We previously addressed the mitogenicity concern by showing that GRFT lacks T-cell mitogenic activity (14). Here, we demonstrated that GRFT does not agglutinate red blood cells from several species, including human, mouse, and sheep. However, we observed that guinea pig erythrocytes were agglutinated by GRFT at high concentrations. The cause of this discrepancy is unclear but may well be associated with the treatment-associated increase in red blood cell width we observed. We conclude that the preliminary toxicity profile of GRFT is acceptable and favors its further development in antiviral prophylaxis and therapy.
GRFT is currently under development as both a topical microbicide and a broad-spectrum antiviral. However, GRFT is not systemically bioavailable after topical administration (our unpublished observations), so parenteral administration is probably necessary to achieve sufficient drug in the systemic concentrations necessary for effective suppression of viral replication. Our data confirm that subcutaneous administration of GRFT is a viable and efficient way to get the drug into systemic circulation to allow a sustained pharmacodynamic effect (37). Our data support the results of published studies on GRFT prophylactic efficacy in murine models of hepatitis C virus and Japanese encephalitis virus infection (17, 18, 21). These studies found that GRFT effectively suppressed viral replication when administered at a 5 mg/kg dose, either subcutaneously or intraperitoneally. We are currently studying whether administrations of GRFT at levels under the 10-mg/kg dosage are sufficient to maintain drug concentrations at potentially therapeutic levels. There is substantial precedent for patient self-administration of drugs via the subcutaneous route; indeed, the peptide HIV fusion inhibitor Enfurtide (T-20) is administered in this fashion. Our data confirm that we can achieve HIV-1 Du156 50% serum neutralization indices in excess of 500 after 14 daily doses of GRFT at 10 mg/kg (Fig. 2), which should be sufficient to inhibit HIV replication and perhaps to promote viral evolution toward enhanced humoral antibody suppression (7, 34, 37). These levels were also more than sufficient to prevent JEV infection in a mouse infection model and HCV infection in in a mouse-humanized liver model (17, 20).
Under selection pressure with CBA, susceptible viruses may evolve toward resistance through loss of key N-linked glycosylation sites (26, 38–41). Interestingly, in the case of HIV-1, CBA resistance correlates with reduced viral fitness and enhanced susceptibility to neutralizing antisera (7, 38, 41). Consequently, it has been suggested that CBA therapy for treatment of chronic viral infections such as those by HIV-1 and HCV may promote viral evolution toward resistance to the CBA concomitant with enhanced susceptibility to host immune control. This concept of CBA-mediated immunotherapy holds considerable appeal as a method to promote durable immune control and perhaps even eradication of HIV infection (7). Given GRFT's antiviral activity in the mid-picomolar range, and our data here that show that the drug accumulates to relevant therapeutic concentrations which are tolerated with minimal toxicity, GRFT is a strong candidate for further experimental testing of this idea first put forward by Balzarini (7).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grant AI076169 to K.E.P. and NIH grant T32-ES011564, DOD grant USAMRMC W81XWH-10-2-CLIN 1, and NIH contract HHSN27201100016C. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
We acknowledge ongoing collaborations with many scientists who have made many key contributions to the development of Griffithsin and other antiviral protein products as broad-spectrum antivirals. We are grateful to Janice Ditslear for help in animal handling and blood work.
Footnotes
Published ahead of print 21 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01407-13.
REFERENCES
- 1.Vigerust DJ, Shepherd VL. 2007. Virus glycosylation: role in virulence and immune interactions. Trends Microbiol. 15:211–218. 10.1016/j.tim.2007.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helle F, Duverlie G, Dubuisson J. 2011. The hepatitis C virus glycan shield and evasion of the humoral immune response. Viruses 3:1909–1932. 10.3390/v3101909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Helle F, Vieyres G, Elkrief L, Popescu CI, Wychowski C, Descamps V, Castelain S, Roingeard P, Duverlie G, Dubuisson J. 2010. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J. Virol. 84:11905–11915. 10.1128/JVI.01548-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doores KJ, Bonomelli C, Harvey DJ, Vasiljevic S, Dwek RA, Burton DR, Crispin M, Scanlan CN. 2010. Envelope glycans of immunodeficiency virions are almost entirely oligomannose antigens. Proc. Natl. Acad. Sci. U. S. A. 107:13800–13805. 10.1073/pnas.1006498107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonomelli C, Doores KJ, Dunlop DC, Thaney V, Dwek RA, Burton DR, Crispin M, Scanlan CN. 2011. The glycan shield of HIV is predominantly oligomannose independently of production system or viral clade. PLoS One 6:e23521. 10.1371/journal.pone.0023521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shan M, Klasse PJ, Banerjee K, Dey AK, Iyer SP, Dionisio R, Charles D, Campbell-Gardener L, Olson WC, Sanders RW, Moore JP. 2007. HIV-1 gp120 mannoses induce immunosuppressive responses from dendritic cells. PLoS Pathog. 3:e169. 10.1371/journal.ppat.0030169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balzarini J. 2007. Targeting the glycans of glycoproteins: a novel paradigm for antiviral therapy. Nat. Rev. Microbiol. 5:583–597. 10.1038/nrmicro1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smee DF, Bailey KW, Wong MH, O'Keefe BR, Gustafson KR, Mishin VP, Gubareva LV. 2008. Treatment of influenza A (H1N1) virus infections in mice and ferrets with cyanovirin-N. Antiviral Res. 80:266–271. 10.1016/j.antiviral.2008.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michelow IC, Lear C, Scully C, Prugar LI, Longley CB, Yantosca LM, Ji X, Karpel M, Brudner M, Takahashi K, Spear GT, Ezekowitz RA, Schmidt EV, Olinger GG. 2011. High-dose mannose-binding lectin therapy for Ebola virus infection. J. Infect. Dis. 203:175–179. 10.1093/infdis/jiq025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mori T, O'Keefe BR, Sowder RC, Jr, Bringans S, Gardella R, Berg S, Cochran P, Turpin JA, Buckheit RW, Jr, McMahon JB, Boyd MR. 2005. Isolation and characterization of griffithsin, a novel HIV-inactivating protein, from the red alga Griffithsia sp. J. Biol. Chem. 280:9345–9353 [DOI] [PubMed] [Google Scholar]
- 11.Moulaei T, Shenoy SR, Giomarelli B, Thomas C, McMahon JB, Dauter Z, O'Keefe BR, Wlodawer A. 2010. Monomerization of viral entry inhibitor griffithsin elucidates the relationship between multivalent binding to carbohydrates and anti-HIV activity. Structure 18:1104–1115. 10.1016/j.str.2010.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ziółkowska NE, O'Keefe BR, Mori T, Zhu C, Giomarelli B, Vojdani F, Palmer KE, McMahon JB, Wlodawer A. 2006. Domain-swapped structure of the potent antiviral protein griffithsin and its mode of carbohydrate binding. Structure 14:1127–1135. 10.1016/j.str.2006.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ziółkowska NE, Shenoy SR, O'Keefe BR, McMahon JB, Palmer KE, Dwek RA, Wormald MR, Wlodawer A. 2007. Crystallographic, thermodynamic, and molecular modeling studies of the mode of binding of oligosaccharides to the potent antiviral protein griffithsin. Proteins 67:661–670. 10.1002/prot.21336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kouokam JC, Huskens D, Schols D, Johannemann A, Riedell SK, Walter W, Walker JM, Matoba N, O'Keefe BR, Palmer KE. 2011. Investigation of griffithsin's interactions with human cells confirms its outstanding safety and efficacy profile as a microbicide candidate. PLoS One 6:e22635. 10.1371/journal.pone.0022635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Keefe BR, Vojdani F, Buffa V, Shattock RJ, Montefiori DC, Bakke J, Mirsalis J, d'Andrea AL, Hume SD, Bratcher B, Saucedo CJ, McMahon JB, Pogue GP, Palmer KE. 2009. Scaleable manufacture of HIV-1 entry inhibitor griffithsin and validation of its safety and efficacy as a topical microbicide component. Proc. Natl. Acad. Sci. U. S. A. 106:6099–6104. 10.1073/pnas.0901506106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferir G, Huskens D, Palmer KE, Boudreaux DM, Swanson MD, Markovitz DM, Balzarini J, Schols D. 2012. Combinations of griffithsin with other carbohydrate-binding agents demonstrate superior activity against HIV type 1, HIV type 2, and selected carbohydrate-binding agent-resistant HIV type 1 strains. AIDS Res. Hum. Retroviruses 28:1513–1523. 10.1089/aid.2012.0026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meuleman P, Albecka A, Belouzard S, Vercauteren K, Verhoye L, Wychowski C, Leroux-Roels G, Palmer KE, Dubuisson J. 2011. Griffithsin has antiviral activity against hepatitis C virus. Antimicrob. Agents Chemother. 55:5159–5167. 10.1128/AAC.00633-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takebe Y, Saucedo CJ, Lund G, Uenishi R, Hase S, Tsuchiura T, Kneteman N, Ramessar K, Tyrrell DL, Shirakura M, Wakita T, McMahon JB, O'Keefe BR. 2013. Antiviral lectins from red and blue-green algae show potent in vitro and in vivo activity against hepatitis C virus. PLoS One 8:e64449. 10.1371/journal.pone.0064449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Keefe BR, Giomarelli B, Barnard DL, Shenoy SR, Chan PK, McMahon JB, Palmer KE, Barnett BW, Meyerholz DK, Wohlford-Lenane CL, McCray PB., Jr 2010. Broad-spectrum in vitro activity and in vivo efficacy of the antiviral protein griffithsin against emerging viruses of the family Coronaviridae. J. Virol. 84:2511–2521. 10.1128/JVI.02322-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexandre KB, Gray ES, Lambson BE, Moore PL, Choge IA, Mlisana K, Karim SS, McMahon J, O'Keefe B, Chikwamba R, Morris L. 2010. Mannose-rich glycosylation patterns on HIV-1 subtype C gp120 and sensitivity to the lectins, Griffithsin, Cyanovirin-N and Scytovirin. Virology 402:187–196. 10.1016/j.virol.2010.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishag HZ, Li C, Huang L, Sun MX, Wang F, Ni B, Malik T, Chen PY, Mao X. 2013. Griffithsin inhibits Japanese encephalitis virus infection in vitro and in vivo. Arch. Virol. 158:349–358. 10.1007/s00705-012-1489-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montefiori DC. 2005. Evaluating neutralizing antibodies against HIV, SIV, and SHIV in luciferase reporter gene assays. Curr. Protoc. Immunol. 64:12.11.1–12.11.17. 10.1002/0471142735.im1211s64 [DOI] [PubMed] [Google Scholar]
- 23.Box GEP, Hunter JS, Hunter WG. 1978. Statistics for experimenters. John Wiley and Sons, Hoboken, NJ [Google Scholar]
- 24.Student B. 1908. The probable error of a mean. Biometrika 6:25 [Google Scholar]
- 25.Wilcoxon F. 1945. Individual comparisons by ranking methods. Biom. Bull. 1:80–83 [Google Scholar]
- 26.Hoorelbeke B, Huskens D, Ferir G, Francois KO, Takahashi A, Van Laethem K, Schols D, Tanaka H, Balzarini J. 2010. Actinohivin, a broadly neutralizing prokaryotic lectin, inhibits HIV-1 infection by specifically targeting high-mannose-type glycans on the gp120 envelope. Antimicrob. Agents Chemother. 54:3287–3301. 10.1128/AAC.00254-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kagiampakis I, Gharibi A, Mankowski MK, Snyder BA, Ptak RG, Alatas K, LiWang PJ. 2011. Potent strategy to inhibit HIV-1 by binding both gp120 and gp41. Antimicrob. Agents Chemother. 55:264–275. 10.1128/AAC.00376-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huskens D, Vermeire K, Vandemeulebroucke E, Balzarini J, Schols D. 2008. Safety concerns for the potential use of cyanovirin-N as a microbicidal anti-HIV agent. Int. J. Biochem. Cell Biol. 40:2802–2814. 10.1016/j.biocel.2008.05.023 [DOI] [PubMed] [Google Scholar]
- 29.Painter RG, White A. 1976. Effect of concanavalin A on expression of cell surface sialyltransferase activity of mouse thymocytes. Proc. Natl. Acad. Sci. U. S. A. 73:837–841. 10.1073/pnas.73.3.837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith CM, Bradding P, Neill DR, Baxendale H, Felici F, Andrew PW. 2011. Novel immunogenic peptides elicit systemic anaphylaxis in mice: implications for peptide vaccines. J. Immunol. 187:1201–1206. 10.4049/jimmunol.1002152 [DOI] [PubMed] [Google Scholar]
- 31.Parker AS, Choi Y, Griswold KE, Bailey-Kellogg C. 2013. Structure-guided deimmunization of therapeutic proteins. J. Comput. Biol. 20:152–165. 10.1089/cmb.2012.0251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi Y, Griswold KE, Bailey-Kellogg C. 2013. Structure-based redesign of proteins for minimal T-cell epitope content. J. Comput. Chem. 34:879–891. 10.1002/jcc.23213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gavrovic-Jankulovic M, Poulsen K, Brckalo T, Bobic S, Lindner B, Petersen A. 2008. A novel recombinantly produced banana lectin isoform is a valuable tool for glycoproteomics and a potent modulator of the proliferation response in CD3+, CD4+, and CD8+ populations of human PBMCs. Int. J. Biochem. Cell Biol. 40:929–941. 10.1016/j.biocel.2007.10.033 [DOI] [PubMed] [Google Scholar]
- 34.Koshte VL, van Dijk W, van der Stelt ME, Aalberse RC. 1990. Isolation and characterization of BanLec-I, a mannoside-binding lectin from Musa paradisiac (banana). Biochem. J. 272:721–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ooi LS, Sun SS, Ooi VE. 2004. Purification and characterization of a new antiviral protein from the leaves of Pandanus amaryllifolius (Pandanaceae). Int. J. Biochem. Cell Biol. 36:1440–1446. 10.1016/j.biocel.2004.01.015 [DOI] [PubMed] [Google Scholar]
- 36.Liu B, Xu XC, Cheng Y, Huang J, Liu YH, Liu Z, Min MW, Bian HJ, Chen J, Bao JK. 2008. Apoptosis-inducing effect and structural basis of Polygonatum cyrtonema lectin and chemical modification properties on its mannose-binding sites. BMB Rep. 41:369–375. 10.5483/BMBRep.2008.41.5.369 [DOI] [PubMed] [Google Scholar]
- 37.Prettyman J. 2005. Subcutaneous or intramuscular? Confronting a parenteral administration dilemma. Medsurg. Nurs. 14:93–98; quiz 99 [PubMed] [Google Scholar]
- 38.Balzarini J, Van Laethem K, Peumans WJ, Van Damme EJ, Bolmstedt A, Gago F, Schols D. 2006. Mutational pathways, resistance profile, and side effects of cyanovirin relative to human immunodeficiency virus type 1 strains with N-glycan deletions in their gp120 envelopes. J. Virol. 80:8411–8421. 10.1128/JVI.00369-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Witvrouw M, Fikkert V, Hantson A, Pannecouque C, O'Keefe BR, McMahon J, Stamatatos L, de Clercq E, Bolmstedt A. 2005. Resistance of human immunodeficiency virus type 1 to the high-mannose binding agents cyanovirin N and concanavalin A. J. Virol. 79:7777–7784. 10.1128/JVI.79.12.7777-7784.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smee DF, Wandersee MK, Checketts MB, O'Keefe BR, Saucedo C, Boyd MR, Mishin VP, Gubareva LV. 2007. Influenza A (H1N1) virus resistance to cyanovirin-N arises naturally during adaptation to mice and by passage in cell culture in the presence of the inhibitor. Antiviral Chem. Chemother. 18:317–327 [DOI] [PubMed] [Google Scholar]
- 41.Huang X, Jin W, Griffin GE, Shattock RJ, Hu Q. 2011. Removal of two high-mannose N-linked glycans on gp120 renders human immunodeficiency virus 1 largely resistant to the carbohydrate-binding agent griffithsin. J. Gen. Virol. 92:2367–2373. 10.1099/vir.0.033092-0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.