

Abstract
We studied the transcriptomic response of Streptococcus pneumoniae to levofloxacin (LVX) under conditions inhibiting topoisomerase IV but not gyrase. Although a complex transcriptomic response was observed, the most outstanding result was the upregulation of the genes of the fatDCEB operon, involved in iron (Fe2+ and Fe3+) uptake, which were the only genes varying under every condition tested. Although the inhibition of topoisomerase IV by levofloxacin did not have a detectable effect in the level of global supercoiling, increases in general supercoiling and fatD transcription were observed after topoisomerase I inhibition, while the opposite was observed after gyrase inhibition with novobiocin. Since fatDCEB is located in a topological chromosomal domain downregulated by DNA relaxation, we studied the transcription of a copy of the 422-bp (including the Pfat promoter) region located upstream of fatDCEB fused to the cat reporter inserted into the chromosome 106 kb away from its native position: PfatfatD was upregulated in the presence of LVX in its native location, whereas no change was observed in the Pfatcat construction. Results suggest that topological changes are indeed involved in PfatfatDCE transcription. Upregulation of fatDCEB would lead to an increase of intracellular iron and, in turn, to the activation of the Fenton reaction and the increase of reactive oxygen species. In accordance, we observed an attenuation of levofloxacin lethality in iron-deficient media and in a strain lacking the gene coding for SpxB, the main source of hydrogen peroxide. In addition, we observed an increase of reactive oxygen species that contributed to levofloxacin lethality.
INTRODUCTION
Streptococcus pneumoniae (the pneumococcus) acts as an opportunistic pathogen. It forms part of the commensal microbiota of the human nasopharynx. Under specific circumstances, it migrates to other niches (ear, lung, bloodstream, and cerebrospinal fluid) causing diverse pathologies. One million children aged <5 years die annually of pneumococcal infections worldwide (1). After the usage of the pneumococcal 7-valent conjugate vaccine, which includes the serotypes more often associated with resistance to antibiotics, the incidence of invasive pneumococcal disease declined (2, 3) coincidentally with a decrease of penicillin resistance rates in many countries (3–5). However, the emergence of serotypes not included in the vaccine has been observed (6, 7). Therefore, knowledge of the molecular bases of antimicrobial action, including the mechanisms of killing, is essential for developing improved therapeutics.
Resistance in S. pneumoniae to antibiotics acting either in cell wall (beta-lactams) or in protein (macrolides) synthesis has spread worldwide in the last 3 decades (8). The fluoroquinolones (FQs) levofloxacin (LVX) and moxifloxacin are used now for treatment of adult patients with pneumonia. FQ resistance in S. pneumoniae is maintained at low prevalence (<3%) in Europe (9, 10), although higher rates have been detected in Asia (11) and in Canada (12). However, an increase in resistance in this bacterium may occur if FQ use is increased (13). FQs target the type II DNA topoisomerases. Despite the functional similarities between topoisomerase IV (topo IV) and gyrase, their susceptibility to FQs varies across bacterial species (14). In S. pneumoniae, the primary target for LVX is topo IV (15–18), while gyrase is the primary target for moxifloxacin (19). Type II topoisomerases maintain DNA topology and solve the topological problems associated with DNA replication, transcription, and recombination (20). Gyrase introduces negative supercoils into DNA (21), whereas topo IV relaxes DNA and participates in chromosome partitioning (22). Chromosomal topology in Escherichia coli is maintained homeostatically by the opposing activities of topoisomerases that relax DNA (topo I and topo IV) and by gyrase. In this bacterium, transcription of the topA gene encoding topo I increases when negative supercoiling increases (23), while that of gyrA and gyrB increases after DNA relaxation (24–26). Changes in DNA supercoiling also have a global effect on genome transcription in E. coli (27, 28) and Haemophilus influenzae (29). We have also shown that relaxation of the S. pneumoniae chromosome with novobiocin (NOV; a GyrB inhibitor) causes upregulation of gyrase genes and downregulation of topo I and IV genes and triggers a global transcriptional response affecting ca. 14% of the genome (30). Most (>68%) responsive genes are closely positioned, forming 15 gene clusters (up- and downregulated topological domains), which showed a coordinated response (30).
The killing effect of FQs has been related to the resolution of reaction intermediates of DNA-FQ-topoisomerase complexes, which generates irreparable double-stranded DNA breaks (31). This could occur in E. coli by two pathways, one dependent on protein synthesis and the other independent of it. It has been shown that hydroxyl radical action contributes to FQ-mediated cell death occurring via a protein-dependent pathway (32). This result agrees with a recent proposal suggesting that, following gyrase poisoning, hydroxyl radical formation utilizing internal iron and the Fenton reaction (33) is generated and contributes to cell killing by FQs (34) as well as by other bactericidal antibiotics (35, 36). In this mechanism, proposed for Enterobacteriaceae (35, 37), the primary drug interactions stimulate oxidation of NADH via the electron transport chain that is dependent on the tricarboxylic acid cycle. Hyperactivation of the electron transport chain stimulates superoxide formation. Superoxide destabilizes the iron-sulfur clusters of enzymes, making Fe2+ available for oxidation by the Fenton reaction. The Fenton reaction leads to the formation of hydroxyl radicals that would damage DNA, proteins, and lipids (38), which results in cell death. Instead of a generalized oxidative damage, a recent study supports that the main action of hydroxyl radicals is the oxidation of guanine (to 8-oxo-guanine) of the nucleotide pool. The incomplete repair of closely spaced 8-oxo-deoxyguanosine lesions caused lethal double-strand DNA breaks, which would underlie much of the cell death caused by beta-lactams and FQs (39). However, recent investigations have questioned the role of hydroxyl radicals and intracellular iron levels in antibiotic-mediated lethality using antibiotic concentrations either similar to (40) or higher than (41) those used previously. The disparate results obtained using diverse antibiotic concentrations and times of treatment emphasize the complexity of the lethal stress response (42).
Given that different antibiotic families have different intracellular targets, it is essential to know the pathway between the initial antibiotic-target interaction and the promotion of hydroxyl radical formation. These pathways are mostly unknown. A model has been proposed for aminoglycosides in E. coli whereby the interference of these drugs with ribosome progression would release incomplete polypeptides, which are translocated to the cell membranes, where they may trigger envelope stress. The Arc regulatory system is perturbed, accelerating respiration and thereby increasing the flux of superoxide and hydrogen peroxide into the cell (43). However, for FQs, the specific pathway has not been established, although a general scheme for stress response regulation in E. coli, which involves the hydroxyl radical cascade, has been proposed (42). The present study was aimed to understand the transcriptional response to levofloxacin in S. pneumoniae at concentrations that inhibited its primary target, topo IV, without inhibiting gyrase, to avoid the opposite effects of these two enzymes on DNA topology. Changes in DNA topology were tested by analyzing the distribution of topoisomers of a replicating plasmid. Global transcription response was analyzed using microarrays technology after cells' exposure to two LVX concentrations. Microarray data were validated by quantitative real-time PCR (qRT-PCR). In addition, transcriptional regulation of the fatDCEB operon, coding for an iron transporter, was analyzed. The relation between iron transport and lethality was also tested. Results provide a pathway between topo IV inhibition and hydroxyl radical production and suggest that S. pneumoniae uses iron accumulation as part of the death process associated with LVX treatment.
MATERIALS AND METHODS
Bacterial strains and growth and transformation of bacteria.
S. pneumoniae was grown in AGCH medium with 0.3% sucrose and transformed as described previously (44). MICs of LVX (Sigma) and chloramphenicol (CHL) for strain R6 were 0.25 μg/ml and 1.25 μg/ml, respectively. To construct the ΔspxB strain, two fragments of 1,481 bp and 1,374 bp flanking spxB were amplified with oligonucleotide pairs SpxBUPF1/SpxBUPR1 and SpxBDOWNF1/SpxBDOWNR1 (see Table S1 in the supplemental material), digested with SphI and XbaI, and ligated to the CHL-acetyltransferase gene (cat) of plasmid pJS3 digested with the same enzymes. R6 was transformed; recombinant colonies were selected in medium containing 2.5 μg/ml CHL and checked by PCR amplification with external oligonucleotides SpxBUPF2/SpxBDOWNR2 (see Table S1 in the supplemental material). Those with the appropriate size (4,574 bp versus 5,352 bp of R6) were sequenced using oligonucleotide CATMED. To construct the R6-Pfatcat strain, five PCR products were obtained. Two from genes spr1793 (1,061 bp) and spr1794 (1,036 bp), flanking the site of insertion, by amplifications with primers 1793F1(XbaI)/spr1793R1 and spr1794F1/spr1794R1(SphI). The third fragment (144 bp) was amplified with UptrcatXba/UptrcatEco and contains the transcriptional terminator that precedes the cat cassette in plasmid pJS3. The fourth fragment (422 bp) containing the 5′-upstream region of the iron transport operon fatDCEB was amplified with UpfatDF1(EcoRI) and phosphorylated UpFatDR1. The fifth fragment (758 bp) contains cat and was amplified with Cat1 phosphorylated and CatDownSph(SphI). Each fragment was digested with the appropriate enzyme, and all fragments were ligated together. The ligation mix was used to transform strain R6, and transformants were selected in medium containing 2.5 μg/ml CHL. This rendered strain R6-Pfatcat (see Fig. 3A), whose genetic structure was checked by PCR with primers Spr1793R2 and Spr1794F2 and by sequencing with CATMED, CAT191, and spr1793R3.
FIG 3.

Transcription of fatD depended on the inhibition of topoisomerase IV by LVX. (A) Genetic structure of strain R6-Pfatcat showing the chromosomal location of PfatfatDCEB and Pfatcat. Topology-reacting gene clusters detected after DNA relaxation with NOV are indicated: U1 to U15, upregulated domains; D1 to D14, downregulated domains. (B) Transcriptional response after NOV treatment measured by qRT-PCR on exponentially growing cultures of strain R6. (C) Transcriptional response of R6, of an LVX-resistant derivative (R6-ParCS79F), and of strain R6-Pfatcat. Cultures were grown in AGCH to an OD620 of 0.4 and treated with LVX at 0.125 μg/ml LVX (0.5× MIC of R6 and R6-Pfatcat; 0.05× MIC of R6-ParCS79F) and at 2.5 μg/ml LVX (10× MIC of R6 and R6-Pfatcat; 0.5× MIC of R6-ParCS79F). Total RNA was isolated; cDNA was synthesized and subjected to qRT-PCR. Data were normalized to time zero min. Transcription represented the means of qRT-PCR values of three independent replicates ± standard errors of the means (SEM).
Analysis of the topology of covalently closed circles.
Plasmid DNA isolation from S. pneumoniae cultures grown on AGCH medium with 1 μg/ml tetracycline (for pLS1selection) was performed as described before (30). Circular DNA molecules were analyzed in neutral/neutral two-dimensional agarose gels, which were subjected to Southern hybridization with a 240-bp specific pLS1 probe as described previously (30). DNA linking number (Lk) was calculated by quantifying the amount of every topoisomer. DNA supercoiling density (σ) was calculated from the equation σ = ΔLk/Lk0. Linking number differences (ΔLk) were determined using the equation Lk = Lk − Lk0, in which Lk0 is N/10.5, where N is the size of the molecule (in bp) and 10.5 is the number of base pairs per complete turn in B-DNA.
RNA extraction and real-time RT-PCR experiments.
Synthesis of cDNAs from 5 μg of total RNA was performed as previously described (45). These cDNAs were subjected to quantitative qRT-PCR (Chromo 4; Bio-Rad) in 20-μl reaction mixtures containing 2 μl of cDNA, 0.3 μM each specific primer, and 10 μl of LightCycler FastStart Universal A SYBR green Master (Roche). Amplification was achieved with 42 cycles of a tree-segment program: denaturation (30 s at 94°C), annealing (30 s at 45 to 56°C), and elongation (30 s at 68°C). To normalize the three independent cDNA replicate samples, values were divided by those obtained from the amplification of internal fragments of rpoB (45) and 16S rRNA genes. The oligonucleotides used are shown in Table S1 in the supplemental material.
Microarray data normalization and analysis.
High-density arrays A6701-00-01 from Roche NimbleGen were used. Double-stranded cDNAs were obtained from total RNA with the SuperScript Double-Stranded cDNA Synthesis kit (Invitrogen). Labeling of cDNAs with Cy3 and hybridization were performed at the Institut de Recerca Biomèdica, Barcelona (Spain). A GenePix 4000B scanner at 5-μm resolution was used, and raw data were extracted and robust multiarray average (RMA) normalized using NimbleScan v2.4. After this normalization, Partek Genomics Suite 6.4 was used to do a principal component analysis and test for significance for differential gene expression using analysis of variance (ANOVA). Each microarray experiment was carried out in duplicate with cDNA prepared from two independent cultures.
Detection of reactive oxygen species.
The intracellular oxidation levels were measured using dihydrorhodamine 123 dye (Sigma-Aldrich), a nonfluorescent compound that diffuses passively across membranes. Oxidation converts it to the fluorescent product rhodamine 123, and this fluorescence is proportional to the level of oxidation (37). In a typical experiment, cells were grown exponentially to an optical density at 620 nm (OD620) of 0.4 before LVX was added. One-milliliter samples were collected, and cells were washed once in 500 μl of 1× phosphate-buffered saline (PBS) (pH 7.2), suspended in 250 μl of 1× PBS containing 2.5 μg/ml of dihydrorhodamine 123, and incubated at 37°C in the dark for 30 min. Cells were washed once in 500 μl of 1× PBS and suspended in 250 μl of 1× PBS. A volume of 200 μl was used to measure fluorescence. The fluorescence signal was analyzed using a Tecan Infinite 2000 with excitation wavelength (λ)/emission λ of 485 nm/535 nm. Results were expressed as relative fluorescence units (RFU) and were normalized according to the number of live cells at each time point (46).
Microarray data accession number.
All microarray data are available at the Array Express (EBI, United Kingdom) database via accession number E-MEXP-3809.
RESULTS
DNA topoisomer distribution did not vary under treatment with LVX.
The effect of LVX was tested in the reference strain R6 carrying plasmid pLS1 at subinhibitory (0.5× MIC) and fully inhibitory (10× MIC) concentrations. The change in OD620 along the 60 min of the experiment was from 0.4 to 0.8. Cell division was inhibited only when the culture was treated with LVX at 10× MIC, with decreases in cell viability to about 70% and 97% at 30 and 60 min, respectively (Fig. 1A). To measure supercoiling alterations, topoisomer distributions of the replicating pLS1 plasmid were analyzed. Under the chloroquine concentration used, the induced ΔLk is −14 (30). Topoisomers appeared distributed in the autoradiograms in a bubble-shaped arc, where negative and positive supercoiled molecules are located to the right and to the left sides, respectively (Fig. 1B). Although we have not measured the supercoiling level of the bacterial chromosome, the values obtained on small plasmids provide a good estimation of chromosomal supercoiling (47). No significant differences in supercoiling densities (σ) were detected under any condition, showing that the inhibition of topo IV by LVX did not have a detectable effect in the level of global supercoiling and that gyrase was not inhibited at the LVX concentrations used.
FIG 1.
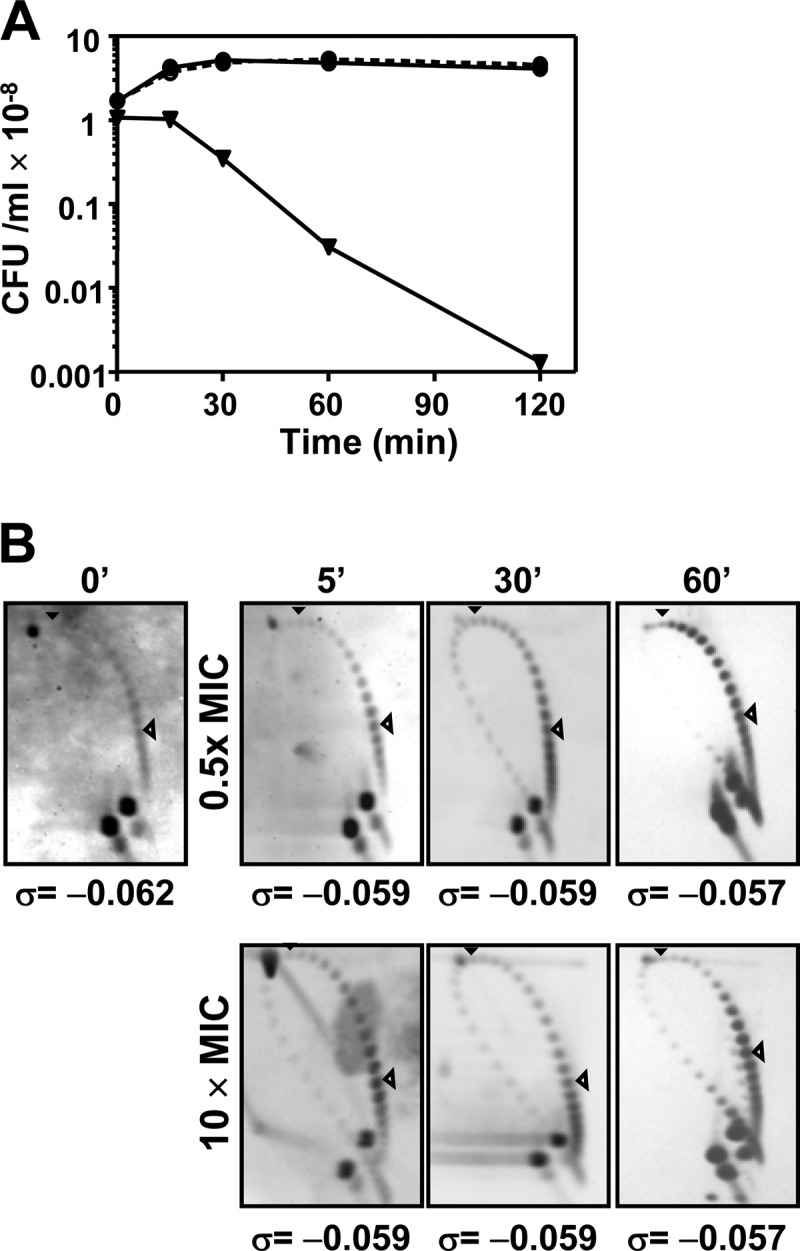
Global supercoiling did not vary under treatment of S. pneumoniae R6 (pLS1) with levofloxacin. (A) Viability. (B) Topoisomer distribution of pLS1. Exponentially growing cultures in AGCH at OD620 of 0.4 were treated with the indicated LVX concentrations. Values of a typical experiment are indicated. Samples were taken before the addition of the drug (time zero min), and at the indicated times, plasmid DNA was isolated and subjected to two-dimensional agarose gel electrophoresis run in the presence of 1 and 2 μg/ml chloroquine in the first and second dimensions, respectively. Supercoiling density (σ) values are indicated. A black arrowhead indicates the topoisomer that migrated with ΔLk of 0 in the second dimension and has a ΔWr of −14. An open arrowhead points to the more abundant topoisomer.
Two kinds of transcriptional responses in LVX-treated cultures: growth related and LVX related.
The transcriptional response was measured in cultures of strain R6 at three time points (5, 15, and 30 min) after treatment with LVX concentrations of 0.5× MIC and 10× MIC. In addition, samples taken at 15 and 30 min of an untreated culture (No-LVX) were also analyzed and used to distinguish those genes varying along the growth curve. Only gene expression variations ≥2 (P values < 0.01) with respect to time zero min were considered. The whole transcriptomic response is shown in Table 1. Based on the results obtained, responsive genes were classified into two categories: growth related and LVX related (Fig. 2; Table 1). Growth-related genes included 108 genes showing transcription variations in the No-LVX culture. Additionally, 10 genes forming parts of operons with these genes were also considered to be growth related. In total, 118 genes (5.8% of the genome) showed variations associated with growth (Fig. 2A).
TABLE 1.
Genes involved in the transcriptomic response of S. pneumoniae R6 to levofloxacin (LVX)a
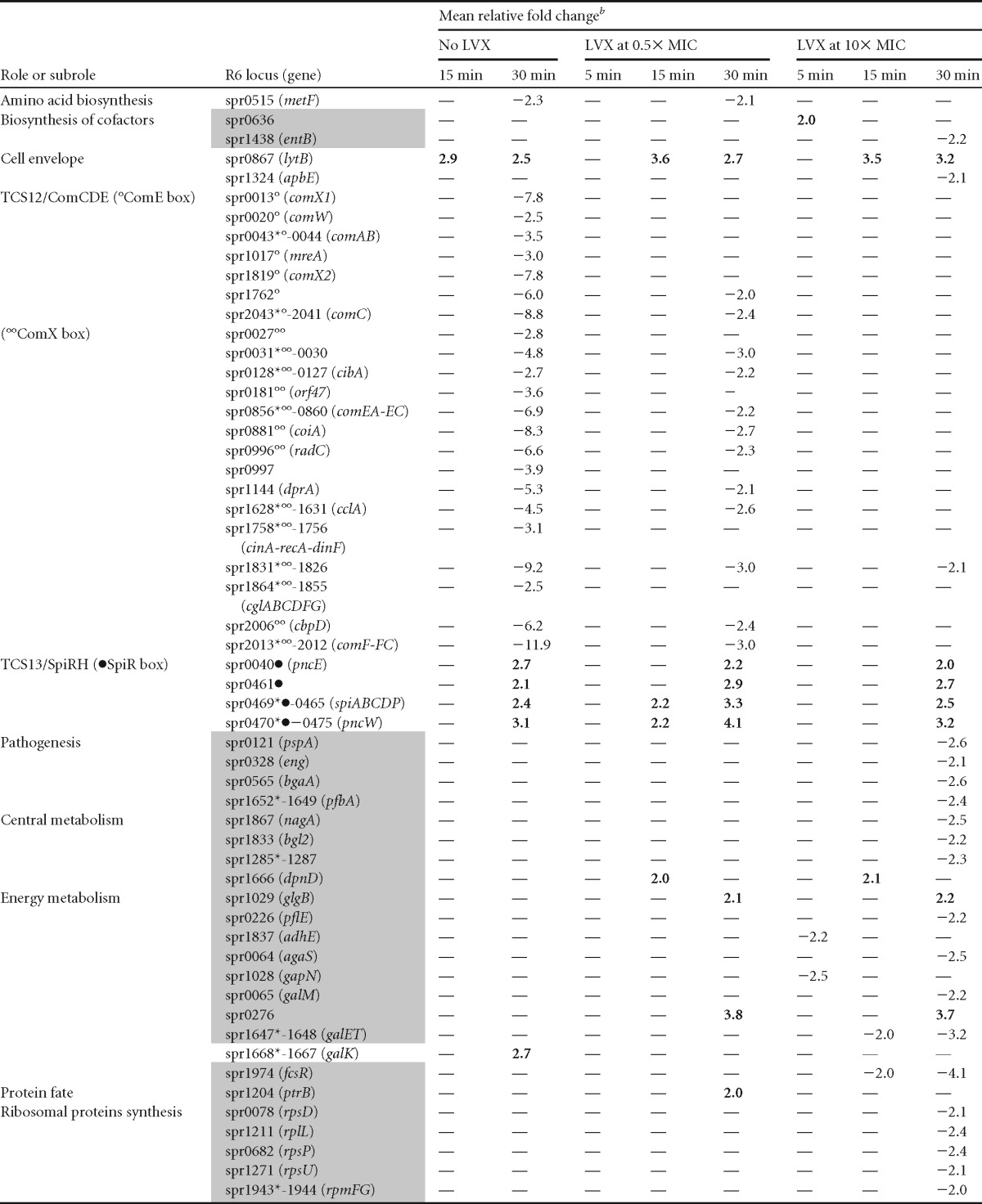
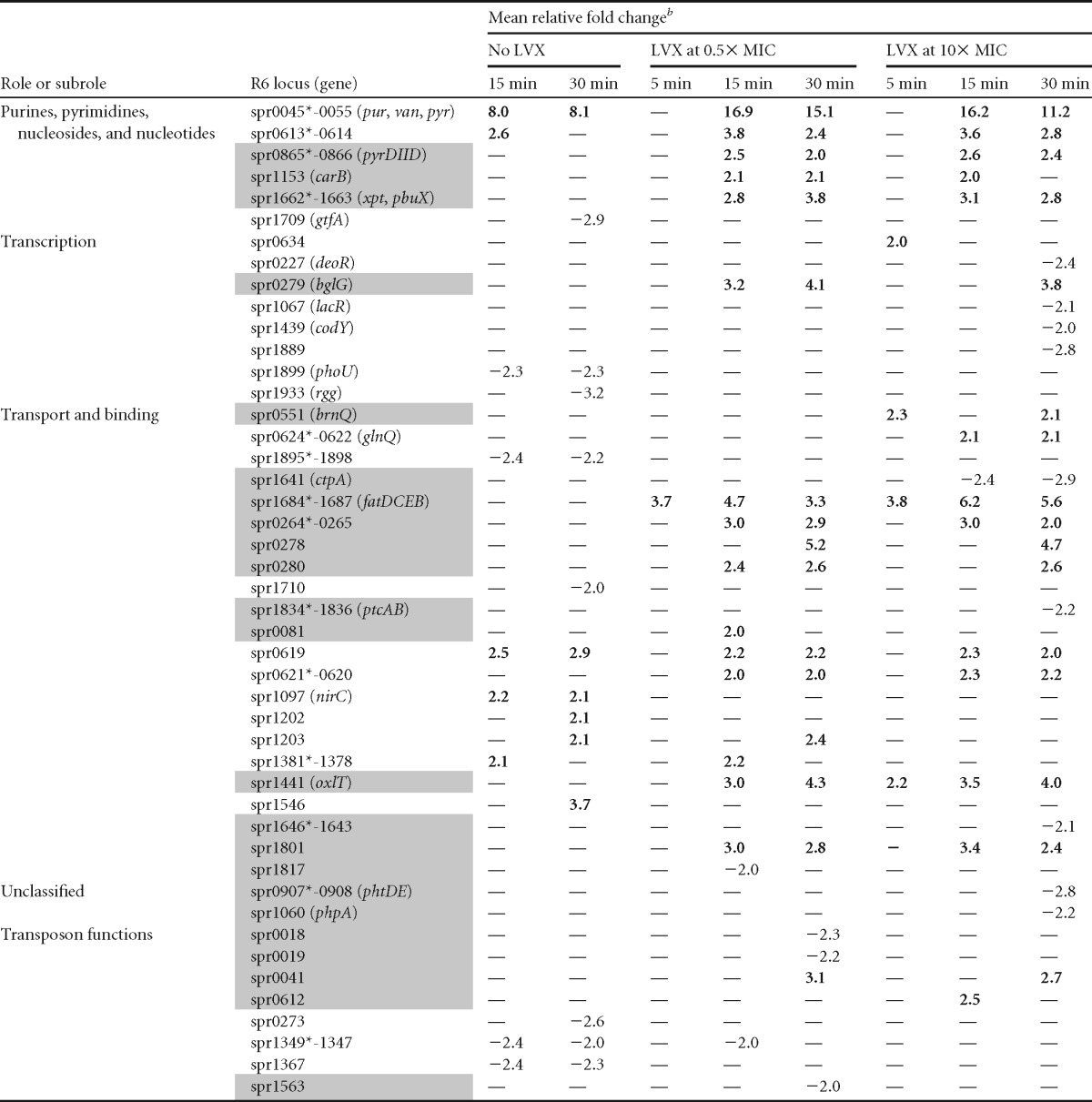
The responsive genes included showed significant fold variations (≥2 and P <0.01). All genes showing variations, with the exception of 47 encoding hypothetical proteins, are included. Genes considered to be involved in the LVX-mediated transcriptomic response (i.e., that did not show variations in the no-LVX culture) are shaded in gray. Nonshaded genes are considered to be involved in the growth-related response. Symbols: *, the first gene of the putative operon; °, gene with a ComE box; °°, gene with a ComX box; •, gene with a SpiR box; —, no change.
In operons, values indicated correspond to those of the first gene of the operon, except in spr0045-0055, spr0613-0614, spr1859-1898, and spr1864-1855, in which variations corresponded to the second gene. Values above 2 are shown in boldface.
FIG 2.

Gene expression analysis under the three conditions assayed. (A and B) Responsive genes represented in Venn diagrams with 3 circles, each one corresponding to one time interval under each condition, showing the differentially expressed genes in microarrays. (B) All genes (left diagram) or only those genes that differed from those present in the No-LVX sample (right diagram) are indicated. A complete list of these genes can be found in Table 1. (C) Classification of responsive genes by functional classes: CiM, central intermediary metabolism; EnM, energy metabolism; Hy, hypothetical proteins; PPy, purines, pyrimidines, nucleosides, and nucleotides; Pat, pathogenesis; PrS, protein synthesis; R-M, restriction-modification; TBP, transport and binding proteins; TCS, two-component systems; Tns, transposon functions; Tr, transcription; U, unclassified; Oth, other (classes with a representation of <2%).
Genes controlled by two-component system 12 (TCS-12) and TCS-13 represented the greatest proportion (50.8%) of responsive genes (Table 1; Fig. 2C). TCS-12 regulates competence for genetic transformation (48). The regulatory cascade begins with the secretion and processing of ComC by the dedicated ABC transporter ComAB. Processed ComC activates TCS-12 ComDE: ComD is the histidine kinase (that senses the stimulus), and ComE is the response regulator (the transcriptional modulator of the responsive genes). Phosphorylated ComE activates the transcription of early genes, including the alternative sigma factor ComX (49), which activates transcription of late competence genes (50, 51). Among competence genes, 53 (11 early and 42 late genes) showed downregulation. These included most (10 of 18) genes of the early competence response (50), which are transcribed from 7 of the 10 promoters (Table 1) containing the binding site of the transcriptional activator ComE (52). Early downregulated genes included those coding for the two transcriptional regulators of competence, comE and two genes coding for the alternative sigma factor ComX (53), required for induction of many late genes. In accordance, 42 of the 81 late-competence genes (50) were downregulated. These 42 genes were transcribed from 13 of 19 promoters containing a ComX box. Concerning TCS-13 (SpiRH), 13 genes containing regulatory sequences for its response regulator SipR (54, 55) were upregulated, including spiP encoding the bacteriocin with a Gly-Gly motif and the dedicated ABC transporter (spiABCD).
The LVX-related response involved 108 of 174 genes that did not show variations in the No-LVX culture (Fig. 2B). Of them, 4 varied only at 5 min after 10× MIC treatment. Among LVX-responsive genes, 24.1% code for hypothetical proteins, and the same proportion code for transport proteins (Fig. 2C). Interestingly, the only genes upregulated at 5 min in 0.5× MIC were the four genes of the fatDCEB operon (56). These genes were upregulated at every time and LVX concentration used. We have previously shown that the fatDCEB operon is located in a topological domain, D14 (Fig. 3A), showing downregulation under NOV treatment, as tested by microarray experiments (30). We validated these results by qRT-PCR, showing that treatment with NOV caused a decrease in fatD transcription at any time tested at 10× MIC and at 5 min at 0.5× MIC. At 0.5× MIC, a recovery in fatDCE transcription was observed (Fig. 3B), as expected from the general supercoiling recovery (30). In contrast, qRT-PCR confirmed the upregulation of fatDCEB at all times regardless of LVX concentration (Fig. 3C), with fold variation values similar to those in microarrays. To test the role of topo IV inhibition in the upregulation of fatD, qRT-PCR determinations in an LVX-resistant R6 mutant containing a ParCS79F change in topo IV (45), treated with the same LVX concentrations that R6, were performed. No increase of fatD transcription in this strain was observed.
fatDCEB transcription is affected by supercoiling levels.
Even when no changes in the general supercoiling levels were detected in the presence of LVX (Fig. 1C), we assumed that local changes in supercoiling could be involved in the regulation of the fatDCEB operon. To check this possibility, a strain (R6-Pfatcat [Fig. 3A]) that contains a 422-bp region located upstream of fatDCEB that includes the promoter of the operon (Pfat), fused to the cat reporter gene, was inserted into the chromosome 106 kb away from fatDCEB (Fig. 3A). The levels of transcription of fatD and cat were tested by qRT-PCR in cultures treated with two LVX concentrations (Fig. 3C). While fatD showed upregulation in the presence of LVX, almost no change was observed in cat transcription. Thus, supercoiling alteration induced by LVX is acting as a regulator of Pfat, given that its transcriptional upregulation is dependent on its location in a topological chromosomal domain.
In addition, the level of transcription of fatDCEB was tested by qRT-PCR in cultures treated either with NOV, an inhibitor of GyrB (21, 57) (Fig. 3B), or with N-methyl-seconeolitsine (a topo I inhibitor) (58). Treatment with N-methyl-seconeolitsine caused, as expected (58), a general increase in supercoiling (Fig. 4A). This increase was accompanied by a rise in the transcription of fatD and fatC at every concentration tested (Fig. 4B).
FIG 4.

Transcription of fatD depended on the general supercoiling level. Cultures were grown as indicated in the legend to Fig. 3 and treated with N-methyl-seconeolitsine at the indicated concentrations. (A) Plasmid DNA was isolated at the indicated times and subjected to two-dimensional agarose gel electrophoresis in the presence of 5 and 15 μg/ml chloroquine in the first and second dimensions, respectively. Supercoiling density (σ) values are indicated. A black arrowhead indicates the topoisomer that migrated with ΔLk of 0 in the second dimension that has a ΔWr of −30 (58). An open arrowhead indicates the more abundant topoisomer. (B) Total RNA was isolated; cDNA was synthesized and subjected to qRT-PCR, and fatD (black bars) and fatC (white bars) values were normalized to time zero min. Transcription represented the means of qRT-PCR values of three independent replicates ± SEM.
Transcriptional activation of iron transport induced by LVX is involved in cell death.
The increase in transcription of the fatDCEB operon would lead to the accumulation of toxic concentrations of iron within the cell. This toxicity would be related to the activation of the Fenton reaction, which utilizes unincorporated intracellular iron and transfers an electron to hydrogen peroxide (Fig. 5A). To test if intracellular iron is an important component of the LVX-mediated killing, R6 was grown in the presence of LVX in three different media: AGCH (containing 1.58 μM SO4Fe), AGCH plus the iron chelator o-phenantroline, or AGCH deficient in SO4Fe (AGCH*). Attenuation of the bactericidal effect of LVX, both in AGCH + o-phenantroline and in AGCH*, was observed, suggesting a role for intracellular iron in LVX lethality (Fig. 5B).
FIG 5.

LVX lethality is related to the level of intracellular iron. (A) Enzymatic reaction of SpxB that renders H2O2, a substrate of the Fenton reaction. P, phosphate. (B) Viability of S. pneumoniae R6 (black symbols) or R6-ΔspxB (red symbols) either in AGCH, in AGCH plus the iron chelator o-phenantroline (AGCH+Ph), or in AGCH deficient in SO4Fe (AGCH*). Cultures were grown in AGCH to an OD620 of 0.4, diluted 100-fold, and treated, when indicated, with LVX at concentrations equivalent to 2.5× MIC. (C) Accumulation of reactive oxygen species. Results are the means ± SEM of three independent replicates. RFU, relative fluorescence units; values were made relative to zero min and divided by the number of viable cells.
The main source of endogenous hydrogen peroxide in S. pneumoniae is SpxB (59), the pyruvate oxidase enzyme (EC 1.2.3.3) that decarboxylates pyruvate to acetyl phosphate plus H2O2 and CO2 (Fig. 5A). To assess that LVX lethality was related to the production of hydroxyl radicals via the Fenton reaction, an SpxB deletion mutant was constructed as detailed in Materials and Methods. The ΔspxB strain was less susceptible to the killing by LVX, the attenuation being similar to that exhibited by R6 grown either in the presence of o-phenantroline or in AGCH* (Fig. 5B). These results provided a relation between LVX lethality and the Fenton reaction via the increase of intracellular iron. In addition, accumulation of reactive oxygen species was measured by the oxidation of dihydrorhodamine 123. Accumulation was observed in R6 cultures treated with LVX (Fig. 5C), with increases with respect to time zero min higher than 35-fold at 3, 4, and 5 h of treatment. Similar increases in reactive oxygen species had been observed in ciprofloxacin-treated S. pneumoniae with a different dye probe (46) and also in norfloxacin-treated E. coli (35). This accumulation was reverted about 10-fold by o-phenantroline. A similar reversion was observed in the ΔspxB strain.
DISCUSSION
The complex transcriptional response observed in our microarray experiments led us to differentiate among those genes whose transcription was altered as a consequence of growth and those that were LVX dependent. We detected that 5.8% of the genome varied as a consequence of growth, involving mainly genes of 2 of the 13 pneumococcal two-component systems, TCS-12 and TCS-13. The major response corresponded to genes dependent on TCS-12 (ComDE), involved in the regulation of competence for genetic transformation. However, in the presence of LVX, competence genes showed decreases in transcription lower (2.7 ± 0.8, average ± standard deviation [SD]) than those observed at 30 min in the untreated culture. These results suggest that two opposed regulation mechanisms are acting over competence development in the LVX-treated cultures: the growth phase caused downregulation of competence genes, while LVX counteracted this downregulation. These results are in agreement with the described transcriptional activation of ssbB, a late competence gene, after 2.5 h of FQ treatment, with the subsequent induction of transformability (60). Thus, in response to FQs, S. pneumoniae, a bacterium lacking an SOS-like system, activates the competence regulon, supporting the hypothesis that competence is a general stress response of S. pneumoniae (60). Conversely, the upregulation of the genes controlled by TCS-13 (SpiRH) was not affected by LVX treatment.
The LVX-related response included 108 genes (5.2% of the genome). The most striking result in the LVX response was the upregulation of the fatDCEB operon at the earliest time analyzed (5 min) and at the subinhibitory (0.5× MIC) concentration, being the only genes varying under this condition. We tested fatD transcription in an LVX-resistant R6 mutant, and no upregulation was observed in the presence of the antibiotic (Fig. 3C). These results indicate that the LVX transcriptional effects were indeed due to the inhibition of topo IV. However, changes in the general supercoiling levels in the presence of LVX were not found (Fig. 1C). Likewise, no changes in general chromosomal supercoiling were observed in E. coli cells treated with oxolinic acid, an inhibitor of gyrase (61).We assumed that local changes in supercoiling could be involved in the regulation of fatDCEB. To test the role of supercoiling in fatDC transcription, we altered global supercoiling in both directions. On one side, we increased global supercoiling by using the DNA topo I inhibitor N-methyl-seconeolitsine (58). On the other, we decreased supercoiling by using the gyrase B inhibitor NOV (57). We observed an increase both in supercoiling (Fig. 4A) and in fatDC transcription with the topo I inhibitor (Fig. 4B). In contrast, treatment with NOV caused a decrease in fatDC transcription, as detected by qRT-PCR (Fig. 3B), in accordance with a general supercoiling decrease and downregulation of fatDCE transcription in microarrays (30). Microarray results have shown that fatDCEB is located in topological cluster D14, which contains genes downregulated when DNA supercoiling decreases (30). We constructed a strain with a copy of the 422-bp fatDCEB-upstream region fused to cat inserted 106 kb away from its native position. Transcription from Pfat in the presence of LVX varied depending on its chromosomal location. It was upregulated in its appropriate chromosomal location in downregulated cluster D14 but was almost not regulated when located 106 kb away (Fig. 3), in a nonregulated domain.
Besides this supercoiling regulation, the fatDCE operon has been shown to be regulated in several ways, as expected for an operon essential for iron homeostasis. Among them, environmental factors, such as high levels of extracellular Mn2+ (62) and low pH (63) caused its transcriptional upregulation, whereas aerobiosis caused its downregulation (64). Other regulators of the operon are the RitR repressor and (56) the CodY repressor, whose DNA binding capacity is modulated by branched-chain amino acids (65, 66). In accordance, we observed codY downregulation in the LVX response (Table 1), probably contributing to the upregulation of fatDCEB.
The genome of S. pneumoniae R6 encodes three operons for iron transport systems (spr0224-spr0220, spr0934-spr0936, and fatDCEB). Of these, only fatDCEB is involved in iron (Fe2+ and Fe3+) uptake (67). In this way, the upregulation of the operon would cause an increased uptake of iron and its intracellular accumulation, which in turn would activate the Fenton reaction (Fig. 5A). We have observed attenuation of the LVX bactericidal effect in media defective in iron (Fig. 5B), confirming that intracellular iron is a component of LVX-mediated killing. In addition, the accumulation of reactive oxygen species (Fig. 5C) was in accordance with this interpretation. These results agree with the proposed mechanism of killing by bactericidal antibiotics, including FQs (35). The stimulation of the Fenton reaction is the final common step. However, there are several differences between this model and the one we propose in this study (Fig. 6). S. pneumoniae is a lactic acid bacterium that obtains its metabolic energy exclusively from the fermentation of carbohydrates via glycolysis. Its genome does not contain genes for the tricarboxylic acid cycle and lacks the cytochromes and heme-containing proteins involved in aerobic respiration. In addition, although genes coding the FoF1-ATPase are present, this proton pump does not synthesize ATP; conversely, it works at the expense of ATP and serves as the major regulator of intracellular pH (68). Consequently, the only enzymes annotated as iron-sulfur dependent in the S. pneumoniae R6 genome are the two subunits of the L-Ser dehydratase (Spr0094 and Spr0095). The main reason for the increase of intracellular Fe2+ in the presence of LVX should be transcriptional activation of the fatDCEB transporter. The importance of iron in the susceptibility to antibiotics has been recently reinforced by the demonstration that overexpression of an iron efflux system in Salmonella enterica serovar Typhimurium protects cells against ampicillin and ciprofloxacin (69). With respect to the other component of the Fenton reaction—hydrogen peroxide—it is produced in S. pneumoniae mainly by the SpxB enzyme (59, 70). We deleted the gene encoding this enzyme (spxB) and observed that the mutant strain was more resistant to the killing by LVX than its spxB+ parental strain, the attenuation being similar to that exhibited by R6 grown in iron-deficient media (Fig. 5B). The difference in lethality between the wild-type R6 strain and the R6-ΔspxB mutant in the presence of 2.5× MIC LVX was in the same range as that observed between E. coli wild-type and mutant strains lacking either superoxide dismutase activities (36) or both catalase and peroxidase activities (36, 40), which accumulate H2O2, in the presence of norfloxacin at 4× to 10× MIC. We have observed protection to FQ lethality using low LVX concentrations (2.5× MIC), in agreement with results of E. coli treatment with norfloxacin at 2× to 4× MIC (41). In conclusion, we have shown for the first time that fatDCEB transcription is regulated by the supercoiling level. The primary effect of the interaction of LVX-topo IV is the upregulation of the operon by local increase in DNA supercoiling. This upregulation would increase the intracellular level of iron, which activates the Fenton reaction, increasing the concentration of hydroxyl radicals. These effects were observed before the inhibition of protein synthesis mediated by LVX. All these effects, together with the DNA damage caused by the inhibition of topo IV, would account for LVX lethality. The possibility to increase FQs' efficacy by elevating the levels of intracellular ferrous iron remains open.
FIG 6.

Oxidative damage cell death pathway. The inhibition of topo IV by levofloxacin (LVX) or of topoisomerase I by N-methyl-seconeolitsine (SCN) would cause a local increase in supercoiling resulting in the upregulation of the fatDCEB operon. The consequent increase in this iron transporter causes an increase of intracellular ferrous iron (Fe2+). This compound and hydrogen peroxide (produced by the activity of the SpxB pyruvate oxidase) are the substrates of the Fenton reaction. The Fenton reaction renders hydroxyl radicals, which oxidatively damage DNA, proteins, and lipids.
Supplementary Material
ACKNOWLEDGMENTS
We thank Cristina Arnanz for invaluable technical assistance. We thank Ernesto García and Jesús Blázquez for critical comments on the manuscript.
This study was supported by grants BIO2011-25343 from Plan Nacional de I+D+i of the Ministerio de Ciencia e Innovación. CIBER de Enfermedades Respiratorias (CIBERES) is an initiative from Instituto de Salud Carlos III.
Footnotes
Published ahead of print 21 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01706-13.
REFERENCES
- 1.World Health Organization 2007. Pneumococcal conjugate vaccine for childhood immunization—WHO position paper. Wkly. Epidemiol. Rec. 82:93–104 http://www.who.int/wer/2007/wer8212.pdf [PubMed] [Google Scholar]
- 2.Whitney CG, Farley MM, Hadler J, Harrison LH, Bennett NM, Lynfield R, Reingold A, Cieslak PR, Pilishvili T, Jackson D, Facklam RR, Jorgensen JH, Schuchat A, the Active Bacterial Core Surveillance of the Emerging Infections Program Network 2003. Decline in invasive pneumococcal disease after the introduction of protein-polysaccharide conjugate vaccine. N. Engl. J. Med. 348:1737–1746. 10.1056/NEJMoa022823 [DOI] [PubMed] [Google Scholar]
- 3.Kyaw MH, Lynfield R, Schaffner W, Craig AS, Hadler J, Reingold A, Thomas AR, Harrison LH, Bennett NM, Farley MM, Facklam RR, Jorgensen JH, Besser J, Zell ER, Schuchat A, Whitney CG, Active Bacterial Core Surveillance of the Emerging Infection Program Network 2006. Effect of introduction of the pneumococcal conjugate vaccine on drug-resistant Streptococcus pneumoniae. N. Engl. J. Med. 354:1455–1463. 10.1056/NEJMoa051642 [DOI] [PubMed] [Google Scholar]
- 4.Pilishvili T, Lexau C, Farley MM, Hadler J, Harrison LH, Bennett NM, Reingold A, Thomas A, Schaffner W, Craig AS, Smith PJ, Beall BW, Whitney CG, Moore MR, the Active Bacterial Core Surveillance/Emerging Infection Program Network 2010. Sustained reductions in invasive pneumococcal disease in the era of conjugate vaccine. J. Infect. Dis. 201:32–41. 10.1086/648593 [DOI] [PubMed] [Google Scholar]
- 5.Fenoll A, Granizo JJ, Aguilar L, Gimenez MJ, Aragoneses-Fenoll L, Hanquet G, Casal J, Tarragó D. 2009. Temporal trends of invasive Streptococcus pneumoniae serotypes and antimicrobial resistance patterns in Spain from 1979 to 2007. J. Clin. Microbiol. 47:1012–1020. 10.1128/JCM.01454-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fenoll A, Gimenez MJ, Vicioso MD, Granizo JJ, Robledo O, Aguilar L. 2009. Susceptibility of pneumococci causing meningitis in Spain and prevalence among such isolates of serotypes contained in the 7-valent pneumococcal conjugate vaccine. J. Antimicrob. Chemother. 64:1338–1340. 10.1093/jac/dkp376 [DOI] [PubMed] [Google Scholar]
- 7.Moore MR, Gertz JRE, Woodbury RL, Barkocy-Gallagher GA, Schaffner W, Lexau C, Gershman K, Reingold A, Farley M, Harrison LH, Hadler JL, Bennett NM, Thomas AR, McGee L, Pilishvili T, Brueggemann AB, Whitney CG, Jorgensen JH, Beall B. 2008. Population snapshot of emergent Streptococcus pneumoniae serotype 19A in the United States, 2005. J. Infect. Dis. 197:1016–1027. 10.1086/528996 [DOI] [PubMed] [Google Scholar]
- 8.Jacobs MR, Felmingham D, Appelbaum PC, Grüneberg RN, the Alexander Project Group 2003. The Alexander Project 1998–2000: susceptibility of pathogens isolated from community-acquired respiratory tract infection to commonly used antimicrobial agents. J. Antimicrob. Chemother. 52:229–246. 10.1093/jac/dkg321 [DOI] [PubMed] [Google Scholar]
- 9.Riedel S, Beekmann SE, Heilmann KP, Richter SS, Garcia-de-Lomas J, Ferech M, Goosens H, Doern GV. 2007. Antimicrobial use in Europe and antimicrobial resistance in Streptococcus pneumoniae. Eur. J. Clin. Microbiol. Infect. Dis. 26:485–490. 10.1007/s10096-007-0321-5 [DOI] [PubMed] [Google Scholar]
- 10.de la Campa AG, Ardanuy C, Balsalobre L, Pérez-Trallero E, Marimón JM, Fenoll A, Liñares J. 2009. Changes in fluoroquinolone-resistant Streptococcus pneumoniae after 7-valent conjugate vaccination, Spain. Emerg. Infect. Dis. 15:905–911. 10.3201/eid1506.080684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuller JD, McGeer A, Low DE. 2005. Drug-resistant pneumococcal pneumonia: clinical relevance and approach to management. Eur. J. Clin. Microbiol. Infect. Dis. 24:780–788. 10.1007/s10096-005-0059-x [DOI] [PubMed] [Google Scholar]
- 12.Adam HJ, Hoban DJ, Gin AS, Zhanel GG. 2009. Association between fluoroquinolone usage and a dramatic rise in ciprofloxacin-resistant Streptococcus pneumoniae in Canada, 1997–2006. Int. J. Antimicrob. Agents 34:82–85. 10.1016/j.ijantimicag.2009.02.002 [DOI] [PubMed] [Google Scholar]
- 13.Chen DK, McGeer A, de Azavedo JC, Low DE. 1999. Decreased susceptibility of Streptococcus pneumoniae to fluoroquinolones in Canada. N. Engl. J. Med. 341:233–239. 10.1056/NEJM199907223410403 [DOI] [PubMed] [Google Scholar]
- 14.Drlica K, Zhao X. 1997. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 61:377–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muñoz R, de La Campa AG. 1996. ParC subunit of DNA topoisomerase IV of Streptococcus pneumoniae is a primary target of fluoroquinolones and cooperates with DNA gyrase A subunit in forming resistance phenotype. Antimicrob. Agents Chemother. 40:2252–2257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janoir C, Zeller V, Kitzis M-D, Moreau NJ, Gutmann L. 1996. High-level fluoroquinolone resistance in Streptococcus pneumoniae requires mutations in parC and gyrA. Antimicrob. Agents Chemother. 40:2760–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernández-Moreira E, Balas D, González I, de la Campa AG. 2000. Fluoroquinolones inhibit preferentially Streptococcus pneumoniae DNA topoisomerase IV than DNA gyrase native proteins. Microb. Drug Resist. 6:259–267. 10.1089/mdr.2000.6.259 [DOI] [PubMed] [Google Scholar]
- 18.Tankovic J, Perichon B, Duval J, Courvalin P. 1996. Contribution of mutations in gyrA and parC genes to fluoroquinolone resistance of mutants of Streptococcus pneumoniae obtained in vivo and in vitro. Antimicrob. Agents Chemother. 40:2505–2510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Houssaye S, Gutmann L, Varon E. 2002. Topoisomerase mutations associated with in vitro selection of resistance to moxifloxacin in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 46:2712–2715. 10.1128/AAC.46.8.2712-2715.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Champoux JJ. 2001. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 70:369–413. 10.1146/annurev.biochem.70.1.369 [DOI] [PubMed] [Google Scholar]
- 21.Gellert M, Mizuuchi K, ODea H, Nash HA. 1976. DNA gyrase: an enzyme that introduces superhelical turns into DNA. Proc. Natl. Acad. Sci. U. S. A. 73:3872–3876. 10.1073/pnas.73.11.3872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kato J, Nishimura Y, Imamura R, Niki H, Hiraga S, Suzuki H. 1990. New topoisomerase essential for chromosome segregation in E. coli. Cell 63:393–404. 10.1016/0092-8674(90)90172-B [DOI] [PubMed] [Google Scholar]
- 23.Tse-Dinh Y-C. 1985. Regulation of the Escherichia coli DNA topoisomerase I gene by DNA supercoiling. Nucleic Acids Res. 13:4751–4763. 10.1093/nar/13.13.4751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menzel R, Gellert M. 1983. Regulation of the genes for E. coli DNA gyrase: homeostatic control of DNA supercoiling. Cell 34:105–113. 10.1016/0092-8674(83)90140-X [DOI] [PubMed] [Google Scholar]
- 25.Menzel R, Gellert M. 1987. Modulation of transcription by DNA supercoiling: a deletion analysis of the Escherichia coli gyrA and gyrB promoters. Proc. Natl. Acad. Sci. U. S. A. 84:4185–4189. 10.1073/pnas.84.12.4185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Menzel R, Gellert M. 1987. Fusions of the Escherichia coli gyrA and gyrB control regions to the galactokinase gene are inducible by coumermycin treatment. J. Bacteriol. 169:1272–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peter BJ, Arsuaga J, Breier AM, Khodursky AB, Brown PO, Cozzarelli NR. 2004. Genomic transcriptional response to loss of chromosomal supercoiling in Escherichia coli. Genome Biol. 5:R87. 10.1186/gb-2004-5-11-r87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeong KS, Xie Y, Hiasa H, Khodursky AB. 2006. Analysis of pleiotropic transcriptional profiles: a case study of DNA gyrase inhibition. PLoS Genet. 2:e152. 10.1371/journal.pgen.0020152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gmüender H, Kuratli KK, Di Padova CP, Gray W, Keck W, Evers S. 2001. Gene expression changes triggered by exposure of Haemophilus influenzae to novobiocin or ciprofloxacin: combined transcription and translation analysis. Genome Res. 11:28–42. 10.1101/gr.157701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferrándiz MJ, Martín-Galiano AJ, Schvartzman JB, de la Campa AG. 2010. The genome of Streptococcus pneumoniae is organized in topology-reacting gene clusters. Nucleic Acids Res. 38:3570–3581. 10.1093/nar/gkq106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drlica K, Malik M, Kerns RJ, Zhao X. 2008. Quinolone-mediated bacterial death. Antimicrob. Agents Chemother. 52:385–392. 10.1128/AAC.01617-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X, Zhao X, Malik M, Drlica K. 2010. Contribution of reactive oxygen species to pathways of quinolone-mediated bacterial cell death. J. Antimicrob. Chemother. 65:520–524. 10.1093/jac/dkp486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imlay JA, Chin SM, Linn S. 1988. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science 240:640–642. 10.1126/science.2834821 [DOI] [PubMed] [Google Scholar]
- 34.Dwyer DJ, Kohanski MA, Hayete B, Collins JJ. 2007. Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Mol. Syst. Biol. 3:91. 10.1038/msb4100135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. 2007. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810. 10.1016/j.cell.2007.06.049 [DOI] [PubMed] [Google Scholar]
- 36.Wang X, Zhao X. 2009. Contribution of oxidative damage to antimicrobial lethality. Antimicrob. Agents Chemother. 53:1395–1402. 10.1128/AAC.01087-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yeom J, Imlay JA, Park W. 2010. Iron homeostasis affects antibiotic-mediated cell death in Pseudomonas species. J. Biol. Chem. 285:22689–22695. 10.1074/jbc.M110.127456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imlay JA. 2013. The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat. Rev. Microbiol. 11:443–454. 10.1038/nrmicro3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC. 2012. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 336:315–319. 10.1126/science.1219192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y, Imlay JA. 2013. Cell death from antibiotics without the involvement of reactive oxygen species. Science 339:1210–1213. 10.1126/science.1232751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keren I, Wu Y, Inocencio J, Mulcahy LR, Lewis K. 2013. Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science 339:1213–1216. 10.1126/science.1232688 [DOI] [PubMed] [Google Scholar]
- 42.Dorsey-Oresto A, Lu T, Mosel M, Wang X, Salz T, Drlica K, Zhao X. 2013. YihE kinase is a central regulator of programmed cell death in bacteria. Cell Rep. 3:528–537. 10.1016/j.celrep.2013.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ. 2008. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 135:679–690. 10.1016/j.cell.2008.09.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lacks SA, López P, Greenberg B, Espinosa M. 1986. Identification and analysis of genes for tetracycline resistance and replication functions in the broad-host-range plasmid pLS1. J. Mol. Biol. 192:753–765. 10.1016/0022-2836(86)90026-4 [DOI] [PubMed] [Google Scholar]
- 45.Balsalobre L, de la Campa AG. 2008. Fitness of Streptococcus pneumoniae fluoroquinolone-resistant strains with topoisomerase IV recombinant genes. Antimicrob. Agents Chemother. 52:822–830. 10.1128/AAC.00731-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fani F, Leprohon P, Legare D, Ouellette M. 2011. Whole genome sequencing of penicillin-resistant Streptococcus pneumoniae reveals mutations in penicillin-binding proteins and in a putative iron permease. Genome Biol. 12:R115. 10.1186/gb-2011-12-11-r115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pruss GJ, Manes SH, Drlica K. 1982. Escherichia coli DNA topoisomerase I mutants: increased supercoiling is corrected by mutations near gyrase genes. Cell 31:35–42. 10.1016/0092-8674(82)90402-0 [DOI] [PubMed] [Google Scholar]
- 48.Claverys JP, Prudhomme M, Martin B. 2006. Induction of competence regulons as a general response to stress in gram-positive bacteria. Annu. Rev. Microbiol. 60:451–475. 10.1146/annurev.micro.60.080805.142139 [DOI] [PubMed] [Google Scholar]
- 49.Luo P, Morrison DA. 2003. Transient association of an alternative sigma factor, ComX, with RNA polymerase during the period of competence for genetic transformation in Streptococcus pneumoniae. J. Bacteriol. 185:349–358. 10.1128/JB.185.1.349-358.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peterson SN, Sung CK, Cline R, Desai BV, Snesrud EC, Luo P, Walling J, Li H, Mintz M, Tsegaye G, Burr PC, Do Y, Ahn S, Gilbert J, Fleischmann RD, Morrison DA. 2004. Identification of competence pheromone responsive genes in Streptococcus pneumoniae by use of DNA microarrays. Mol. Microbiol. 51:1051–1070. 10.1046/j.1365-2958.2003.03907.x [DOI] [PubMed] [Google Scholar]
- 51.Dagkessamanskaia A, Moscoso M, Henard V, Guiral S, Overweg K, Reuter M, Martin B, Wells J, Claverys JP. 2004. Interconnection of competence, stress and CiaR regulons in Streptococcus pneumoniae: competence triggers stationary phase autolysis of ciaR mutant cells. Mol. Microbiol. 51:1071–1086. 10.1111/j.1365-2958.2003.03892.x [DOI] [PubMed] [Google Scholar]
- 52.Ween O, Gaustad P, Havarstein LS. 1999. Identification of DNA binding sites for ComE, a key regulator of natural competence in Streptococcus pneumoniae. Mol. Microbiol. 33:817–827. 10.1046/j.1365-2958.1999.01528.x [DOI] [PubMed] [Google Scholar]
- 53.Lee MS, Morrison DA. 1999. Identification of a new regulator in Streptococcus pneumoniae linking quorum sensing to competence for genetic transformation. J. Bacteriol. 181:5004–5016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Saizieu A, Gardes C, Flint N, Wagner C, Kamber M, Mitchell TJ, Keck W, Amrein KE, Lange R. 2000. Microarray-based identification of a novel Streptococcus pneumoniae regulon controlled by an autoinduced peptide. J. Bacteriol. 182:4696–4703. 10.1128/JB.182.17.4696-4703.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reichmann P, Hakenbeck R. 2000. Allelic variation in a peptide-inducible two-component system of Streptococcus pneumoniae. FEMS Microbiol. Lett. 190:231–236. 10.1111/j.1574-6968.2000.tb09291.x [DOI] [PubMed] [Google Scholar]
- 56.Ulijasz AT, Andes DR, Glasner JD, Weisblum B. 2004. Regulation of iron transport in Streptococcus pneumoniae by RitR, an orphan response regulator. J. Bacteriol. 186:8123–8136. 10.1128/JB.186.23.8123-8136.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Muñoz R, Bustamante M, de la Campa AG. 1995. Ser-127-to-Leu substitution in the DNA gyrase B subunit of Streptococcus pneumoniae is implicated in novobiocin resistance. J. Bacteriol. 177:4166–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.García MT, Blázquez MA, Ferrándiz MJ, Sanz MJ, Silva-Martín N, Hermoso JA, de la Campa AG. 2011. New alkaloid antibiotics that target the DNA topoisomerase I of Streptococcus pneumoniae. J. Biol. Chem. 286:6402–6413. 10.1074/jbc.M110.148148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spellerberg B, Cundell DR, Sandros J, Pearce BJ, Idanpaan-Heikkila I, Rosenow C, Masure HR. 1996. Pyruvate oxidase, as a determinant of virulence in Streptococcus pneumoniae. Mol. Microbiol. 19:803–813 [DOI] [PubMed] [Google Scholar]
- 60.Prudhomme M, Attaiech L, Sanchez G, Martin B, Claverys JP. 2006. Antibiotic stress induces genetic transformability in the human pathogen Streptococcus pneumoniae. Science 313:89–92. 10.1126/science.1127912 [DOI] [PubMed] [Google Scholar]
- 61.Snyder M, Drlica K. 1979. DNA gyrase on the bacterial chromosome: DNA cleavage induced by oxolonic acid. J. Mol. Biol. 131:287–302 [DOI] [PubMed] [Google Scholar]
- 62.Rosch JW, Gao G, Ridout G, Wang YD, Tuomanen EI. 2009. Role of the manganese efflux system mntE for signalling and pathogenesis in Streptococcus pneumoniae. Mol. Microbiol. 72:12–25. 10.1111/j.1365-2958.2009.06638.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martín-Galiano AJ, Overweg K, Ferrándiz MJ, Reuter M, Wells JM, de la Campa AG. 2005. Transcriptional analysis of the acid tolerance response in Streptococcus pneumoniae. Microbiology 151:3935–3946. 10.1099/mic.0.28238-0 [DOI] [PubMed] [Google Scholar]
- 64.Bortoni ME, Terra VS, Hinds J, Andrew PW, Yesilkaya H. 2009. The pneumococcal response to oxidative stress includes a role for Rgg. Microbiology 155:4123–4134. 10.1099/mic.0.028282-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hendriksen WT, Bootsma HJ, Estevao S, Hoogenboezem T, de Jong A, de Groot R, Kuipers OP, Hermans PW. 2008. CodY of Streptococcus pneumoniae: link between nutritional gene regulation and colonization. J. Bacteriol. 190:590–601. 10.1128/JB.00917-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Caymaris S, Bootsma HJ, Martin B, Hermans PW, Prudhomme M, Claverys JP. 2010. The global nutritional regulator CodY is an essential protein in the human pathogen Streptococcus pneumoniae. Mol. Microbiol. 78:344–360. 10.1111/j.1365-2958.2010.07339.x [DOI] [PubMed] [Google Scholar]
- 67.Manzor ISS, Klosterman TG, Kuipers OP. 2013. Transcriptional response of Streptococcus pneumoniae to varying sources of iron and the regulatory mechanism of iron uptake system PiuBCDA, abstr OGE02. XI Eur. Meet. Mol. Biol. Pneumococcus (EuroPneumo 2013), Madrid, Spain [Google Scholar]
- 68.Martín-Galiano AJ, Ferrándiz MJ, de la Campa AG. 2001. The promoter of the operon encoding the F0F1 ATPase of Streptococcus pneumoniae is inducible by pH. Mol. Microbiol. 41:1327–1338. 10.1046/j.1365-2958.2001.02597.x [DOI] [PubMed] [Google Scholar]
- 69.Frawley ER, Crouch ML, Bingham-Ramos LK, Robbins HF, Wang W, Wright GD, Fang FC. 2013. Iron and citrate export by a major facilitator superfamily pump regulates metabolism and stress resistance in Salmonella typhimurium. Proc. Natl. Acad. Sci. U. S. A. 110:12054–12059. 10.1073/pnas.1218274110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pericone CD, Overweg K, Hermans PWM, Weiser JN. 2000. Inhibitory and bactericidal effects of hydrogen peroxide production by Streptococcus pneumoniae on other inhabitants of the upper respiratory tract. Infect. Immun. 68:3990–3997. 10.1128/IAI.68.7.3990-3997.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.