

Abstract
Cyclooxygenase-2 (COX-2), the rate-limiting enzyme for prostanoid biosynthesis, plays a key role in gastrointestinal carcinogenesis. Among various prostanoids, prostaglandin E2 (PGE2) appears to be most responsible for cancer development. To investigate the role of PGE2 in gastric tumorigenesis, we constructed transgenic mice simultaneously expressing COX-2 and microsomal prostaglandin E synthase (mPGES)-1 in the gastric epithelial cells. The transgenic mice developed metaplasia, hyperplasia and tumorous growths in the glandular stomach with heavy macrophage infiltrations. Although gastric bacterial counts in the transgenic mice were within the normal range, treatment with antibiotics significantly suppressed activation of the macrophages and tumorous hyperplasia. Importantly, the antibiotics treatment did not affect the macrophage accumulation. Notably, treatment of the transgenic mice with lipopolysaccharides induced proinflammatory cytokines through Toll-like receptor 4 in the gastric epithelial cells. These results indicate that an increased level of PGE2 enhances macrophage infiltration, and that they are activated through epithelial cells by the gastric flora, resulting in gastric metaplasia and tumorous growth. Furthermore, Helicobacter infection upregulated epithelial PGE2 production, suggesting that the COX-2/mPGES-1 pathway contributes to the Helicobacter-associated gastric tumorigenesis.
Keywords: COX-2, gastric hyperplasia, Helicobacter, macrophages, mPGES-1
Introduction
Regular use of nonsteroidal anti-inflammatory drugs (NSAIDs) is associated with a reduced risk of cancer development in the gastrointestinal tract (Thun et al, 1993; Farrow et al, 1998). The best-known targets of NSAIDs are cyclooxygenase (COX)-1 and COX-2 isoenzymes for prostaglandin (PG)H2 synthesis. It has been established that inhibition of COX-2 is an effective chemoprevention strategy for colorectal cancer (Taketo, 1998a, 1998b). Using ApcΔ716 mice, a model for familial adenomatous polyposis, we have demonstrated that COX-2 plays a key role in the intestinal polyposis (Oshima et al, 1996, 2001). Accumulating evidence shows that COX-2 expression is also induced in the gastric cancer tissues (Ristimaki et al, 1997; Soydan et al, 1997). Gastric cancer is the second most common cancer in the world (Correa, 2003), and Helicobacter pylori infection is listed as a ‘class I carcinogen' (IARC, 1994). In the H. pylori-associated gastritis and adenocarcinoma, COX-2 is induced significantly, whereas its level is decreased dramatically upon H. pylori eradication (Fu et al, 1999; Sung et al, 2000). Furthermore, treatment of the H. pylori-infected mice with NSAIDs or COX-2 inhibitors suppresses the gastric hyperplasia (Kim et al, 2001; Xiao et al, 2001). These results suggest that COX-2 plays a pivotal role in the H. pylori-associated gastric tumorigenesis.
PGE2 is one of the key prostanoids responsible for tumorigenesis. In the ApcΔ716 polyps, PGE2 signaling through its receptor EP2 is responsible for angiogenesis and construction of the basement membrane (Sonoshita et al, 2001; Seno et al, 2002). Other effects of PGE2 on tumorigenesis have also been suggested, such as increased cell survival and motility (Sheng et al, 1998, 2001), inhibition of host immune responses (Huang et al, 1998) and activation of the EGF receptor signal (Pai et al, 2002). Moreover, it has been demonstrated that a long-term treatment of rats with 16,16-dimethyl PGE2 increases mucosal thickness in the stomach, accompanied by elevated numbers of mucosal cells (Reinhart et al, 1983). However, the biological mechanism(s) of how PGE2 is involved in tumorigenesis remains to be elucidated.
It has been reported that microsomal PGE synthase (mPGES)-1, one of the PGE2 synthases, is co-localized and functionally coupled with COX-2 (Murakami et al, 2000; Lazarus et al, 2002). Furthermore, induction of mPGES-1 is also accompanied by COX-2 induction in colon cancer tissues (Yoshimatsu et al, 2001). Therefore, it is possible that a simultaneous induction of both COX-2 and mPGES-1 stimulates tumorigenesis synergistically.
To investigate the precise mechanism of the PGE2 action on the gastric epithelial cell growth, we have constructed transgenic mice simultaneously expressing both COX-2 and mPGES-1 in the gastric mucosa. Here we demonstrate that the transgenic mice develop hyperplastic gastric tumors, and that the increased levels of PGE2 cause these phenotypes through recruitment of mucosal macrophages and their stimulation mediated through epithelial Toll-like receptor (TLR) 4 activated by the indigenous bacterial flora.
Results
The transgenic mice express COX-2 and mPGES-1 in glandular stomach
To investigate the effects of PGE2 on the gastric mucosa, we constructed transgenic mice that express both COX-2 and mPGES-1 by co-microinjection of two expression vectors (Figure 1A). We used cytokeratin 19 (K19) promoter to target the epithelial cells of gastric mucosa (Brembeck et al, 2001). Two transgenic lines, K19-C2mE-2 and K19-C2mE-8, showed high expression levels of the transduced genes and essentially the same phenotypes (data not shown). Accordingly, we present the results with K19-C2mE-8 (hereafter K19-C2mE) mice. The K19-C2mE mice expressed the transgene-encoded COX-2 and mPGES-1 in the forestomach and glandular stomach, whereas expression of the endogenous COX-2 was undetectable and mPGES-1 was weakly expressed in the whole digestive tract (Figure 1B). The PGE2 levels in the glandular stomach of K19-C2mE mice were approximately twice as high as those in the wild-type mice (Figure 1C). These results indicate that forced expression of COX-2 and mPGES-1 efficiently stimulates PGE2 biosynthesis in the gastric epithelial cells.
Figure 1.
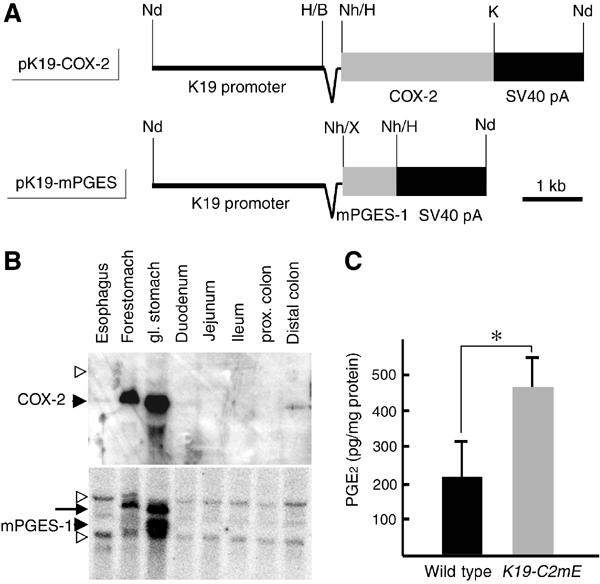
Expression of COX-2 and mPGES-1, and production of PGE2 in K19-C2mE stomach. (A) Transgenic vectors for COX-2 and mPGES-1 expression, respectively. Synthetic chimeric intron is indicated as a wedge-shaped line between the K19 promoter and each cDNA. Fragments of cDNAs and SV40 pA cassettes are indicated as gray and black boxes, respectively. Nd, NdeI; H, HindIII; B, BamHI; K, KpnI; Nh, NheI; and X, XbaI. (B) Northern blots for COX-2 and mPGES-1 mRNAs in the K19-C2mE mouse digestive tract. Filled arrowheads indicate the mRNA sizes for transgenic genes, whereas open arrowheads indicate the sizes for endogenous genes. The arrow on the bottom panel indicates the residual COX-2 band because of the reprobing of the same filter as the top. (C) PGE2 levels in the glandular stomach are presented as the mean±s.d. *P<0.05.
The K19-C2mE mice show aberrant differentiation of the gastric mucosa
In the transgenic mice, COX-2 and mPGES-1 were detected essentially in the surface epithelial cells of glandular stomach (Figure 2A). At 12 weeks of age, the K19-C2mE mice showed abnormal gastric histology of the glandular stomach, with elongated gastric pits. Furthermore, the mucous cell population was found expanded in the whole gland, as detected by Helix pometia-lectin staining (Figure 2B). The numerous mucous cells in K19-C2mE mice contained acidic mucins stained with Alcian blue, which were not produced in the normal gastric mucosa. These results indicate that the increased level of PGE2 disturbs normal differentiation of epithelial cells of the gastric mucosa.
Figure 2.
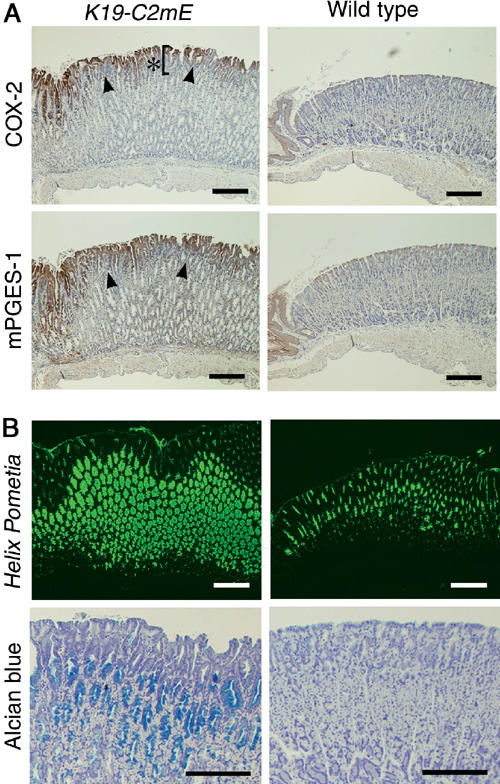
Histological analyses of the glandular stomach at 12 weeks of age, in the K19-C2mE mice (left) and wild type (right). (A) Immunohistochemistry for COX-2 (top) and mPGES-1 (bottom). Arrowheads indicate the positive staining on mucosal surface. Asterisk in the top left panel indicates elongated pit. (B) Mucous metaplasia in the gastric gland. Helix pometia-lectin staining (top) and Alcian blue staining (bottom). Bars in (A) and (B), 200 μm.
K19-C2mE mice develop gastric hyperplasia and tumorous growths
We next determined the cell proliferation rate by bromodeoxyuridine (BrdU) incorporation. In the wild-type mice, proliferating cells were detected only in the neck of the gastric gland (Figure 3A). In the K19-C2mE mice, however, the BrdU-labeled cells were found in the gland bottom as well as in the neck. Moreover, the labeling index per gland in the K19-C2mE stomach was twice higher than that in the wild-type mice (Figure 3B). At 48 weeks of age, large tumorous growths were found in the proximal glandular stomach (Figure 3C). Histopathology of the tumors showed benign metaplastic hyperplasia consisting of Alcian blue-positive mucous cells and other types of differentiated epithelial cells. We confirmed expression of both COX-2 and mPGES-1 in the tumor epithelial cells by immunohistochemistry.
Figure 3.
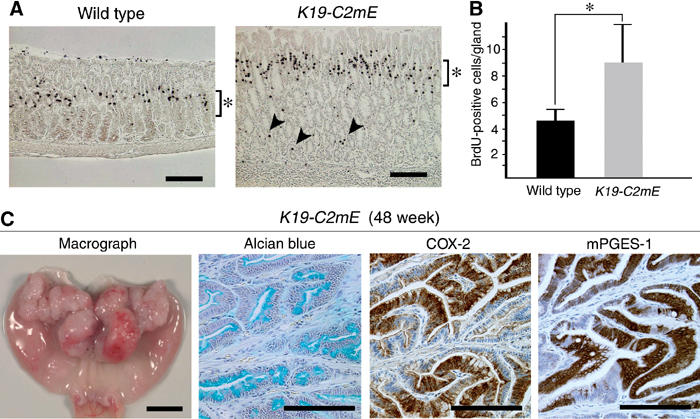
Hyperplasia and tumor development in the K19-C2mE glandular stomach. (A) BrdU staining after 1 h labeling at 12 weeks of age. Asterisks indicate BrdU-labeled cells in the gland neck. Arrowheads in the K19-C2mE stomach indicate BrdU-positive cells in the gland bottom. Bars, 500 μm. (B) BrdU labeling indices are presented as the mean±s.d. *P<0.05. (C) Tumor development at 48 weeks. From left to right: macroscopic photograph, Alcian blue staining, immunostaining for COX-2 and mPGES-1. Bars in macrograph and histological sections, 5 mm and 200 μm, respectively.
To examine whether the gastric tumorous hyperplasia is dependent on the COX-2 activity, we treated the mice with a COX-2 selective inhibitor NS-398. In the no-drug control mice, the gastric mucosal thickness at the proximal stomach increased gradually with age, to more than twice of that in the wild type by 20 weeks (Figure 4A and B). However, treatment of K19-C2mE mice with NS-398 for 4 weeks completely suppressed the gastric hypertrophy, reducing the mucosal thickness to that in the age-matched wild type. These results clearly indicate that the increased levels of PGE2 are essential for the gastric pathology in K19-C2mE mice.
Figure 4.
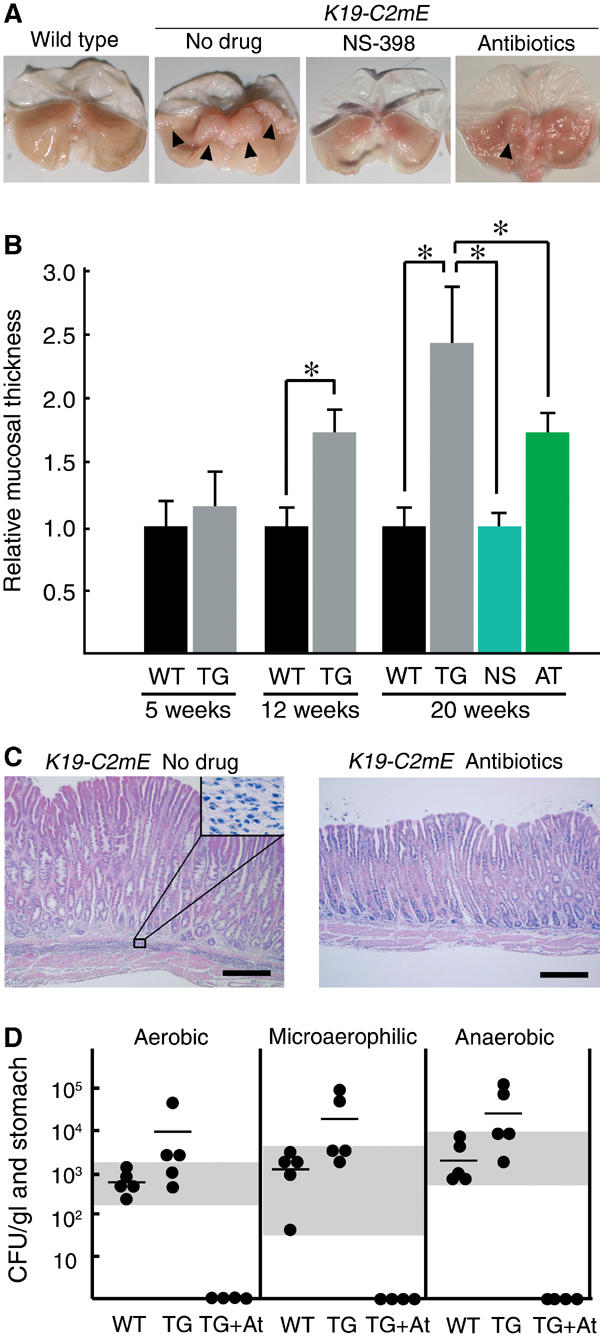
Suppression of gastric hyperplasia either by treatment with a COX-2 inhibitor (NS-398) or antibiotics. (A) Macroscopic photographs of the stomach at 20 weeks of age of the wild-type (left) and K19-C2mE (right) mice, with respective treatments as indicated. Arrowheads in ‘no-drug' indicate the hypertrophic lesion. In the antibiotics-treated stomach, a mild hypertrophic lesion is still found (arrowhead). (B) Relative mucosal thickness at 5, 12 and 20 weeks of age is presented as the mean±s.d. (i.e., the mean thickness divided by that of the wild type). WT, wild type; TG, nontreated K19-C2mE; NS and AT, K19-C2mE treated with NS-398 and antibiotics, respectively. *P<0.05. (C) Histopathology of the glandular stomach in the untreated (left) and antibiotics-treated (right) K19-C2mE mice, respectively (H&E staining). Inset in the left panel shows a higher magnification of the mononuclear cell-infiltrated submucosa. Bars, 300 μm. (D) Bacterial counts in the glandular stomach at 20 weeks of age. CFUs per mouse glandular stomach are indicated as closed circles. Horizontal bars indicate the mean CFU. Culture conditions are shown on top. WT, wild type; TG, K19-C2mE; TG+At, K19-C2mE with antibiotics. Gray horizontal zones indicate the ranges of bacterial counts in wild-type mice.
Bacterial infection triggers gastric hyperplasia in K19-C2mE mice
Numerous mononuclear cells were found infiltrating in the submucosa in K19-C2mE mice (Figure 4C). To determine whether gastric bacterial flora was responsible for these inflammatory responses and gastric hyperplasia, we treated K19-C2mE mice with antibiotics, streptomycin and cefoperazone for 3 weeks. The treatment eliminated gastric bacteria to undetectable levels (Figure 4D) and cleared the submucosal infiltration of mononuclear cells (Figure 4C). Furthermore, such a treatment decreased the mucosal thickness remarkably from 2.4 to 1.7 times of the wild type (Figure 4A and B). It is established that the same antibiotics with the same dosing protocol as in this study do not affect cell proliferation in the mouse gastric mucosa (Zavros et al, 2002a), excluding the possibility that the suppressive effect was caused directly by the antibiotics against the mucous cells.
We next determined gastric bacterial counts under aerobic, microaerophilic and anaerobic conditions, respectively. The mean bacterial counts of the K19-C2mE mice were higher than those in the wild type under all culture conditions. However, the bacterial counts were still within the normal range in nearly half of the transgenic mice (gray zones in Figure 4D), although all transgenic mice developed gastric hypertrophy regardless of their bacterial counts (data not shown). These results, taken together, indicate that host inflammatory responses in K19-C2mE mice can be triggered by the normal gastric flora and cause the gastric hyperplasia and hypertrophy. Increased bacterial counts in some transgenic mice are possibly caused by the weakened gastric acidity in the metaplastic epithelial cells (Figure 2B).
Lipopolysaccharide (LPS) stimulates gastric epithelial cells through TLR4
Upon close histological examination of the K19-C2mE transgenic mice, we could not find any signs of bacterial invasion or colonization in the gastric mucosa (data not shown). Accordingly, it was expected that epithelial cells, rather than stromal cells, were stimulated directly by the gastric bacteria. TLR2 and TLR4 recognize such bacterial pathogens as peptidoglycan (PGN) and LPS, respectively, and play important roles in innate immune responses (Kaisho and Akira, 2002). We found expression of both TLR2 and TLR4 mRNAs by RT–PCR in the total RNA from the whole glandular stomach (data not shown). Interestingly, we detected only TLR4 mRNA in the gastric mucosa excised by the laser microdissection (LMD) method, and in the primary culture of the gastric epithelial cells (Figure 5A). Consistent with the results, treatment of the gastric epithelial cells with PGN did not induce tumor necrosis factor (TNF)-α significantly (Figure 5B). In contrast, LPS stimulation induced TNF-α in a dose-dependent manner, although the expression level was ∼1000-fold lower than that detected in the LPS-stimulated mouse monocyte line, RAW264 (Figure 5C). We also detected expression of interleukin (IL)-1β in the LPS-stimulated epithelial cells by RT–PCR (data not shown). Induction of TNF-α was efficiently suppressed by treatment with TLR4-blocking antibody in both the gastric epithelia and RAW264, although the antibody itself could induce TNF-α slightly (Figure 5C). Interestingly, the levels of TLR4 expression and TNF-α induction were essentially the same in K19-C2mE and wild-type gastric epithelial cells. Therefore, it is possible that normal gastric flora stimulates the epithelial cells through TLR4 weakly but continuously both in the wild-type and transgenic mice, and that the TLR4 activation in the epithelial cells indirectly induces inflammatory responses in the macrophage-accumulated K19-C2mE stomach. We did not detect any TNF-α or IL-1β in the epithelial cells by immunohistochemistry, possibly because of low expression levels.
Figure 5.
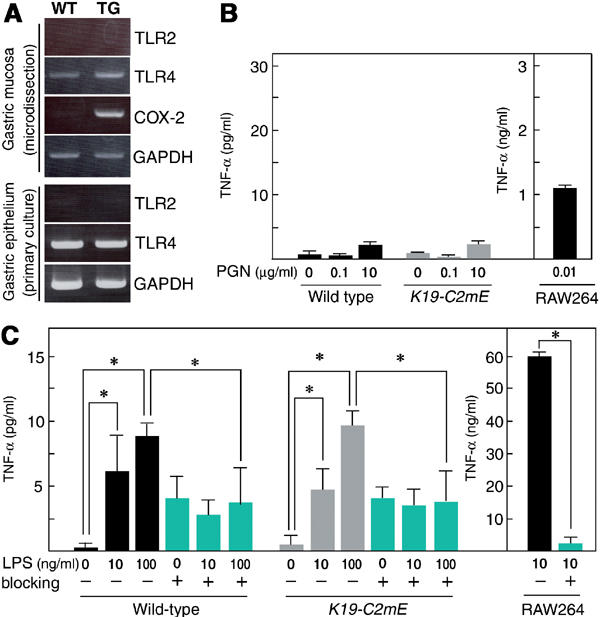
Stimulations of TLR on the gastric epithelial cells with bacterial PGN or LPS. (A) RT–PCR for TLR2 and TLR4 mRNAs using total RNA from gastric mucosa sampled by LMD (top) and primary culture of the epithelial cells (bottom), respectively. COX-2 in LMD-sample RT–PCR was used as positive control. GAPDH was used as endogenous controls. WT, wild type; TG, K19-C2mE. Gastric epithelial cells were treated with PGN (B) and LPS (C) for 20 h. Concentrations of TNF-α in the supernatants are presented as the mean±s.d. RAW264 mouse monocyte cells were used for positive control. For TLR4 blocking, anti-TLR4 antibody was added at 10 μg/ml (blue bars). *P<0.05.
Inflammatory cytokines, chemokines and growth factors are induced in K19-C2mE mice
To characterize the tissue microenvironment that caused gastric hyperplasia and hypertrophy, we next determined mRNA levels for various proinflammatory cytokines, chemokines and growth factors in the whole glandular stomach. Among them, only TNF-α and CXCL14 (BRAK) were increased significantly at 3 weeks of age (Figure 6A). At 20 weeks, the mRNA levels for proinflammatory cytokines IL-1β and IL-6 and chemokine MIP-2 (murine homolog of CXCL2/3) were increased significantly in addition to TNF-α and CXCL14 (Figure 6B). These results were consistent with the inflammatory histopathology (Figure 4C). Expression of these cytokines and chemokines was found in the stroma by immunohistochemistry (Figures 7C and 8A; data not shown). The levels of granulocyte-macrophage colony-stimulating factor (GM-CSF), hepatocyte growth factor (HGF) and vascular endothelial growth factor (VEGF) were also increased significantly at 20 weeks of age.
Figure 6.
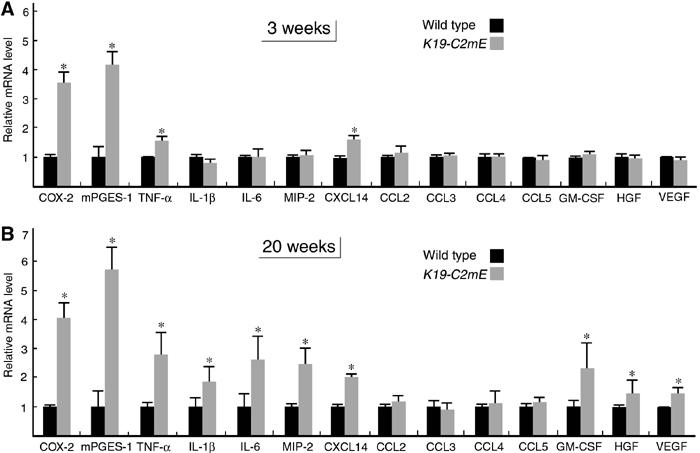
mRNA levels for cytokines, chemokines and growth factors in the glandular stomach at 3 weeks (A) and 20 weeks (B) of age. Relative mRNA levels in K19-C2mE mice compared with those in the wild type are presented as the mean±s.d. *P<0.05 versus wild type.
Figure 7.
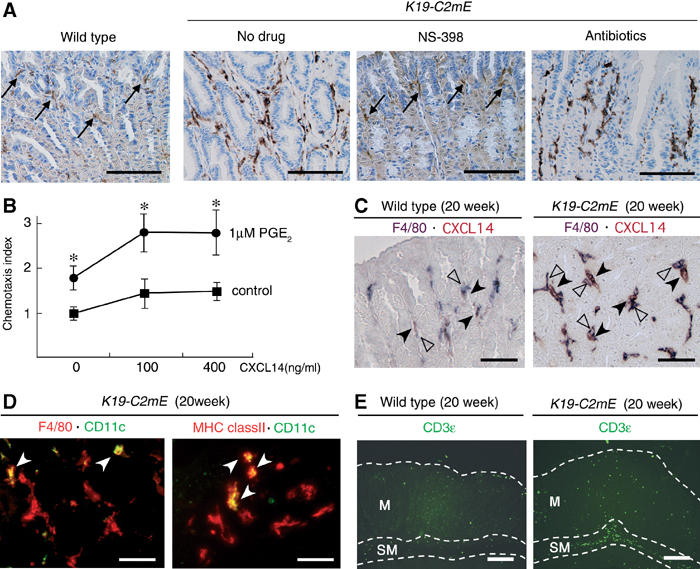
Macrophage infiltration in the glandular stomach of K19-C2mE mice. (A) Immunostaining for macrophage marker F4/80 at 20 weeks of age. Arrows in wild type and NS-398-treated K19-C2mE indicate scarce macrophages. Numerous macrophages are found infiltrating in the ‘no-drug' control K19-C2mE mucosa. Note that a similar macrophage infiltration is still found even after the antibiotics treatment. Bars, 200 μm. (B) Relative chemotaxis index of RAW264 cells to CXCL14. Cells were cultured in the presence (circles) or absence (squares) of 1 μM PGE2 for 20 h prior to migration assays. *P<0.05 versus untreated control cells. (C) Double immunostaining for F4/80 (purple) and CXCL14 (brown) in the wild type (left) and K19-C2mE (right) at 20 weeks of age. Closed arrowheads indicate the CXCL14-expressing stromal cells, whereas open arrowheads show F4/80-positive macrophages. Bars, 100 μm. (D) Merged images of the double fluorescence immunostaining for F4/80 (red) and CD11c (green) (left), and MHC class II (red) and CD11c (green) (right). White arrowheads indicate double-positive cells (yellow). Bars, 50 μm. (E) Fluorescence immunostaining for CD3ɛ (green). Boundaries for mucosa (lamina propria) (M) and submucosa (SM) are drawn with a white dashed line. Bars, 200 μm.
Figure 8.
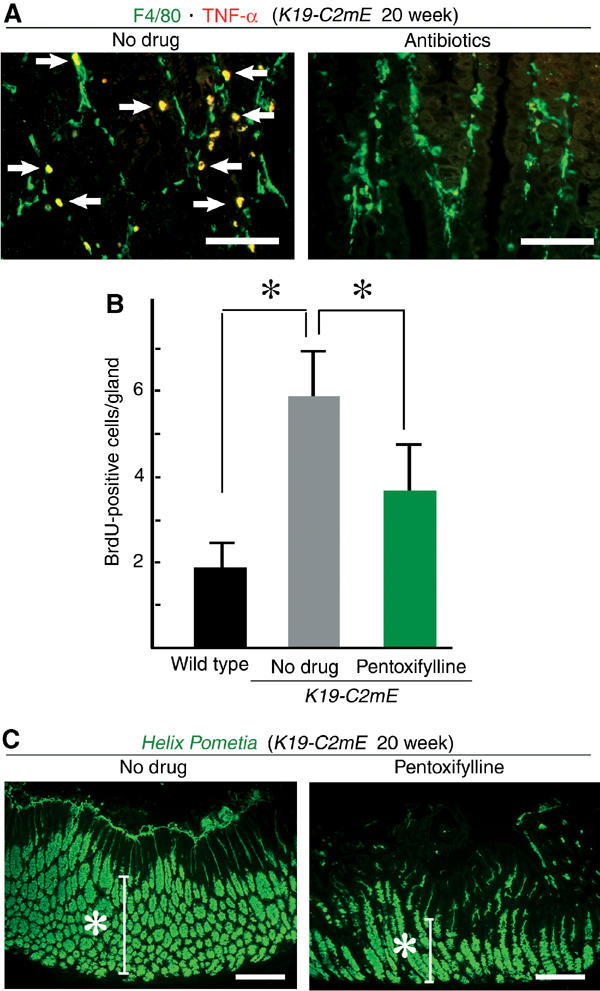
Macrophage TNF-α in gastric hyperplasia. (A) Merged images of double immunostaining for F4/80 (green) and TNF-α (red) in K19-C2mE mice at 20 weeks of age. No-drug control (left) and antibiotics-treated (right). White arrows indicate double-positive cells (yellow). Bars, 200 μm. (B) BrdU labeling indices in the glandular stomach of the wild type, no-drug K19-C2mE and pentoxifylline-treated K19-C2mE mice are presented as the mean±s.d. *P<0.05. (C) Helix pometia-lectin staining in a K19-C2mE mouse at 20 weeks of age. No-drug control (left) and pentoxifylline-treated (right). Bars, 200 μm.
Increased PGE2 levels help recruit macrophages in the glandular stomach
To investigate whether mucosal macrophages were involved in inflammatory responses in K19-C2mE, we next examined the glandular stomach immunohistochemically with a macrophage marker F4/80 (Figure 7A). In the wild-type mice, tissue macrophages were sparsely scattered in the mucosal stroma. In contrast, a heavy macrophage infiltration to mucosal stroma was found in K19-C2mE mice. Treatment of K19-C2mE mice with NS-398 effectively suppressed the macrophage infiltration to the wild-type level. However, numerous macrophages were still found in the stomach of the antibiotics-treated mice, despite the fact that the bacterial infection was eliminated (Figure 4D). Accordingly, the increased PGE2 level is the likely primary cause of the enhanced macrophage infiltration, regardless of the epithelial responses to gastric bacteria.
CXCL14 is a homeostatic chemokine that plays a role in monocyte recruitment to normal tissues (Muller, 2001), and cultured monocytes become more responsive to CXCL14 in the presence of PGE2 (Kurth et al, 2001). We confirmed that treatment of RAW264 cells with PGE2 significantly enhanced chemotaxis to CXCL14 (Figure 7B). Consistent with the RT–PCR results, many stromal cells of the K19-C2mE glandular stomach showed strong staining for CXCL14, whereas only a small number of cells were stained weakly in the wild-type stomach (Figure 7C). Numerous CXCL14-positive stromal cells were also found in the antibiotics-treated stomach (data not shown), suggesting that production of CXCL14 was stimulated by PGE2, rather than bacterial infection. Importantly, we found F4/80-positive macrophages adjacent to the CXCL14-producing cells, which suggests that CXCL14 helps recruit macrophages in the K19-C2mE gastric mucosa. Notably, treatment of RAW264 cells with PGE2 enhanced cell migration even in the absence of CXCL14 (Figure 7B). As RAW264 does not express CXCL14 (data not shown), other factors may also be involved in PGE2-stimulated macrophage infiltration. Although CCL2 (monocyte chemotactic protein (MCP)-1), CCL3 (MIP-1α), CCL4 (MIP-1β) and CCL5 (RANTES) can also recruit monocytes or macrophages (Homey et al, 2002), these chemokines are unlikely to have caused macrophage infiltration in K19-C2mE mice because their expression levels were not increased (Figure 6B).
We also characterized other types of infiltrating cells in the K19-C2mE gastric mucosa. When double stained with an antibody for dendritic cell marker CD11c, together with that for F4/80 (Figure 7D), only a small subpopulation of the F4/80-positive cells were stained for CD11c. Interestingly, there were numerous cells expressing the MHC class II antigen, but only a few of them were CD11c+ dendritic cells (Figure 7D), suggesting that the majority of the F4/80-positive cells consisted of activated macrophages. Consistent with the histopathology (Figure 4C), most T cells visualized by CD3ɛ were found in the submucosa of K19-C2mE mice (Figure 7E). In the gastric mucosa (lamina propria), however, the number of T cells was slightly higher in K19-C2mE mice than in the wild type. Moreover, we found B220+ B cells and Gr-1+ granulocytes mainly in the submucosa, and only rarely in the mucosa (data not shown).
Suppression of proinflammatory cytokines reduces proliferation of the gastric epithelial cells
We next determined the cell type(s) that expresses TNF-α in the mucosal stroma by fluorescence double immunostaining with F4/80 antibody. Abundant TNF-α was detected primarily in the mucous macrophages in K19-C2mE mice (Figure 8A). In contrast, macrophages in the antibiotics-treated mice seldom produced TNF-α, indicating that gastric bacterial flora indirectly induced TNF-α in the infiltrating macrophages.
TNF-α is a key mediator of inflammation, and also plays important roles in the early stages of tumorigenesis (Moore et al, 1999; Knight et al, 2000). In addition, activated macrophages produce other proinflammatory cytokines, such as IL-1 and IL-6 (Cavaillon, 1994), levels of which were elevated also in K19-C2mE mice (Figure 6). To investigate the effects of these cytokines on the gastric hyperplasia, we treated K19-C2mE mice with pentoxifylline, which inhibits production of TNF-α, IL-1 and IL-6 (Voisin et al, 1998). Only after a short-term treatment (5 days), pentoxifylline substantially reduced the BrdU-labeling index of the epithelial cells in K19-C2mE mice compared with that in the untreated controls (Figure 8B). The number of metaplastic mucous cells in the glandular stomach was also decreased in the pentoxifylline-treated mice (Figure 8C). Therefore, the proinflammatory cytokines produced by activated macrophages should significantly contribute to the gastric metaplasia and hyperplasia in the transgenic mice.
Helicobacter infection induces PGE2 production in gastric epithelial cells
It has been reported that infection of mice with H. pylori or its close relative H. felis results in mucous metaplasia and hyperplasia of the glandular stomach (Lee et al, 1990, 1997; Fox et al, 1996). Therefore, we examined whether the COX-2/mPGES-1 pathway is involved in the gastric phenotypes of Helicobacter infection models, using frozen infected tissues and primary culture of the gastric epithelial cells. In the surface epithelial cells of mouse glandular stomach, in vivo infection of H. felis induced COX-2 mRNA, while the mPGES-1 mRNA increased only slightly (Figure 9A). In the primary gastric epithelial cells, H. felis infection also increased both COX-2 and mPGES-1 (Figure 9A). Consistently, the PGE2 level in the primary culture medium of H. felis-infected cells was two times higher than that of the control (Figure 9B). Moreover, H. felis infection stimulated expression of TNF-α in both wild-type and K19-C2mE epithelial cells at the same level (Figure 9C). These results are consistent with a recent report that COX-2 is induced in the mouse gastric epithelial cells in vivo by H. pylori infection (Jüttner et al, 2003). On the other hand, it has been reported that H. pylori LPS stimulates TLR4 of gastric pit cells (Kawahara et al, 2001), whereas H. pylori LPS and flagellin stimulate TLR2 and TLR5 of gastric cancer cells, respectively (Smith et al, 2003). Moreover, H. pylori proteins, membrane protein 1 and urease B, can induce TNF-α (Suganuma et al, 2001). While multiple mechanisms may be involved, our present results establish that H. felis infection induces TNF-α in the normal gastric epithelial cells. Therefore, activation of the COX-2/mPGES-1 pathway, together with induction of TNF-α in the gastric epithelial cells, contributes to the gastric phenotypes by the Helicobacter infection.
Figure 9.
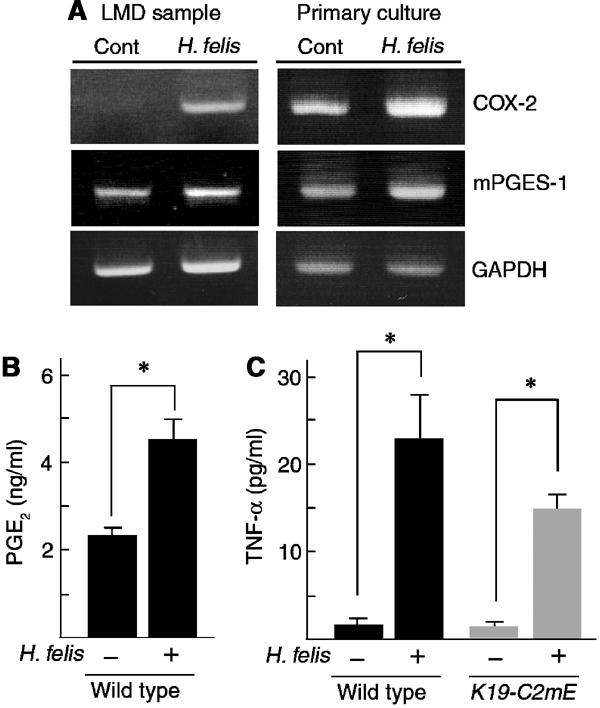
Infection of H. felis to gastric epithelial cells. (A) RT–PCR for COX-2 and mPGES-1 mRNA in surface epithelial cells of the glandular stomach of H. felis-infected and control mice sampled by LMD (left) and those of H. felis-infected and control primary culture of gastric epithelial cells (right). GAPDH was used for endogenous control. Note that the expression of COX-2 and mPGES-1 is increased by H. felis infection both in vivo and in vitro. (B) PGE2 levels in the culture medium of H. felis-infected and control gastric epithelial cells are presented as the mean±s.d. *P<0.05. (C) Gastric epithelial cells of wild-type and K19-C2mE were infected with H. felis. Concentrations of induced TNF-α in supernatants are presented as the mean±s.d. *P<0.05 versus uninfected cells.
Discussion
Increased PGE2 level in the gastric mucosa enhances recruitment of tissue macrophages
It is widely accepted that a functional relationship exists between inflammation and tumor cell growth (Balkwill and Mantovani, 2001; Coussens and Werb, 2002). It is estimated that over 15% of cancer in the world is caused by infections that often induce persistent chronic inflammations in the hosts (Kuper et al, 2000). Activated macrophages in the inflammatory sites may supply cytokines and growth factors to the tumor epithelial cells. These tumor-associated macrophages (TAM) are derived from monocytes that have been recruited largely by chemokine MCPs (Balkwill and Mantovani, 2001; Coussens and Werb, 2002). Here we have demonstrated that an increased PGE2 level, which is characteristic of inflammatory responses, plays an important role in macrophage infiltration into the gastric stroma. As enhanced macrophage infiltration was suppressed by COX-2 inhibition, but not by elimination of the gastric bacterial flora, we conclude that increased levels of PGE2 play a crucial role in recruiting monocytes into the gastric mucosa of K19-C2mE mice.
It has been demonstrated that circulating monocytes migrate to the normal (i.e. un-inflamed) tissues guided by constitutively expressed chemokine CXCL14 (Kurth et al, 2001; Muller, 2001). Furthermore, PGE2 is shown to upregulate dramatically the monocyte responsiveness to CXCL14 (Kurth et al, 2001). Considering the facts that CXCL14 expression is increased in K19-C2mE mice (Figure 6 and 7C) and that macrophages are found adjacent to CXCL14-expressing cells (Figure 7C), it is conceivable that CXCL14 is one of the key factors for mucosal macrophage infiltration. As PGE2 treatment stimulates migration of RAW264 cells in the absence of CXCL14 (Figure 7B), additional factors may also be involved in the PGE2-dependent monocyte recruitment.
Macrophage activation is responsible for gastric hyperplasia
It has been well documented that infections of H. pylori and its close relative H. felis cause chronic inflammation and hyperplasia in the mouse glandular stomach (Lee et al, 1990, 1997; Fox et al, 1996). Infection of mice with other bacterial species, H. heilmannii or Acinetobacter lwoffi, can induce gastric metaplasia and hyperplasia (Peterson et al, 2001; Zavros et al, 2002a). Importantly, such gastric mucosal hypertrophy/nodular hyperplasia by H. felis was not induced in T-cell-deficient mice, RAG-1−/− or TCRβδ−/− mutants (Roth et al, 1999). These results indicate that host immune responses are essential for the gastric pathology. In the present study, we have provided direct evidence that host inflammatory responses to the normal gastric flora can cause similar phenotypes. Moreover, we have demonstrated that bacterial LPS stimulates TLR4 on the gastric epithelial cells to induce proinflammatory cytokines activating mucosal macrophages of K19-C2mE mice. Accordingly, it is conceivable that PGE2-dependent heavy macrophage infiltrations together with cytokine signals from the epithelial cells, are important for inflammatory responses and gastric hypertrophy triggered by gastric bacteria.
Proinflammatory cytokines are key molecules in gastric hyperplasia
It has been reported that TNF-α plays a key role in the early stages of carcinogen-induced tumorigenesis in the skin and liver (Moore et al, 1999; Knight et al, 2000). Inhibition of proinflammatory cytokine production by pentoxifylline decreased the epithelial cell proliferation rate in K19-C2mE mice. This effect was found in only 5 days, when immune cells were still present in the submucosa (data not shown). Thus, it is possible that macrophage-derived proinflammatory cytokines participate in the gastric tumorigenesis. It remains to be determined which particular molecule is most responsible for the gastric hyperplastic tumorigenesis.
It is also possible that upregulation of HGF and VEGF in the K19-C2mE glandular stomach is mediated by macrophage-derived proinflammatory cytokines. HGF has been implicated in gastric tumorigenesis because of its upregulation in gastric cancer (Konturek et al, 2001), whereas VEGF is a key angiogenic factor in tumor tissues (Seno et al, 2002). Thus, these growth factors induced by proinflammatory cytokines can contribute to the gastric phenotypes in K19-C2mE mice.
H. pylori-associated gastric tumorigenesis may be suppressed more efficiently by COX-2 inhibition coupled with antibiotics
A chemoprevention trial showed that antimicrobial therapy against H. pylori improved the regression of gastric cancer precursor lesions (Correa et al, 2000). Moreover, accumulating evidence suggests that COX-2 plays a key role in H. pylori-associated gastric pathology (Fu et al, 1999; Sung et al, 2000). Here, we have demonstrated two important steps in gastric tumorigenesis that are triggered by increased levels of PGE2. First, PGE2 induces macrophage infiltration in the stomach, which can be stimulated by gastric infectious agents activating epithelial TLR4. Second, macrophage activation accelerates gastric epithelial growth through upregulation of proinflammatory cytokines and growth factors. It has been well established that expression of COX-2 as well as proinflammatory cytokines, such as TNF-α, IL-1β, IL-6 and IL-8 (CXCL8), is induced in H. pylori-associated gastric cancer (Crabtree et al, 1991; Noach et al, 1994). We have demonstrated that H. felis infection induces expression of both COX-2 and mPGES-1 in the gastric epithelial cells, suggesting that mucosal macrophages are also recruited in the Helicobacter-infected stomach. In addition, H. felis infection induced epithelial expression of TNF-α, although the molecular mechanism for TNF-α induction remains to be investigated further. Accordingly, this two-step mechanism triggered by PGE2 (i.e., macrophage infiltration followed by their activation) can also explain the H. pylori-associated gastric tumorigenesis. Therefore, COX-2 inhibition combined with antibiotics administration can be an effective chemopreventive strategy for H. pylori-associated gastric cancer.
In conclusion, inhibition of the PGE2 production should be an effective strategy in suppressing macrophage infiltration and hyperplastic cell growth in the gastric mucosa through cytokine networks.
Materials and methods
Transgenic mice
A 2.1-kb promoter fragment K19 (GenBank, AF237661) amplified by genomic PCR, a 1.0-kb SV40 polyA cassette and a synthetic chimeric intron excised from pCI (Promega, Madison, WI, USA) were cloned into pBluescript vector (Stratagene, La Jolla, CA, USA) to construct pK19. Full-length cDNAs for COX-2 and mPGES-1 were amplified by RT–PCR. After sequence confirmation, the cDNA fragments were subcloned into pK19 to construct pK19-COX-2 and pK19-mPGES, respectively. Two expression vectors were co-microinjected into fertilized eggs of the F1 (C3 H and C57BL/6) hybrid females crossed with C57BL/6 male. Two of the six constructed transgenic lines, K19-C2mE-2 and K19-C2mE-8, showed high expression levels of COX-2 and mPGES-1, and N2-backcrossed mice with C57BL/6 of these lines were used for further analysis. Wild-type littermates were used as controls.
Northern blotting
Total RNA (15 μg) extracted from glandular stomach was electrophoresed in 1% agarose, transferred to Hybond-N+ nylon filters (Amersham, Little Chalfont, UK), hybridized with the [32P]-labeled COX-2 or mPGES-1 cDNA probe, and autoradiographed.
Histopathology and immunohistochemistry
Tissues were fixed in 4% paraformaldehyde, embedded and sectioned at 4-μm thickness. These sections were stained with H&E and processed for further staining. Mucins were visualized by staining with Alcian blue (pH 2.5) or FITC-labeled Helix pometia-lectin (Sigma, St. Louis, MO, USA). For immunohistochemistry, rabbit polyclonal antibodies for COX-2 and mPGES-1 (Cayman Chemical, Ann Arbor, MI, USA), rat monoclonal antibodies for F4/80 (Serotec, Oxford, UK), CD11c, CD3ɛ and class II antigen (BD: Becton Dickinson, Franklin Lakes, NJ, USA), mouse monoclonal antibody for CXCL14 (BD) and goat polyclonal antibody for TNF-α (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used as the primary antibodies. Staining signals were visualized using the Vectorstain Elite Kit (Vector, Burlingame, CA, USA). The MOM. Kit (Vector Laboratories, Burlingame, CA, USA) was used to minimize the background staining signals. For immunofluorescence staining, Alexa Fluor anti-goat IgG (Molecular Probes, Eugene, OR, USA) or FITC-conjugated anti-rat IgG (Jackson Immunoresearch, West Grove, PA, USA) was used for the secondary antibody.
PGE2 analysis
The PGE2 levels were measured at SRL (SRL, Inc., Tokyo, Japan). The gastric mucosa was homogenized in a lysis buffer, extracted with ethanol and the PGE2 levels were measured by radioimmunoassay (RIA) using [125I]-labeled PGE2 (Perkin Elmer, Boston, MA, USA). Culture supernatants were used directly after appropriate dilutions.
BrdU-labeling index
Mice were injected i.v. with 200 μl of BrdU solution (Roche Diagnostics, IN, USA) 1 h before euthanasia. Tissue samples were fixed in 70% ethanol, embedded and sectioned at 5-μm thickness. These sections were stained with anti-BrdU antibody (Roche). The labeling index was calculated by dividing the number of BrdU-positive cells with the total number of nucleated cells.
Treatment of mice with chemicals
For COX-2 inhibition, mice were injected subcutaneously with 10 mg/kg/day of NS-398 (Sigma) in 5% Arabia gum for 4 weeks starting at 16 weeks of age. For antibiotics, mice were treated for 3 weeks from 17 weeks of age with streptomycin (5 mg/ml in drinking water) and cefoperazone (100 mg/kg/day, s.c. injection) (Sigma) as previously described (Zavros et al, 2002b). For inhibition of proinflammatory cytokines, such as TNF-α, IL-1 and IL-6, mice of 20 weeks of age were injected i.p. with 80 mg/kg of pentoxifylline (Sigma) every 12 h for 5 days (Voisin et al, 1998).
Bacterial counts
Glandular stomach was homogenized in sterilized saline. Serially diluted homogenates were spread on trypticase soy agar plates with 5% sheep blood (BBL: Becton Dickinson Labware, Sparks, MD, USA) and incubated under aerobic, microaerophilic and anaerobic conditions at 37°C for 1–3 days. For microaerophilic and anaerobic conditions, Campy Pouch and GasPak Pouch (BBL) were used, respectively. The colony-forming units (CFU)/glandular stomach were calculated from triplicated experimental results.
Cell culture experiments
Gastric epithelial cells were prepared from mice at 3 weeks of age and cultured as described (Fujikawa et al, 2003). Mouse monocyte cell line RAW264 was used for positive control (RIKEN BioResource Center, Tsukuba, Japan). Cells were treated with LPS from Salmonella typhimurium (Sigma) or PGN from Staphylococcus aureus (Fluka) for 20 h, and TNF-α concentration in culture supernatants was measured using an ELISA kit (Biosource, Camarillo, CA, USA). For blocking the TLR4 signaling, monoclonal antibody to mouse TLR4/MD2 (Clone MTS510; HyCult biotechnology, Uden, Netherlands) was used at 10 μg/ml. H. felis (ATCC 49179) was cultured as described (Fox et al, 1996). For H. felis infection, 0.5% horse serum was used to reduce nonspecific basal COX-2 expression. Bacteria were inoculated to epithelial cells at moi 100, and co-cultured for 20 h. For RT–PCR analysis of COX-2 expression, primary epithelial cells were cultured in 0.5% horse serum-containing medium.
LMD system
Tissues were frozen immediately after euthanasia and embedded in O.C.T. compound (Tissue-Tek, Torrance, CA, USA). Gastric mucosal regions were excised from frozen sections using LMD (Leica Microsystems, Wetzler, Germany). Total RNA was extracted and processed for RT–PCR analysis. For H. felis infection experiments, female C57BL/6 mice were infected orally with 5.0 × 108 cfu per mice. Tissues were sampled 6 h after infection and surface epithelial layers of glandular stomach were excised with LMD.
RT–PCR analysis
Total RNA was extracted from the glandular stomach and primary culture epithelial cells, using ISOGEN (Nippon Gene, Tokyo, Japan). Extracted RNA was reverse-transcribed and PCR-amplified. Band intensities of the RT–PCR products were quantified in a densitometer, using Image J (NIH, Bethesda, MD, USA). Specific GAPDH primers were used for the internal control to normalize the sample amounts. RT–PCR was carried out using the following primers: COX-2 (F-5′-CAAACTCAAGTTTGACCCAG-3′, R-5′-GCCGGGATCCTTTTACAGCTCAGTTGAACG-3′), mPGES-1 (F-5′-CCGAATTCTTGAAGTCCAGGCCGGCTAG-3′, R-5′-TAATGTCGACACCAAGTCCGCAAGTTC3′), TLR2 (F-5′-TAAGCTGTGTCTCCACAAGC-3′, R-5′-CTCCAGGTAGGTCTTGGTGT-3′), TLR4 (F-5′-GTGTGTCAGTGATCAGTGTG-3′, R-5′-GTCTTCTCCAGAAGATGTGC-3′), TNF-α (F-5′-GTGACAAGCCTGTAGCCCA-3′, R-5′-AAAGTAGACCTGCCCGGAC-3′), IL-1β (F-5′-ATTAGACAACTGCACTACAGGCTC-3′, R-5′-GGATTCCATGGTGAAGTCAATTAT-3′), IL-6 (F-5′-CATGTTCTCTGGGAAATCGTGG-3′, R-5′-TGCCGAGTAGATCTCAAAGTG-3′), MIP-2 (F-5′-CTGTTGTGGCCAGTGAACTGCG-3′, R-5′-GGCTCCTCCTTTCCAGGTCAGT-3′), CXCL14 (F-5′-GCGTTGGACGGGTCCAAGTGT-3′, R-5′-TTCGTAGACCCTGCGCTTCT-3′), CCL2 (F-5′-TGTCATGCTTCTGGGCCTGCT-3′, R-5′-TTCACTGTCACACTGGTCACT-3′), CCL3 (F-5′-GGTCTCCACACTGCCCTT-3′, R-5′-TCAGGCATTCAGTTCCAGGTC-3′), CCL4 (F-5′-GAAGCTCTGCGTGTCTGCCCT-3′, R-5′-ACTCCAAGTCACTCATGTACT-3′), CCL5 (F-5′-ATGAAGATCTCTGCAGCTGCC-3′, R-5′-CTAGCTCATCTCCAAATAGTT-3′), GM-CSF (F-5′-GCATTGTGGTCTACAGCCTCT-3′, R-5′-GCTGTCTATGAAATCCGCATA-3′), HGF (F-5′-GGCTTGGCATCCACGATGTTC3′, R-5′-CCAGGACGATTTGGGATGGCA-3′) and VEGF (F-5′-GCCAAGTGGTCCCAGGCTGC-3′, R-5′-CTGTGCTGTAGGAAGCTCAT-3′).
Chemotaxis assay
Cell migration was assayed in Transwell culture chambers (5-μm-pore membranes, Coster, Cambridge, MA, USA). RAW264 cells were cultured in the presence or absence of 1 μM PGE2 for 20 h. Then, cells (5 × 104) were added to the upper chamber, whereas recombinant mouse BRAK/CXCL14 (R&D systems, Inc., Minneapolis, MN, USA) was added to the lower chamber at 0, 100 and 400 ng/ml. After incubation for 1 h, cells attached on the lower surface of the membrane were counted in at least five different fields (original magnification, × 200).
Statistical analysis
Statistical analyses were carried out by unpaired Student's T-test, and P-value <0.05 was considered significant.
Acknowledgments
We thank H Seno for discussion, and A Shiomi, E Kaifu and S Toyota for technical assistance. This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to MO and MMT), the Organization of Pharmaceutical Safety and Research, Japan and Ground-based Research Announcement for Space Utilization prompted by Japan Space Forum.
References
- Balkwill F, Mantovani A (2001) Inflammation and cancer: back to Virchow? Lancet 357: 539–545 [DOI] [PubMed] [Google Scholar]
- Brembeck FH, Moffett J, Wang TC, Rustgi AK (2001) The keratin 19 promoter is potent for cell-specific targeting of genes in transgenic mice. Gastroenterology 120: 1720–1728 [DOI] [PubMed] [Google Scholar]
- Cavaillon JM (1994) Cytokines and macrophages. Biomed Pharmacother 48: 445–453 [DOI] [PubMed] [Google Scholar]
- Correa P (2003) Helicobacter pylori infection and gastric cancer. Cancer Epidemiol Biomarkers Prev 12: 238s–241s [PubMed] [Google Scholar]
- Correa P, Fontham ETH, Bravo JC, Bravo LE, Ruiz B, Zarama G, Realpe JL, Malcom GT, Li D, Johnson WD, Mera R (2000) Chemoprevention of gastric dysplasia: randomized trial of antioxidant supplements and anti-Helicobacter pylori therapy. J Natl Cancer Inst 92: 1881–1888 [DOI] [PubMed] [Google Scholar]
- Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420: 860–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree JE, Shallcross TM, Heatley RV, Wyatt JI (1991) Mucosal tumor necrosis factor alpha and interleukin-6 in patients with Helicobacter pylori associated gastritis. Gut 32: 1473–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrow DC, Vaughan TL, Hansten PD, Stanford JL, Risch HA, Gammon MD, Chow W-H, Dubrow R, Ahsan H, Mayne ST, Schoenberg JB, West AB, Rotterdam H, Fraumeni JF Jr, Blot WJ (1998) Use of aspirin and other nonsteroidal anti-inflammatory drugs and risk of esophageal and gastric cancer. Cancer Epidemiol Biomarkers Prev 7: 97–102 [PubMed] [Google Scholar]
- Fox JG, Li X, Cahill RJ, Andrutis K, Rustgi AK, Odze R, Wang TC (1996) Hypertrophic gastropathy in Helicobacter felis-infected wild-type C57BL/6 mice and p53 hemizygous transgenic mice. Gastroenterology 110: 155–166 [DOI] [PubMed] [Google Scholar]
- Fu S, Ramanujam KS, Wong A, Fantry GT, Drachenberg CB, James SP, Meltzer SJ, Wilson KT (1999) Increased expression and cellular localization of inducible nitric oxide synthase and cyclooxygenase 2 in Helicobacter pylori gastritis. Gastroenterology 116: 1319–1329 [DOI] [PubMed] [Google Scholar]
- Fujikawa A, Shirasaka D, Yamamoto S, Ota H, Yahiro K, Fukada M, Shintani T, Wada A, Aoyama N, Hirayama T, Fukamachi H, Noda M (2003) Mice deficient in protein tyrosine phosphatase receptor type Z are resistant to gastric ulcer induction by VacA of Helicobacter pylori. Nat Genet 33: 375–381 [DOI] [PubMed] [Google Scholar]
- Homey B, Muller A, Zlotnik A (2002) Chemokines: agents for the immunotherapy of cancer? Nat Rev Immunol 2: 175–184 [DOI] [PubMed] [Google Scholar]
- Huang M, Stolina M, Sharma S, Mao JT, Zhu L, Miller PW, Wollman J, Herschman H, Dubinett SM (1998) Non-small cell lung cancer cyclooxygenase-2-dependent regulation of cytokine balance in lymphocytes and macrophages: up-regulation of interleukin 10 and down-regulation of interleukin 12 production. Cancer Res 58: 1208–1216 [PubMed] [Google Scholar]
- IARC (1994) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Schistosomes, Liver Flukes and Helicobacter pylori, Vol 61, Lyon, France: IARC [PMC free article] [PubMed] [Google Scholar]
- Jüttner S, Cramer T, Wessler S, Walduck A, Gao F, Schmitz F, Wunder C, Weber M, Fischer SM, Schmidt WE, Wiedenmann B, Meyer TF, Naumann M, Höcker M (2003) Helicobacter pylori stimulates host cyclooxygenase-2 gene transcription: critical importance of MEK/ERK-dependent activation of USF1/-2 and CREB transcription factors. Cell Microbiol 5: 821–834 [DOI] [PubMed] [Google Scholar]
- Kaisho T, Akira S (2002) Toll-like receptors as adjuvant receptors. Biochim Biophys Acta 1589: 1–13 [DOI] [PubMed] [Google Scholar]
- Kawahara T, Teshima S, Oka A, Sugiyama T, Kishi K, Rokutan K (2001) Type I Helicobacter pylori lipopolysaccharide stimulates toll-like receptor 4 and activates mitogen oxidase 1 in gastric pit cells. Infect Immun 69: 4382–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TI, Lee YC, Lee KH, Han JH, Chon CY, Moon YM, Kang JK, Park IS (2001) Effects of nonsteroidal anti-inflammatory drugs on Helicobacter pylori-infected gastric mucosae of mice: apoptosis, cell proliferation, and inflammatory activity. Infect Immun 69: 5056–5063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight B, Yeoh GCT, Husk KL, Ly T, Abraham LJ, Yu C, Rhim JA, Fausto N (2000) Impaired preneoplastic changes and liver tumor formation in tumor necrosis factor receptor type 1 knockout mice. J Exp Med 192: 1809–1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konturek PC, Konturek SJ, Sulekova Z, Meixner H, Bielanski W, Starzynska T, Karczewska E, Marlicz K, Stachura J, Hahn EG (2001) Expression of hepatocytes growth factor, transforming growth factor alpha, apoptosis related proteins Bax and Bcl-2, and gastrin in human gastric cancer. Aliment Pharmacol Ther 15: 989–999 [DOI] [PubMed] [Google Scholar]
- Kuper H, Adami H-O, Trichopoulos D (2000) Infections as a major preventable cause of human cancer. J Intern Med 248: 171–183 [DOI] [PubMed] [Google Scholar]
- Kurth I, Willimann K, Schaerli P, Hunziker T, Clark-Lewis I, Moser B (2001) Monocyte selectivity and tissue localization suggests a role for breast and kidney-expressed chemokine (BRAK) in macrophage development. J Exp Med 194: 855–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus M, Kubata BK, Eguchi N, Fujitani Y, Urade Y, Hayaishi O (2002) Biochemical characterization of mouse microsomal prostaglandin E synthase-1 and its colocalization with cyclooxygenase-2 in peritoneal macrophages. Arch Biochem Biophys 397: 336–341 [DOI] [PubMed] [Google Scholar]
- Lee A, Fox JG, Otto G, Murphy J (1990) A small animal model of human Helicobacter pylori active chronic gastritis. Gastroenterology 99: 1315–1323 [DOI] [PubMed] [Google Scholar]
- Lee A, O'Rourke J, de Ungria MC, Robertson B, Daskalopoulos G, Dixon MF (1997) A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology 112: 1386–1397 [DOI] [PubMed] [Google Scholar]
- Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, East N, Holdsworth H, Turner L, Rollins B, Pasparakis M, Kollias G, Balkwill F (1999) Mice deficient in tumor necrosis factor-α are resistant to skin carcinogenesis. Nat Med 5: 828–831 [DOI] [PubMed] [Google Scholar]
- Muller WA (2001) New mechanisms and pathways for monocyte recruitment. J Exp Med 194: F47–F51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, Ikeda T, Fueki M, Ueno A, Oh-ishi S, Kudo I (2000) Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem 275: 32783–32792 [DOI] [PubMed] [Google Scholar]
- Noach LA, Bosma NB, Jansen J, Hoek FJ, van Deventer SJH, Tytgat GNJ (1994) Mucosal tumor necrosis factor-α, interleukin-1β, and interleukin-8 production in patients with Helicobacter pylori infection. Scand J Gastroenterol 29: 425–429 [DOI] [PubMed] [Google Scholar]
- Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM (1996) Suppression of intestinal polyposis in ApcΔ716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 87: 803–809 [DOI] [PubMed] [Google Scholar]
- Oshima M, Murai N, Kargman S, Arguello M, Luk P, Kwong E, Taketo MM, Evans JF (2001) Chemoprevention of intestinal polyposis in the ApcΔ716 mouse by rofecoxib, a specific cyclooxygenase-2 inhibitor. Cancer Res 61: 1733–1740 [PubMed] [Google Scholar]
- Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS (2002) Prostaglandin E2 transactivates EGF receptor: a novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat Med 8: 289–293 [DOI] [PubMed] [Google Scholar]
- Peterson RA, Danon SJ, Eaton KA (2001) Comparison of gastritis and gastric epithelial proliferation in Helicobacter heilmannii-infected nude and BALB/c mice. Vet Pathol 38: 173–183 [DOI] [PubMed] [Google Scholar]
- Reinhart WH, Muller O, Halter F (1983) Influence of long-term 16, 16-dimethyl prostaglandin E2 treatment on the rat gastrointestinal mucosa. Gastroenterology 85: 1003–1010 [PubMed] [Google Scholar]
- Ristimaki A, Honkanen N, Jankala H, Sipponen P, Harkonen M (1997) Expression of cyclooxygenase-2 in human gastric carcinoma. Cancer Res 57: 1276–1280 [PubMed] [Google Scholar]
- Roth KA, Kapadia SB, Martin SM, Lorenz RG (1999) Cellular immune responses are essential for the development of Helicobacter felis-associated gastric pathology. J Immunol 163: 1490–1497 [PubMed] [Google Scholar]
- Seno H, Oshima M, Ishikawa T, Oshima H, Takaku K, Chiba T, Narumiya S, Taketo MM (2002) Cyclooxygenase 2- and prostaglandin E2 receptor EP2-dependent angiogenesis in ApcΔ716 mouse intestinal polyps. Cancer Res 62: 506–511 [PubMed] [Google Scholar]
- Sheng H, Shao J, Morrow JD, Beauchamp RD, DuBois RN (1998) Modulation of apoptosis and Bcl-2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res 58: 362–366 [PubMed] [Google Scholar]
- Sheng H, Shao J, Washington MK, DuBois RN (2001) Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J Biol Chem 276: 18075–18081 [DOI] [PubMed] [Google Scholar]
- Sonoshita M, Takaku K, Sasaki N, Sugimoto Y, Ushikubi F, Narumiya S, Oshima M, Taketo MM (2001) Acceleration of intestinal polyposis through prostaglandin receptor EP2 in ApcΔ716 knockout mice. Nat Med 7: 1048–1051 [DOI] [PubMed] [Google Scholar]
- Soydan AS, Gaffen JD, Weech PK, Tremblay NM, Kargman S, O'Neill G, Bennett A, Tavares IA (1997) Cytosolic phospholipase A2, cyclo-oxygenases and arachidonate in human stomach tumours. Eur J Cancer 33: 1508–1512 [DOI] [PubMed] [Google Scholar]
- Smith MF Jr, Mitchell A, Li G, Ding S, Fitzmaurice AM, Ryan K, Crowe S, Goldberg JB (2003) Toll-like receptor (TLR)2 and TLR5, but not TLR4, are required for Helicobacter pylori-induced NF-κB activation and chemokine expression by epithelial cells. J Biol Chem 278: 32552–32560 [DOI] [PubMed] [Google Scholar]
- Suganuma M, Kurusu M, Okabe S, Sueoka N, Yoshida M, Wakatsuki Y, Fujiki H (2001) Helicobacter pylori membrane protein 1: a new carcinogenic factor of Helicobacter pylori. Cancer Res 61: 6356–6359 [PubMed] [Google Scholar]
- Sung JJY, Leung WK, Go MYY, To KF, Cheng ASL, Ng EKW, Chan FKL (2000) Cyclooxygenase-2 expression in Helicobacter pylori-associated premalignant and malignant gastric lesions. Am J Pathol 157: 729–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taketo MM (1998a) Cyclooxygenase-2 inhibitors in tumorigenesis (part I). J Natl Cancer Inst 90: 1529–1536 [DOI] [PubMed] [Google Scholar]
- Taketo MM (1998b) Cyclooxygenase-2 inhibitors in tumorigenesis (part II). J Natl Cancer Inst 90: 1609–1620 [DOI] [PubMed] [Google Scholar]
- Thun MJ, Namboodiri MM, Calle EE, Flanders WD, Heath CW Jr (1993) Aspirin use and risk of fatal cancer. Cancer Res 53: 1322–1327 [PubMed] [Google Scholar]
- Voisin L, Breuille D, Ruot B, Ralliere C, Rambourdin F, Dalle M, Obled C (1998) Cytokine modulation by PX differently affects specific acute phase proteins during sepsis in rats. Am J Physiol 275: R1412–R1419 [DOI] [PubMed] [Google Scholar]
- Xiao F, Furuta T, Takashima M, Shirai N, Hanai H (2001) Involvement of cyclooxygenase-2 in hyperplastic gastritis induced by Helicobacter pylori infection in C57BL/6 mice. Aliment Pharmacol Ther 15: 875–886 [DOI] [PubMed] [Google Scholar]
- Yoshimatsu K, Golijanin D, Paty PB, Soslow RA, Jakobsson P-J, DeLellis RA, Subbaramaiah K, Dannenberg AJ (2001) Inducible microsomal prostaglandin E synthase is overexpressed in colorectal adenomas and cancer. Clin Cancer Res 7: 3971–3976 [PubMed] [Google Scholar]
- Zavros Y, Rieder G, Ferguson A, Merchant JL (2002b) Gastritis and hypergastrinemia due to Acinetobacter Iwoffii in mice. Infect Immun 70: 2630–2639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavros Y, Rieder G, Ferguson A, Samuelson LC, Merchant JL (2002a) Genetic or chemical hypochlorhydria is associated with inflammation that modulates parietal and G-cell populations in mice. Gastroenterology 122: 119–133 [DOI] [PubMed] [Google Scholar]