

Abstract
Polyomavirus BK (BKV) causes polyomavirus-associated nephropathy (PyVAN) and hemorrhagic cystitis (PyVHC) in renal and bone marrow transplant patients, respectively. Antiviral drugs with targeted activity against BKV are lacking. Since the antimalarial drug artesunate was recently demonstrated to have antiviral activity, the possible effects of artesunate on BKV replication in human primary renal proximal tubular epithelial cells (RPTECs), the host cells in PyVAN, were explored. At 2 h postinfection (hpi), RPTECs were treated with artesunate at concentrations ranging from 0.3 to 80 μM. After one viral replication cycle (approximately 72 hpi), the loads of extracellular BKV DNA, reflecting viral progeny production, were reduced in a concentration-dependent manner. Artesunate at 10 μM reduced the extracellular BKV load by 65%; early large T antigen mRNA and protein expression by 30% and 75%, respectively; DNA replication by 73%; and late VP1 mRNA and protein expression by 47% and 64%, respectively. Importantly, the proliferation of RPTECs was also inhibited in a concentration-dependent manner. At 72 hpi, artesunate at 10 μM reduced cellular DNA replication by 68% and total metabolic activity by 47%. Cell impedance and lactate dehydrogenase measurements indicated a cytostatic but not a cytotoxic mechanism. Flow cytometry and 5-ethynyl-2′-deoxyuridine incorporation revealed a decreased number of cells in S phase and suggested cell cycle arrest in G0 or G2 phase. Both the antiproliferative and antiviral effects of artesunate at 10 μM were reversible. Thus, artesunate inhibits BKV replication in RPTECs in a concentration-dependent manner by inhibiting BKV gene expression and genome replication. The antiviral mechanism appears to be closely connected to cytostatic effects on the host cell, underscoring the dependence of BKV on host cell proliferative functions.
INTRODUCTION
The ubiquitous human polyomavirus BK (BKV) is linked to the two major diseases polyomavirus-associated nephropathy (PyVAN), affecting 1 to 10% of kidney transplant recipients, and polyomavirus-associated hemorrhagic cystitis (PyVHC), affecting 5 to 15% of allogeneic hematopoietic stem cell transplant recipients (1, 2). The pathogenesis of PyVAN is characterized by high-level BKV replication in renal tubular epithelial cells of the transplant, leading to cytopathic loss of the cell monolayer, followed by tubular atrophy and interstitial fibrosis (1). Importantly, there is also a high level of BKV replication in the urothelial cells, which may influence the progression of PyVAN (3–5). The pathogenesis of PyVHC is not fully understood but has been suggested to result from a sequence of events involving cytotoxicity from the conditioning protocol received by the patients before transplantation, high-level BKV replication in the urothelial cells of the bladder mucosa, and subsequent inflammation (1, 6, 7).
Unfortunately, antiviral drugs with specific activity against polyomavirus replication are still lacking. For PyVAN, the mainstay of therapy is to improve BKV-specific immunity by reducing or discontinuing immunosuppressive drugs, but this approach is not always applicable or sufficient for the treatment of PyVAN (8) and cannot be used for the treatment of PyVHC. The development of a drug specifically targeting BKV replication is complicated, since the virus has a small genome encoding only a few targetable proteins and is heavily reliant on host cell proteins, for instance, DNA polymerase for genome replication. Some patients have been treated with the nucleotide analogue cidofovir or the pyrimidine synthesis inhibitor leflunomide, but there are no randomized controlled studies, and the graft survival benefit is questionable (9–11). Our in vitro studies with cidofovir and leflunomide concluded that their anti-BKV activities were related to nonspecific cytostatic effects (12, 13).
Artesunate, a semisynthetic derivative of an extract (artemisinin) from the traditional Chinese medicinal herb Artemisia annua, is the preferred drug for the treatment of severe malaria (14–16). In 2001, artesunate was, for the first time, reported to have antiviral activity against human cytomegalovirus (CMV) in vitro (17), and a few years later it was also reported to have activity against rat CMV in vivo (18). In 2008, a patient with recurrent multiresistant CMV infection was successfully treated with artesunate (19), and since then 7 more transplant patients with CMV infections were treated with varying success (20, 21). Recently, a patient with multidrug-resistant herpes simplex virus 2 (HSV-2) infection (22) and a child with human herpesvirus 6B (HHV-6B) myocarditis (23) were successfully treated with artesunate. In addition, antiviral activity in vitro has also been found against other herpesviruses, including herpes simplex virus 1 (17), Epstein-Barr virus (24), and human herpesvirus 6A (25), and also to some extent against nonherpesviruses, such as hepatitis B virus (26), hepatitis C virus (27), HIV-1 (17), and bovine viral diarrhea virus (28). Moreover, artesunate has been reported to be active against cancer cells and parasites (reviewed in reference 27).
The reported broad antiviral activity, coupled with high bioavailability (29) and limited adverse effects (16), made it an interesting candidate for antiviral therapy of BKV infections. Thus, the aim of our study was to perform a detailed investigation of the effects of artesunate on BKV replication in human primary renal proximal tubular epithelial cells (RPTECs), the host cells for BKV during PyVAN.
Here we show that artesunate inhibits BKV replication in RPTECs in a concentration-dependent manner by a mechanism closely connected to its cytostatic effects. Our results also underscore the close relationship between BKV replication and the host cells.
MATERIALS AND METHODS
Cells and virus.
Human RPTECs (ScienCell) were propagated in renal epithelial growth medium (REGM; Lonza) containing 0.5% fetal bovine serum. No latent BKV was detected by quantitative PCR (qPCR) of intracellular DNA. All experiments were performed with RPTECs at passage 4 and BKV Dunlop supernatants or CsCl gradient-purified virus, both obtained from Vero cells. BKV Dunlop has been shown to have a high replication capacity in RPTECs (30) and has been used for several antiviral studies in these cells (12, 13, 31, 32).
Infection and drug treatment.
Artesunate (Saokim, Hanoi, Vietnam) was dissolved in dimethyl sulfoxide (DMSO) to a concentration of 473 mM and immediately stored in aliquots at −70°C. Prior to use, the stock was diluted in the growth medium to working concentrations. RPTECs at about 50% confluence were incubated with BKV Dunlop for 2 h before surplus infectious units were removed. The RPTECs were washed once with phosphate-buffered saline (PBS), and the medium with or without artesunate was added, unless indicated otherwise. A DMSO control at a concentration matching the DMSO concentration in the 10 μM artesunate dilution was included in all experiments.
Extraction of RNA and intracellular DNA.
A NucleoSpin TriPrep (total DNA, RNA, and protein isolation) kit (Macherey-Nagel GmbH) was used to isolate RNA and intracellular DNA from the cell lysates prepared from untreated and artesunate-treated cells at 24 and 48 h postinfection (hpi). In brief, the cells were washed once with PBS before a mixture of 350 μl RP1 buffer (provided in the kit) and 3.5 μl β-mercaptoethanol was added to lyse the cells. The cell lysates were collected and stored at −70°C until the isolation of RNA and DNA following the manufacturer's instructions. The RNA and DNA concentrations were measured by use of a NanoDrop apparatus.
cDNA synthesis and quantification of BKV mRNA levels.
cDNA was synthesized from 150 ng RNA using a high-capacity reverse transcription (RT) kit (Applied Biosystems), and the level of BKV mRNA was quantified in duplicate by RT-qPCR, as described previously (12). Of note, truncated large T antigen but no small T antigen (sT-ag) transcripts were also intercepted by the LT-ag RT-qPCR, but for simplicity, we refer to these as LT-ag transcripts. The housekeeping gene human hypoxanthine phosphoribosyltransferase (huHPRT) was found to be insignificantly affected by artesunate treatment for 24 and 48 h and was therefore used for normalization by the 2−ΔΔCT threshold cycle (CT) method (33). The expression at 24 hpi was also normalized to the level of expression of beta-actin (ACTB), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and TATA box binding protein (TBP).
Quantification of extracellular and intracellular BKV loads.
Cell culture supernatants were harvested and stored at −70°C until the extracellular BKV DNA load was quantified by qPCR using primers and a probe targeting the BKV LT-ag gene (34). Shortly before qPCR, supernatants were diluted in distilled H2O (1:100) and boiled for 5 min. The intracellular BKV DNA load was determined by performing the same qPCR on DNA extracted from cells. To express the amount of intracellular BKV DNA as the number of genome equivalents (GEq) per cell, each sample was simultaneously analyzed by qPCR for the gene aspartoacylase (ACY) (12, 35). All experiments had duplicate samples, and each sample was measured in triplicate.
Western blotting.
To prepare cell lysates for Western blotting, cells in 48-well plates were solubilized in 50 μl cell disruption buffer (mirVana microRNA isolation kit; Ambion) at 24 hpi or 100 μl buffer at 48 hpi and stored at −70°C until use. For each sample, 24 μl of cell lysate was separated by SDS-polyacrylamide gel electrophoresis (PAGE) and proteins were blotted onto a polyvinylidene difluoride (PVDF) membrane. Viral and cellular proteins were detected and quantified by the use of a Li-Cor Odyssey imager as previously described (31). The following primary antibodies were used: polyclonal rabbit antiserum directed against BKV VP1 (36), BKV agnoprotein (37, 38), and BKV N-terminal LT-ag (37). Antiserum directed against BKV N-terminal LT-ag detected both LT-ag and sT-ag. In addition, a monoclonal mouse antibody directed against the housekeeping protein GAPDH (6C5; Abcam) was used. The protein standard marker was the Odyssey two-color marker (Li-Cor).
Immunofluorescence staining, microscopy, and digital image processing.
Immunofluorescence staining was performed as previously described (31) using as primary antibodies a polyclonal rabbit antiserum directed against BKV agnoprotein (37, 38) and a mouse monoclonal anti-simian virus 40 (anti-SV40) LT-ag antibody (PAb416; Abcam). DNA was labeled with Draq5 dye (Biostatus). Images were captured using a Nikon TE2000 microscope (×10 objective) and processed with NIS-Elements BR (version 3.2) software (Nikon Corporation).
Infectious progeny virus release.
The supernatants harvested from untreated and artesunate-treated BKV-infected RPTECs at 72 hpi were used to infect new RPTECs for 2 h before removing the inoculum, washing the cells, and adding fresh growth medium. At 72 hpi, the cells were washed, methanol fixed, and immunostained as described above.
Cell viability and cell proliferation assay.
The viability of mock- and BKV-infected RPTECs with or without artesunate treatment was monitored in real time using an xCELLigence RTCA DP instrument as previously described (31). This system measures the adhesion, proliferation, and size of the cells and expresses these together as an arbitrary unit, the cell index (CI). In short, at 25 h after seeding, half of the medium was replaced with fresh medium with or without purified BKV Dunlop and with or without artesunate. The CI was measured every 15 min for the first 6 h after seeding and thereafter every 30 min.
In addition, cellular DNA replication in mock- and BKV-infected RPTECs with or without artesunate treatment was quantified by colorimetric measurement of bromodeoxyuridine (BrdU) incorporation into DNA (for 20 h) using a cell proliferation enzyme-linked immunosorbent assay (Roche). Total cellular metabolic activity was monitored by colorimetric measurement of the reduction of resazurin (Res) dye (for 3 h) by mitochondrial, microsomal, and cytosolic enzymes using a TOX-8 kit (Sigma).
The potential cytotoxic effects of artesunate on BKV-infected RPTECs were quantified by use of a Cytotoxicity Detection KitPLUS (Roche) by following the manufacturer's instructions. In short, the lactate dehydrogenase (LDH) released from dead cells was measured in supernatants. Similarly, the total LDH (in supernatants and cells) of parallel wells was measured after prior addition of a potent lysis solution provided in the kit. Percent cytotoxicity was calculated by dividing the amount of LDH in the supernatant by the total amount of LDH for the corresponding concentration of artesunate. Each experiment had triplicate samples.
Analysis of cell cycle by flow cytometry.
Untreated and artesunate-treated RPTECs cultured in 75-cm2 flasks were trypsinized, washed twice with PBS, and fixed in 75% ice-cold ethanol at 4°C for at least 24 h. Next, the cells were centrifuged at 300 × g for 5 min, and the ethanol was completely removed without disturbing the cell pellet. The cells were resuspended in PBS with 1% bovine serum albumin (BSA) and incubated at room temperature for 5 min. Thereafter, the cells were centrifuged again at 300 × g for 5 min, washed once with PBS with 1% BSA, resuspended in PBS, stained with Draq5 (1:500) for 15 to 20 min, and passed through cell strainer filters. Using a Becton, Dickinson FACSCalibur flow cytometer (BD Biosciences), about 15,000 cells per sample were recorded, and cell cycle analysis was performed by using FlowJo software (Tree Star, Inc.).
Analysis of cell proliferation by EdU incorporation.
Actively proliferating RPTECs were visualized by incorporation of the thymidine analogue 5-ethynyl-2′-deoxyuridine (EdU) into newly synthesized DNA using a Click-iT EdU imaging kit (Invitrogen). In short, EdU was added to untreated or artesunate-treated cells at a final concentration of 10 μM, and the cells were incubated for 2 h, before they were fixed by 4% paraformaldehyde and permeabilized by addition of 0.5% Triton X-100. EdU was fluorescently labeled according to the manufacturer's instructions before immunofluorescence and phase-contrast images were acquired with a Nikon TE2000 microscope (×10 objective).
SI50 of artesunate.
Extracellular BKV loads and cellular DNA replication (mock-infected RPTECs) measured at 72 hpi were analyzed by the XLfit program (fit model 210; dose-response one site, five-parameter logistic model, fit = {A + [(B − A)/(1 + x/C)^D]}^E; IDBS) to determine the artesunate effective concentration for 50% inhibition of the BKV load (EC50) and for 50% cell cytotoxicity (CC50). The selectivity index (SI50) was calculated by dividing the CC50 by the EC50.
Statistical analysis.
When appropriate, the mean and standard deviation (SD) were calculated and P values were determined by the use of the unpaired t test (GraphPad). For the t test, the mean value of the untreated samples was compared to the mean value of the artesunate-treated samples.
RESULTS
Artesunate inhibits BKV replication in RPTECs.
BKV Dunlop requires approximately 48 to 72 h to complete one replication cycle in RPTECs (12). In order to determine the effect of artesunate on BKV replication, cells were infected for 2 h, artesunate was added at 0.3 to 80 μM, and the BKV DNA load in the supernatants was measured by qPCR at 72 hpi. Artesunate decreased the extracellular BKV DNA load in a concentration-dependent manner, with a reduction of up to 1.2 log (i.e., a 93% reduction) (Fig. 1A). Artesunate at 10 μM gave a 65% reduction, and the DMSO control (which gave 13% inhibition) indicated that part of this reduction was due to the DMSO.
FIG 1.
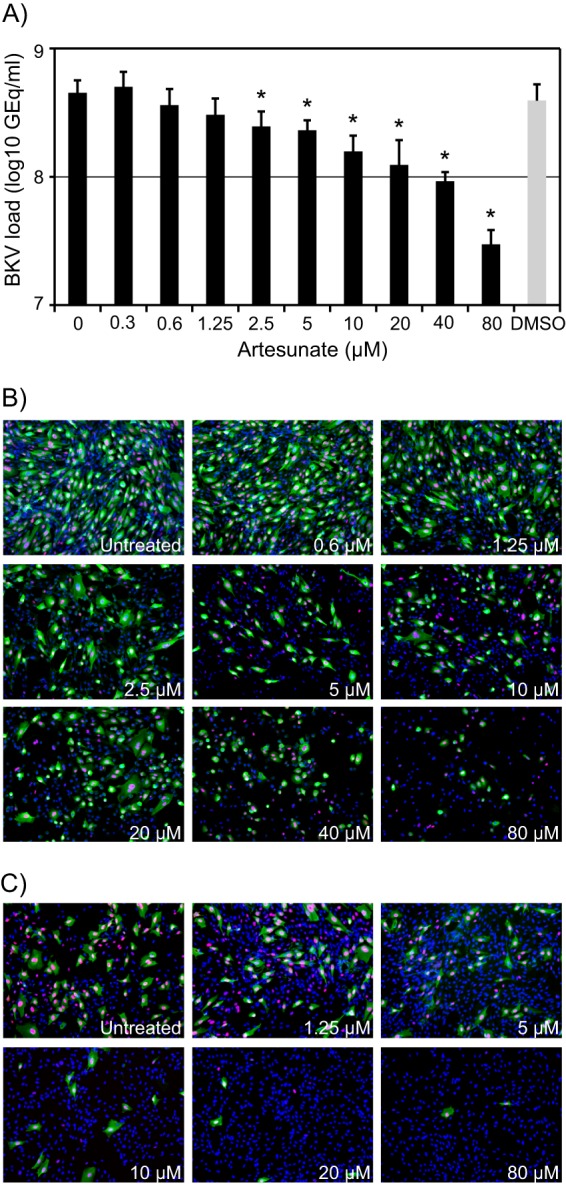
Effect of increasing artesunate concentrations on BKV replication in RPTECs. (A) Extracellular BKV DNA load. At 72 hpi, supernatants were harvested from BKV-infected RPTECs treated with artesunate at the indicated concentrations from 2 hpi, and BKV DNA loads were measured by qPCR. Mean values of the number of GEq/ml ± SDs of four experiments (each experiment was performed in two wells) are presented. *, P <0.05, determined by the t test. (B) Expression of BKV proteins. At 72 hpi, BKV-infected RPTECs treated with the indicated concentrations of artesunate from 2 hpi were fixed and stained with polyclonal rabbit anti-agnoprotein serum for visualization of the BKV late agnoprotein (green) and with the SV40 LT-ag monoclonal antibody PAb416 for visualization of the BKV early protein LT-ag (red). Cell nuclei (blue) were stained with Draq5. The pictures were taken with a fluorescence microscope (×10 objective). (C) Infectious progeny release. The supernatants from untreated and artesunate-treated BKV-infected RPTECs harvested at 72 hpi were also seeded onto new RPTECs. At 72 hpi, these cells were fixed and indirect immunofluorescence staining was performed, as described for panel B. The pictures were taken with a fluorescence microscope (×10 objective).
Immunofluorescence staining of cells fixed at 72 hpi showed a concentration-dependent reduction in cells expressing the BKV early protein LT-ag (red) and the late agnoprotein (green) (Fig. 1B). Of note, the staining also revealed a gradual reduction in cell numbers (blue) when increasing concentrations of artesunate were used. However, the morphology of the cells seemed to be unchanged with concentrations up to 10 μM (data not shown).
To investigate whether the inhibition of the BKV DNA load in the supernatants also corresponded to a decline in the amount of infectious BKV progeny released, the collected supernatants were used to infect new RPTECs. Again, cells were fixed at 72 hpi and immunofluorescence staining was performed with antibodies directed against LT-ag and agnoprotein. A significant concentration-dependent reduction in the number of cells expressing LT-ag (red) and agnoprotein (green) was seen when supernatants from cells treated with artesunate at 1.25 μM or higher concentrations were used as inocula (Fig. 1C). These results confirmed that artesunate decreased the release of infectious progeny in a concentration-dependent manner. We conclude that artesunate inhibits BKV replication in RPTECs. Since artesunate at 10 μM gave a 65% BKV load reduction without changing the cellular morphology, this concentration was chosen for use in the next experiments, unless otherwise stated.
Artesunate reduces BKV early and late gene expression.
To investigate the effect of artesunate on BKV early and late gene expression, we measured LT-ag and VP1 mRNA in untreated and artesunate-treated RPTECs by RT-qPCR at 24 and 48 hpi. The results were normalized to the expression of the housekeeping gene huHPRT and presented as the fold expression relative to that for the untreated sample at 24 hpi. Artesunate reduced LT-ag mRNA expression from 1.0- to 0.7-fold (30% reduction) and from 16- to 14-fold (12.5% reduction) at 24 hpi and 48 hpi, respectively (Fig. 2A). At 24 hpi, the normalization was also performed with other housekeeping genes (ACTB, GAPDH, and TBP), and this gave similar results (data not shown). At 24 hpi, the mRNA expression of VP1 was, as expected, very low, and no significant differences were found between untreated and artesunate-treated samples. At 48 hpi, artesunate reduced the level of VP1 mRNA expression from 509- to 269-fold, i.e., a reduction of 47% (Fig. 2B). The DMSO control did not affect LT-ag mRNA expression but reduced VP1 mRNA expression by 13%.
FIG 2.
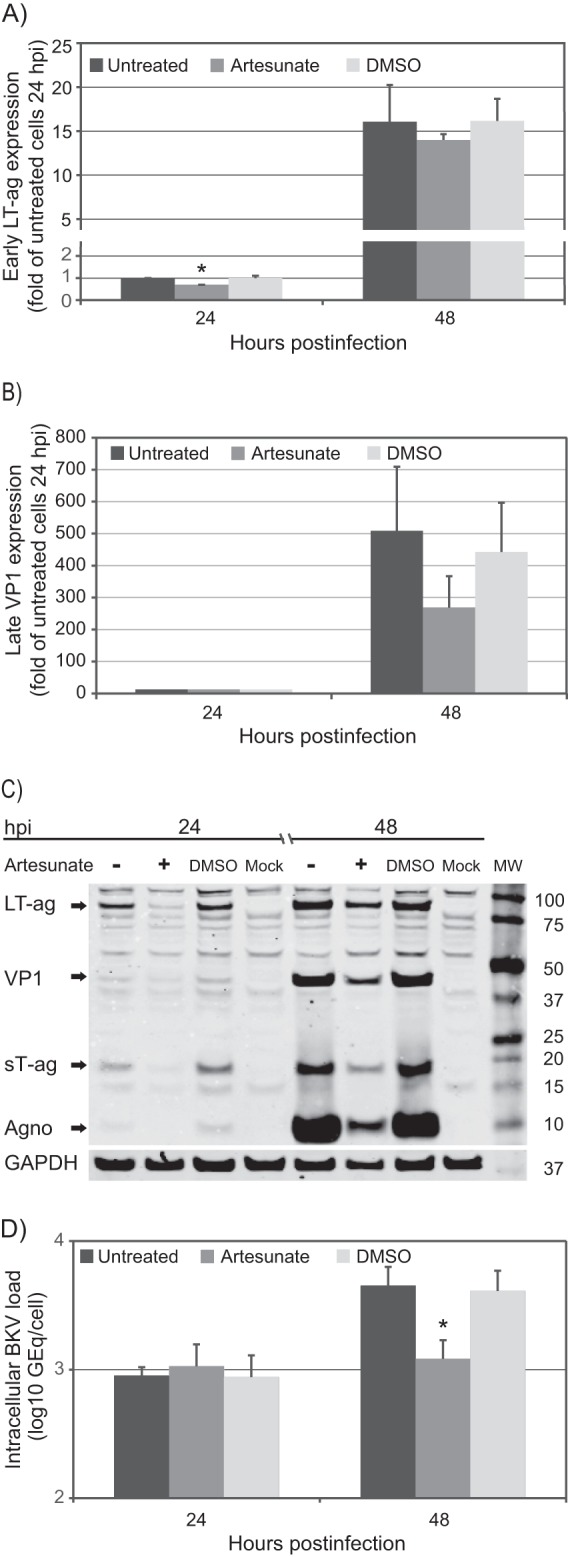
Effect of artesunate at 10 μM on different phases of the BKV replication cycle. (A) BKV early mRNA expression. RNA was isolated from untreated and artesunate-treated BKV-infected RPTECs at 24 and 48 hpi, and LT-ag mRNA levels were measured by RT-qPCR and normalized to the levels of the huHPRT transcripts. Mean fold expression values ± SDs of three experiments (each sample was prepared from two wells) are presented, where the level of untreated cells at 24 hpi was arbitrarily set to 1. *, P < 0.05, determined by the t test. (B) BKV late mRNA expression. VP1 mRNA levels were measured by RT-qPCR, normalized to the levels of the huHPRT transcripts, and presented as described for panel A. (C) BKV early and late protein expression. Cell extracts were made from untreated and artesunate-treated BKV-infected RPTECs at 24 and 48 hpi, and Western blotting was performed with polyclonal rabbit anti-N-terminal LT-ag, anti-VP1, and anti-agnoprotein (Agno) serum and a monoclonal antibody directed against the housekeeping protein GAPDH. The anti-N-terminal LT-ag serum recognizes LT-ag and sT-ag. Lane MW, molecular weight markers (indicated on the right, in thousands). (D) BKV genome replication. Untreated and artesunate-treated BKV-infected RPTECs were harvested at 24 and 48 hpi, DNA was extracted, and qPCR for BKV and the cellular gene ACY was performed. Intracellular BKV DNA was expressed as number of GEq per cell. Mean values of the number of GEq/cell ± SDs of four experiments (each of two experiments was performed in two wells) are presented.
Next, we investigated the effect of artesunate on BKV protein expression by Western blot analysis. The same volume of cell lysate from all samples was investigated, and the results were normalized to the expression of the housekeeping protein GAPDH. Compared to untreated samples, 75% and 25% reductions of LT-ag expression were found at 24 and 48 hpi, respectively (Fig. 2C), and a similar reduction was found for sT-ag expression (Fig. 2C). At 24 hpi, very low levels of BKV late proteins were observed, and presumably, VP1 mainly represents input virus. At 48 hpi, both VP1 and agnoprotein were strongly expressed in untreated cells, while artesunate treatment reduced their expression by 64 and 49%, respectively. Of note, artesunate also reduced the expression of GAPDH by 40 and 70% at 24 and 48 hpi, respectively, probably reflecting the reduced cell numbers observed by Draq5 staining after artesunate treatment (Fig. 1B). The DMSO control did not affect LT-ag protein expression but increased VP1 expression by 13% and reduced agnoprotein expression by 3%. We conclude that artesunate has a significant inhibitory effect on the expression of early and late BKV proteins. For the early proteins, the inhibition is more pronounced at 24 than at 48 hpi.
Artesunate reduces BKV genome replication.
To study the effect of artesunate on BKV genome replication in RPTECs starting at about 36 hpi (12), we measured intracellular BKV DNA and cellular ACY loads in untreated and artesunate-treated RPTECs by qPCR at 24 and 48 hpi and expressed the results as the number of GEq per cell. At 24 hpi, the intracellular BKV DNA measured mainly represents input genomes. In agreement with this, no significant difference in the intracellular BKV DNA load was found between untreated and artesunate-treated cells (Fig. 2D). From 24 to 48 hpi, the intracellular BKV DNA load in untreated cells was found to increase by 0.7 log. However, in artesunate-treated cells, the increase was only 0.1 log. On the basis of this, artesunate was found to reduce the replication of BKV DNA by 73% compared to the level of replication for untreated cells (Fig. 2D). Of note, the cellular ACY load was 17% lower in artesunate-treated cells than untreated cells at 48 hpi, probably reflecting the lower cell numbers. The DMSO control decreased BKV replication by 9%. We conclude that artesunate inhibits BKV genome replication in RPTECs.
Artesunate induces cytostatic effects in RPTECs.
Although phase-contrast microscopy of mock- and BKV-infected cells exposed to artesunate at a concentration up to 10 μM for 70 h (72 hpi) did not reveal changes in morphology compared to the morphology of untreated cells, the nuclear staining (blue) unveiled a reduction in cell numbers when more than 1.25 μM artesunate was used (Fig. 1B). We therefore decided to investigate the effect of artesunate on the viability of mock- and BKV-infected RPTECs in more detail. First, we examined the influence of artesunate concentrations ranging from 1.25 to 40 μM in real time for 96 h using the xCELLigence system. In order to keep the disturbance of the measurements to a minimum, artesunate was added together with the virus. Artesunate influenced the cell index (CI) in a similar manner in both mock- and BKV-infected cells (Fig. 3A). All concentrations tested completely stopped cell proliferation. With artesunate at 40 μM, cell proliferation stopped at 7 h posttreatment, while this effect was delayed up to 17 h posttreatment when lower concentrations were used. For artesunate at 1.25 and 2.5 μM, the inhibition of proliferation was partly abolished at 48 h posttreatment. Also, for artesunate at 5 and 10 μM, a partial reversal of inhibition was seen, though to a lesser extent than with the lower concentrations. For artesunate at 20 and 40 μM, no reversal of inhibition was seen during observation for 96 h posttreatment. The DMSO control did not seem to affect proliferation. In summary, addition of artesunate completely stopped the proliferation of both mock-infected and BKV-infected RPTECs from about 7 h after addition. However, the inhibition was partly released after about 40 h for concentrations up to 10 μM but not for higher concentrations.
FIG 3.
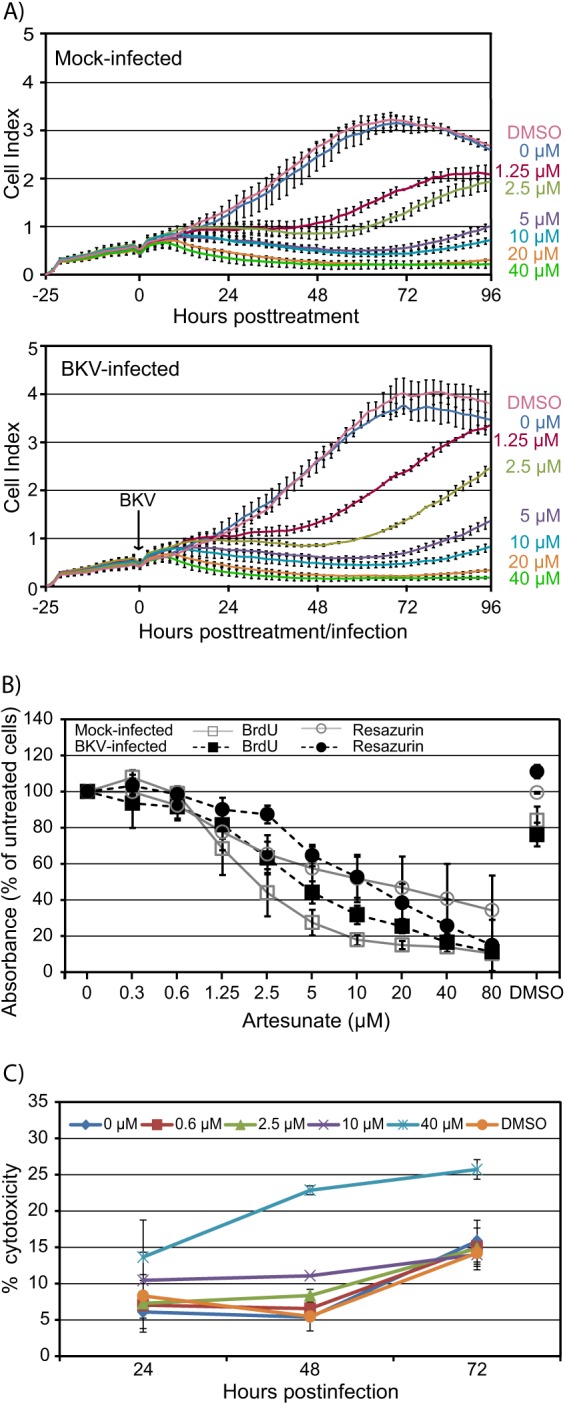
Effect of increasing concentrations of artesunate on viability of mock- and BKV-infected RPTECs. (A) Real-time cell proliferation. RPTECs were seeded in an E plate 16, and 25 h later, half of the medium in the wells was replaced by growth medium with artesunate to reach the indicated concentrations (top). In some wells, purified BKV Dunlop was also added (bottom). The CI, a combined measure of cell adhesion, proliferation, and size, was monitored from the time of seeding until 96 h posttreatment and infection by an xCELLigence RTCA DP instrument. The CIs, normalized to the value for the cell-free background, are shown as mean values ± SDs of 2 wells. The results of one representative experiment out of two are shown. (B) DNA replication and metabolic activity. Cellular DNA replication (BrdU) and the total metabolic activity (resazurin) of artesunate-treated mock- and BKV-infected RPTECs were measured at 72 hpi (i.e., 70 h posttreatment). Mean values of the percent absorbance of untreated cells ± SDs of three experiments (each experiment was performed in three wells) are presented. (C) Cytotoxicity. BKV-infected RPTECs were treated with the indicated concentrations of artesunate from 2 hpi. The LDH levels in the supernatant and the LDH levels in the well after complete lysis of the cell layer (total LDH) were measured at 24, 48, and 72 hpi. Cytotoxicity was calculated by dividing the amount of LDH in the supernatant by the total amount of LDH in the wells with the corresponding artesunate concentrations. Mean values of the percent cytotoxicity ± SDs from two to three experiments (each experiment was performed in three wells) are presented.
We next examined the effect of artesunate at concentrations ranging from 0.3 to 80 μM on cellular DNA replication (measured by the BrdU assay) and on total metabolic activity (measured by the resazurin assay) at 72 hpi. As before, artesunate was added at 2 hpi and left on the cells until 72 hpi (i.e., 70 h posttreatment), when measurements were performed. Artesunate was found to reduce both cellular DNA replication and total metabolic activity in a concentration-dependent manner. The DNA replication in mock- and BKV-infected cells was reduced between 0 and 90% (Fig. 3B). Of note, the DNA replication was slightly more affected in mock-infected cells than in infected cells. For instance, artesunate at 10 μM reduced cellular DNA replication in mock- and BKV-infected cells by 82 and 68%, respectively. The DMSO control reduced cellular DNA replication in mock- and BKV-infected cells by 16 and 24%, respectively.
The total metabolic activity in mock- and BKV-infected cells was reduced by between 0 and 85% (Fig. 3B). While mock-infected cells were slightly more affected by concentrations below 10 μM, infected cells were most affected by concentrations above 10 μM. Artesunate at 10 μM reduced total metabolic activity in both mock- and BKV-infected cells by approximately 50%. The DMSO control gave no reduction in total metabolic activity.
After having shown that artesunate leads to significantly decreased cell numbers (Fig. 1B), cellular DNA replication (Fig. 3B), total metabolic activity (Fig. 3B), and cell proliferation (Fig. 3A), we investigated whether artesunate also induced cytotoxic effects by measuring the LDH activity in supernatants. Since the half-life of released LDH has been found to be only 9 h in cell culture (39), we decided to measure LDH at 24, 48, and 72 hpi. The results are presented as percent cytotoxicity, where 100% cytotoxicity was the total LDH level measured in supernatants after complete lysis of the cells. In untreated cells, a cytotoxicity of 6% was found at 24 hpi, and presumably due to viral cytopathic effects, it increased up to 16% at 72 hpi. In artesunate-treated cells, concentrations from 0.6 to 10 μM induced slightly higher relative cytotoxicity at 24 and 48 hpi, while no difference from untreated cells was observed at 72 hpi (Fig. 3C). Artesunate at 40 μM, on the other hand, increased the cytotoxicity at every time point investigated. At 48 hpi the cytotoxicity was 4 times higher than in untreated cells and it increased to a maximum of 26% at 72 hpi. The DMSO control did not affect the cytotoxicity. In summary, artesunate concentrations up to 10 μM gave only a weakly increased cytotoxic effect at 24 and 48 hpi, while 40 μM increased the cytotoxicity at all time points but especially at 48 hpi.
The finding of a mainly cytostatic effect of artesunate made us ask how the cell cycle distribution in RPTECs was affected by artesunate. We decided to investigate the effect of 10 and 40 μM artesunate after 24 and 72 h of treatment by performing flow cytometry of ethanol-fixed and Draq5-stained mock-infected RPTECs. After 24 h of treatment with artesunate at 10 μM, the cell population in S phase was decreased by 42% compared to the S-phase population of untreated cells. At the same time, the cell population in G0/G1 phase was increased 5% and that in G2/M phase was increased almost 2% (Fig. 4A and B). Artesunate at 40 μM gave exactly the same decrease of the cell population in S phase, but here the cell population in G0/G1 phase was decreased by 17%, while the cell population in G2/M phase was increased by 56% (Fig. 4A and B). At 72 h posttreatment, artesunate at both 10 and 40 μM decreased the cell population in S phase by approximately 50%, while the cell population in G2/M phase was increased by 9 and 132% at concentrations of 10 and 40 μM, respectively (Fig. 4C). In summary, artesunate dramatically decreased the cell population in S phase, but it appeared that the different concentrations of artesunate did this by arresting the cells at different points in the cell cycle.
FIG 4.

Effect of artesunate on cell cycle distribution of RPTECs. (A) Untreated RPTECs and RPTECs treated with 10 or 40 μM artesunate for 24 h were collected, fixed, and stained with nuclear stain Draq5 immediately before flow cytometric analysis was performed. Representative results of one of three independent experiments are shown. (B) As for panel A. The area histogram was used to determine the percentage of cells in G0/G1, S, and G2/M phase after 24 h of artesunate treatment. The results show the means ± SDs of two experiments. *, P < 0.05, determined by the t test. (C) As for panel A. The area histogram was used to determine the percentage of cells in G0/G1, S, and G2/M phase after 72 h of artesunate treatment. The results show the means ± SDs of four experiments. *, P < 0.05, determined by the t test. (D) RPTECs were labeled with EdU (blue) for the last 2 h before the cells were fixed by 4% paraformaldehyde at 24 h posttreatment. Images were taken by fluorescence and phase-contrast microscopy (×10 objective).
To study the effect of artesunate on the cell cycle distribution on the single-cell level, the Click-iT EdU cell proliferation assay was used. This assay allowed us to detect cells with DNA replication during the time span when EdU was present. First, artesunate at 10 μM or 40 μM was added to mock-infected RPTECs, and then, 22 h later, EdU was added for 2 h before the cells were fixed at 24 h posttreatment. In agreement with our flow cytometry data, a 24-h artesunate exposure resulted in an approximate halving of the number of cells going through S phase independent of the artesunate concentration used (Fig. 4D). Similar results were also obtained after 72 h of artesunate exposure (data not shown). In summary, artesunate decreased the cell population in S phase.
We conclude that artesunate has a rapid and strong cytostatic effect on both mock- and BKV-infected RPTECs. For artesunate concentrations of 10 μM or less, inhibition is short-lived and seems to be caused by cell cycle arrest in G0. However, for higher artesunate concentrations, the inhibition is longer lasting and seems to also involve G2 arrest.
The cytostatic and the antiviral effects of artesunate in BKV-infected RPTECs are reversible.
The data demonstrate that artesunate at 10 μM inhibits BKV replication and host cell proliferation without overt cytotoxicity. In order to assess whether these effects of artesunate may be reversed by removal of artesunate, mock- and BKV-infected cells were treated with artesunate at 10 μM for 10 h before the artesunate was removed, the cells were washed, and medium without artesunate was added. A 10-h treatment was chosen since adverse effects on cell proliferation had been found from approximately 7 h posttreatment. Subsequently, the BKV load was measured at 72 hpi, cell proliferation was monitored in real time until 72 hpi (i.e., 72 h posttreatment), and the cell cycle distribution was analyzed by flow cytometry at 72 h posttreatment. By interrupting the treatment after 10 h, the BKV DNA load was reduced by only 0.1 log unit, whereas it was decreased by 0.6 log after 70 h of treatment (i.e., a 24% versus a 75% reduction) (Fig. 5A). The real-time viability assay showed that BKV-infected cells (Fig. 5B) and mock-infected cells (data not shown) almost returned to normal proliferation approximately 3 h after artesunate was removed. In agreement with this, the flow cytometry data showed that the cell cycle distribution 62 h after the removal of artesunate was similar to that of untreated cells (Fig. 5C).
FIG 5.
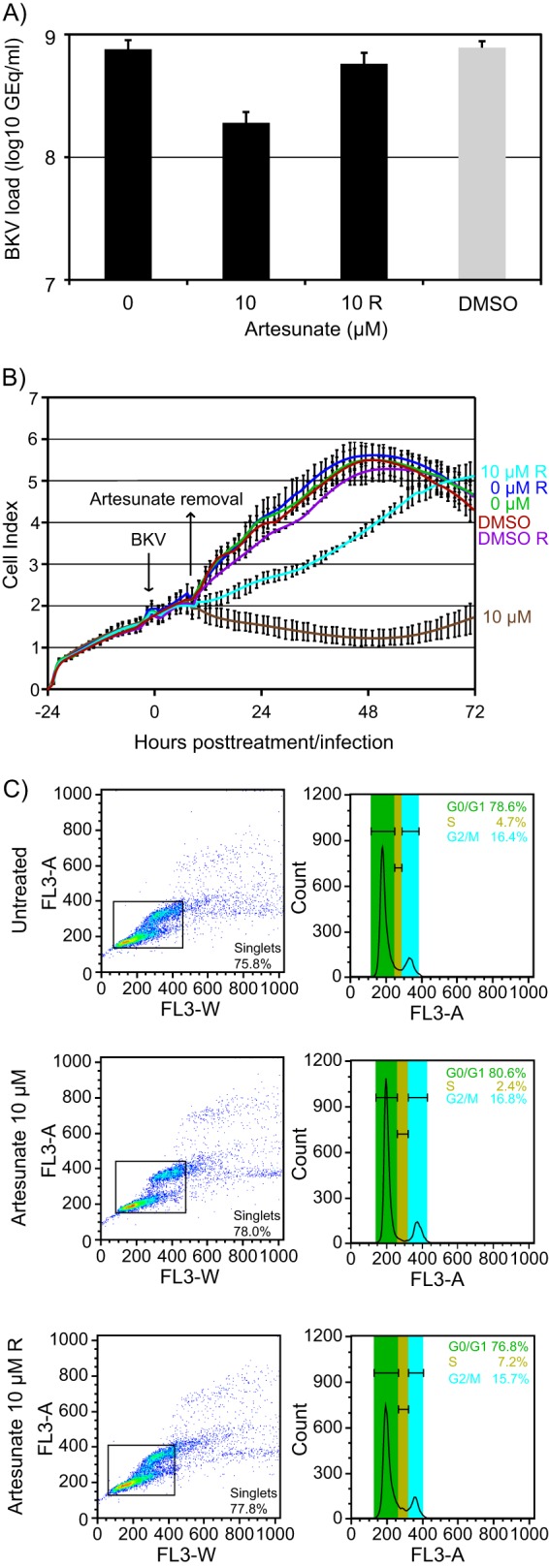
Reversible effects of artesunate. Mock- and BKV-infected RPTECs were left untreated or treated with the indicated concentrations of artesunate or DMSO. Bars or graphs labeled with R (removal) represent wells where artesunate was removed from the cells at approximately 10 h posttreatment and replaced with fresh medium. (A) Supernatants were harvested at 72 hpi from BKV-infected RPTECs treated with the indicated concentrations of artesunate from 2 hpi, and BKV DNA loads were measured by qPCR. Mean numbers of GEq/ml ± SDs of four experiments (each experiment was performed in two wells) are presented. (B) The CI was monitored by the xCELLigence RPTEC DP instrument from the time of cell seeding until 72 h posttreatment/infection. The CIs are shown as mean values ± SDs of 2 wells. The results of one representative experiment out of three independent experiments are shown. (C) Cell cycle distribution of mock-infected RPTECs determined by flow cytometric analysis at 72 h posttreatment.
We conclude that the strong cytostatic effects of artesunate on RPTECs can be almost completely reversed by removal of artesunate. However, by restoring the proliferation potential of the cells, most of the antiviral activity of artesunate was lost.
The SI of artesunate in RPTECs is low, likely reflecting the dependence of BKV on host cell proliferation.
The relative efficacy of an antiviral drug may be defined by the selectivity index (SI), usually expressed as SI50 (CC50/EC50). To find the EC50, we calculated the 50% reduction in the BKV DNA load at 72 hpi on the basis of the data in Fig. 1A, and this was found to be 4.2 μM (Fig. 6A). Since the cytotoxic effects of artesunate found here were far less than 50%, we decided to determine the CC50 on the basis of cellular DNA replication, as previously described for CMX001 (31), ofloxacin, and levofloxacin (32). The CC50 was therefore calculated on the basis of the BrdU incorporation data in Fig. 3A, and this was found to be 2.4 μM (Fig. 6B). These results suggested an SI50 (CC50/EC50) of ∼0.6 for artesunate on RPTECs. We conclude that the selectivity index determined for proliferating RPTECs was low; however, the present calculation was based on a method much more sensitive than that usually employed for cell toxicity.
FIG 6.
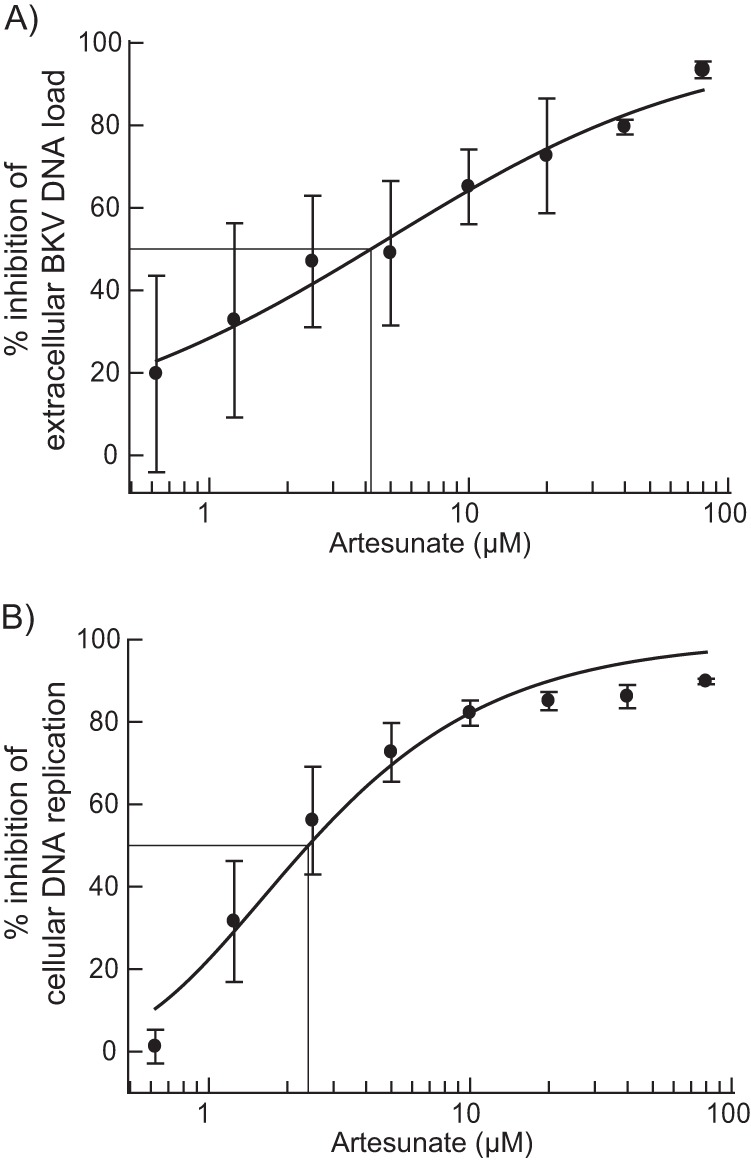
Determination of artesunate EC50 and CC50 by curve fitting. To determine the EC50 and CC50, the effect of increasing artesunate concentrations on extracellular BKV DNA loads and on cellular DNA replication (BrdU incorporation in uninfected cells) was analyzed by curve fitting using the XLfit program. Mean values ± SDs of at least three independent experiments are presented.
DISCUSSION
An antiviral treatment option for PyVAN is urgently needed, but drugs efficiently inhibiting BKV replication are lacking. We have explored the antiviral activity of artesunate, an antimalarial drug recently shown to have antiviral activity, on BKV replication in human primary renal proximal tubular epithelial cells. The data presented here indicate that artesunate inhibits BKV replication in a concentration-dependent manner. With artesunate at 10 μM, a 65% reduction of the extracellular BKV DNA load at 72 hpi was found, reflecting a significant decrease in infectious progeny release. Several steps in the BKV replication cycle were inhibited, such as early BKV mRNA and protein expression, as well as BKV genome replication and late mRNA and protein expression. In addition, artesunate inhibited BKV proliferation in a concentration-dependent manner. With artesunate at 10 μM, cellular DNA replication was reduced 68%, while metabolic activity was reduced 50%. In mock-infected cells, the reduction in DNA replication was 82%, while metabolic activity was the same as that for infected cells. By real-time monitoring of cell proliferation, a reduction was registered as early as 7 h after artesunate addition. The effect was mainly cytostatic and was transient when concentrations up to 10 μM were used. The mechanism seems to involve a concentration-dependent arrest in G0 or G2, reducing the cell population in S phase. Removal of the drug almost completely reversed the cytostatic effects but also reduced the inhibition of BKV replication. Taken together, these data suggest that the antiviral effect of artesunate on BKV replication in RPTECs is closely related to its strong but partly reversible cytostatic effect.
In a productive BKV infection, transcription of early pre-mRNA is the first step taking place after uncoating of the BKV genome. This is followed by splicing to 3 functional early mRNAs encoding LT-ag, st-ag, and truncated LT-ag. Our detailed study with 10 μM artesunate demonstrated a 30% reduction of normalized LT-ag mRNA levels at 24 hpi. In agreement with this, an approximately 75% reduction of both LT-ag and st-ag protein expression was found by Western blotting at 24 hpi. Of note, this finding is different from the finding in our previous studies with cidofovir and CMX001, where no reduced LT-ag mRNA and protein expression was found as early as 24 hpi (12, 31). Since LT-ag functions as the replicative helicase and initiator of viral DNA replication by melting the double-stranded DNA strand to initiate fork unwinding and by recruiting cellular enzymes like DNA polymerase α primase, topoisomerase I, and replication protein A (40, 41), the low LT-ag level detected could be responsible for the 73% reduced viral DNA replication observed in artesunate-treated cells at 48 hpi. However, part of the inhibition was probably caused by other mechanisms, like cell cycle arrest (see below), since cellular DNA replication, which is completely independent of LT-ag, was also reduced.
In addition to the initiation and coordination of DNA replication, LT-ag stimulates late gene expression from newly replicated BKV DNA by activating transcription from the late promoter (42). At 48 hpi, a 47% reduction in VP1 mRNA transcripts and corresponding 64% and 49% reductions of VP1 and agnoprotein, respectively, were found. Both the lack of LT-ag and the reduced BKV genome replication, with the latter leading to fewer templates for late transcription, probably contributed to the reduced late mRNA and protein expression. We cannot exclude the possibility of an additional direct inhibitory effect on late transcription and translation. Not unexpectedly, the inhibition of all investigated steps of the BKV replication cycle culminated in a 65% reduction of the BKV DNA load and a corresponding reduction of infectious progeny. The reason that some cells expressed both early and late BKV proteins and therefore seemed to be resistant to even high artesunate concentrations is unknown.
With regard to the effect on host cells, the RPTECs, artesunate induced a significant concentration-dependent inhibition of cell proliferation. The effect was already apparent 7 to 17 h after the start of treatment, when artesunate concentrations from 1.25 μM completely stopped cell proliferation. Both cellular DNA replication and metabolic activity were significantly reduced, but the LDH assay revealed that the effect was cytostatic and not cytotoxic. The cytostatic effect was characterized in more detail by flow cytometry analysis. Artesunate decreased the numbers of cells in S phase by more than 50%. However, while artesunate at 40 μM increased the numbers of cells in G2/M phase, artesunate at 10 μM slightly increased the numbers of cells in G0/G1 phase. The results therefore suggest that the mechanism inhibiting cell proliferation depends on the artesunate concentration. This is also supported by our real-time proliferation analysis, where only cells treated with artesunate at concentrations of 10 μM or less, which were possibly arrested in G0 phase, spontaneously reinitiated proliferation after 40 to 50 h. Of note, artesunate and dihydroartemisinin have previously been described to decrease the numbers of cells in S phase in A431 human epidermoid carcinoma cells (43), cells of the human leukemia cell line K562 (44), and cells of the murine myeloma cell line SP2/0 (45). Interestingly, while some found simultaneously increased numbers of cells in G0/G1 phase (43) (45), others described increased numbers of cells in G2/M phase (46). Apparently, artesunate also induced apoptosis in many cancer cell lines (43, 45, 46). Since we did not detect any sub-G1-phase fraction when performing flow cytometry, we concluded that artesunate at 10 and 40 μM does not induce apoptosis in RPTECs.
Interestingly, the reduction of cellular DNA replication was more pronounced in mock-infected than in BKV-infected RPTECs; i.e., artesunate at 10 μM reduced cellular DNA replication in mock- and BKV-infected cells by 82 and 68%, respectively. One explanation for this could be that the BKV infection counteracted the inhibitory effect of artesunate. It is well-known that polyomaviruses can override the cell cycle, as recently also demonstrated for BKV (47). The mechanism has been studied in detail for simian virus 40 (SV40) (41). Early in infection, LT-ag interacts with retinoblastoma (Rb) proteins to free the transcription factor E2F. This drives the infected cells into the beneficial replicative S phase, which induces a DNA damage response. However, LT-ag binds and inactivates p53, thereby preventing apoptosis or cellular senescence. In agreement with this, we have previously demonstrated that BKV infection increases cellular DNA replication in RPTECs (12).
In our study, cultured cells infected with BKV received a single dose of artesunate, and this gave a transient effect, at least at concentrations up to 10 μM. It is possible that repetitive smaller doses would have given a stronger effect, as seen in an in vitro study of CMV (48). For clinical use, frequent dosing would probably be necessary, since the drug is rapidly metabolized in the liver and has a half-life of only 0.36 or 2.14 h, depending on the route of administration (reviewed in reference 29). Due to the short half-life, the current World Health Organization-recommended treatment of malaria is artesunate in combination with other antimalaria drugs, and both drugs are usually administered for 3 to 7 days (49). However, a 7-day treatment regime would probably not be sufficient for a successful treatment of PyVAN or PyVHC. Mathematical modeling has shown that a >90% reduction of renal BKV replication must be maintained for up to 10 weeks in order to clear plasma and urine viral loads (3). Long-term treatment might become a problem, since both animal and human studies suggest that long-term rather than short-term peak concentrations of artesunate may cause toxicity (reviewed in reference 50).
Concerning the present study, only minimal cytotoxic effects were found following 72 h of artesunate treatment, but significant cytostatic effects were revealed as early as 7 h after treatment start. Interestingly, cytostatic effects have not been reported in other antiviral studies on artesunate. There may be several reasons for this, for instance, the use of different cells or the treatment of fully confluent cells, but it is also possible that mainly cytotoxic effects were investigated. For instance, the effect on host cells was investigated by measuring residual cell layers by the LDH assay at 4 or 7 days after artesunate addition to confluent cells (17, 51) or by measuring the LDH release in the supernatant 7 days after artesunate addition (18). Although short-term artesunate treatment is considered safe, there have been some reports on hemolytic anemia after treatment of severe malaria, but this may actually be a consequence of the severe malaria and not of artesunate (52). Even though the cytostatic effects will probably not afflict the slowly cycling epithelial cells of healthy kidneys or the urinary bladder, it may jeopardize the regeneration of epithelium damaged by PyVAN or PyVHC.
In order to kill the malaria parasites, only nanomolar concentrations of artemisinins are needed (reviewed in reference 50). However, to inhibit the replication of BKV in RPTECs by 50% (i.e., the EC50), a concentration of 4.2 μM (1.6 μg/ml) artesunate was needed, and it is still unclear if this concentration can be attained in kidney epithelial cells in vivo. Following intravenous administration of artesunate in healthy volunteers, plasma concentrations up to 217 μM (83.34 μg/ml) have been described (29, 53, 54), indicating that the EC50 could at least be achieved in the plasma.
Adverse side effects of drugs may be acceptable if they occur at concentrations higher than the concentration required to achieve the therapeutic effect. We calculated a SI50 of only 0.6. Since the EC50 was found to be in the same range as that reported for herpesviruses (1.5 to 6.4 μM) (24, 25, 48, 51), the low SI50 mainly resulted from the low CC50 of only 2.4 μM. However, unlike most researchers, we calculate the CC50 using the very sensitive BrdU assay. Nevertheless, comparing it to the SI50 of ofloxacin (6.48) and levofloxacin (7.13), which were obtained using the same assay and cells (32), the SI50 of artesunate was found to be at least 10 times lower.
In conclusion, artesunate inhibits BKV replication in RPTECs in a concentration-dependent manner by impairing BKV gene expression and genome replication. Notably, the antiviral effect is closely connected to the cytostatic effect of the drug on the host cell. Importantly, both BKV inhibition and cytostatic effects are reversible. Whether artesunate will be useful for the treatment of BKV disease can be answered only by performing carefully designed clinical studies also addressing long-term effects. Such studies need to take the short half-life of artesunate into account and might consider combination therapy which is also indicated when using artesunate in the treatment of malaria.
ACKNOWLEDGMENTS
We thank Garth D. Tylden at the Department of Microbiology and Infection Control at the University Hospital of North Norway for critical reading of the manuscript and helpful comments. We also thank a former member of our research group, Solrun Finstad, for excellent technical assistance and Tore J. Gutteberg for support.
The project and B.N.S. are financially supported by the Northern Norway Regional Health Authority Medical Research Program, and M.M. is funded in part by the Deutsche Forschungsgemeinschaft (grant DFG MA 1289/7-1).
Footnotes
Published ahead of print 21 October 2013
REFERENCES
- 1.Hirsch HH. 2010. Polyoma and papilloma virus infections after hematopoietic stem cell or solid organ transplantation, p 465–482 In Bowden P, Ljungman P, Snydman DR. (ed), Transplant infections, 3rd ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Hirsch HH, Steiger J. 2003. Polyomavirus BK. Lancet Infect. Dis. 3:611–623. 10.1016/S1473-3099(03)00770-9 [DOI] [PubMed] [Google Scholar]
- 3.Funk GA, Gosert R, Comoli P, Ginevri F, Hirsch HH. 2008. Polyomavirus BK replication dynamics in vivo and in silico to predict cytopathology and viral clearance in kidney transplants. Am. J. Transplant. 8:2368–2377. 10.1111/j.1600-6143.2008.02402.x [DOI] [PubMed] [Google Scholar]
- 4.Nickeleit V, Hirsch HH, Binet IF, Gudat F, Prince O, Dalquen P, Thiel G, Mihatsch MJ. 1999. Polyomavirus infection of renal allograft recipients: from latent infection to manifest disease. J. Am. Soc. Nephrol. 10:1080–1089 [DOI] [PubMed] [Google Scholar]
- 5.Hirsch HH. 2002. Polyomavirus BK nephropathy: a (re-)emerging complication in renal transplantation. Am. J. Transplant. 2:25–30. 10.1034/j.1600-6143.2002.020106.x [DOI] [PubMed] [Google Scholar]
- 6.Azzi A, Cesaro S, Laszlo D, Zakrzewska K, Ciappi S, de Santis R, Fanci R, Pesavento G, Calore E, Bosi A. 1999. Human polyomavirus BK (BKV) load and haemorrhagic cystitis in bone marrow transplantation patients. J. Clin. Virol. 14:79–86. 10.1016/S1386-6532(99)00055-4 [DOI] [PubMed] [Google Scholar]
- 7.Binet I, Nickeleit V, Hirsch HH. 2000. Polyomavirus infection in transplant recipients. Curr. Opin. Organ Transplant. 5:210–216. 10.1097/00075200-200009000-00007 [DOI] [Google Scholar]
- 8.Hirsch HH, Randhawa P. 2009. BK virus in solid organ transplant recipients. Am. J. Transplant. 9(Suppl 4):S136–S146. 10.1111/j.1600-6143.2009.02904.x [DOI] [PubMed] [Google Scholar]
- 9.Krisl JC, Taber DJ, Pilch N, Chavin K, Bratton C, Thomas B, McGillicuddy J, Baliga P. 2012. Leflunomide efficacy and pharmacodynamics for the treatment of BK viral infection. Clin. J. Am. Soc. Nephrol. 7:1003–1009. 10.2215/CJN.12531211 [DOI] [PubMed] [Google Scholar]
- 10.Johnston O, Jaswal D, Gill JS, Doucette S, Fergusson DA, Knoll GA. 2010. Treatment of polyomavirus infection in kidney transplant recipients: a systematic review. Transplantation 89:1057–1070. 10.1097/TP.0b013e3181d0e15e [DOI] [PubMed] [Google Scholar]
- 11.Rinaldo CH, Hirsch HH. 2007. Antivirals for the treatment of polyomavirus BK replication. Expert Rev. Anti Infect. Ther. 5:105–115. 10.1586/14787210.5.1.105 [DOI] [PubMed] [Google Scholar]
- 12.Bernhoff E, Gutteberg TJ, Sandvik K, Hirsch HH, Rinaldo CH. 2008. Cidofovir inhibits polyomavirus BK replication in human renal tubular cells downstream of viral early gene expression. Am. J. Transplant. 8:1413–1422. 10.1111/j.1600-6143.2008.02269.x [DOI] [PubMed] [Google Scholar]
- 13.Bernhoff E, Tylden GD, Kjerpeseth LJ, Gutteberg TJ, Hirsch HH, Rinaldo CH. 2010. Leflunomide inhibition of BK virus replication in renal tubular epithelial cells. J. Virol. 84:2150–2156. 10.1128/JVI.01737-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klayman DL. 1985. Qinghaosu (artemisinin): an antimalarial drug from China. Science 228:1049–1055. 10.1126/science.3887571 [DOI] [PubMed] [Google Scholar]
- 15.Dondorp AM, Fanello CI, Hendriksen IC, Gomes E, Seni A, Chhaganlal KD, Bojang K, Olaosebikan R, Anunobi N, Maitland K, Kivaya E, Agbenyega T, Nguah SB, Evans J, Gesase S, Kahabuka C, Mtove G, Nadjm B, Deen J, Mwanga-Amumpaire J, Nansumba M, Karema C, Umulisa N, Uwimana A, Mokuolu OA, Adedoyin OT, Johnson WB, Tshefu AK, Onyamboko MA, Sakulthaew T, Ngum WP, Silamut K, Stepniewska K, Woodrow CJ, Bethell D, Wills B, Oneko M, Peto TE, von Seidlein L, Day NP, White NJ. 2010. Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomised trial. Lancet 376:1647–1657. 10.1016/S0140-6736(10)61924-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dondorp A, Nosten F, Stepniewska K, Day N, White N. 2005. Artesunate versus quinine for treatment of severe falciparum malaria: a randomised trial. Lancet 366:717–725. 10.1016/S0140-6736(05)67176-0 [DOI] [PubMed] [Google Scholar]
- 17.Efferth T, Marschall M, Wang X, Huong SM, Hauber I, Olbrich A, Kronschnabl M, Stamminger T, Huang ES. 2002. Antiviral activity of artesunate towards wild-type, recombinant, and ganciclovir-resistant human cytomegaloviruses. J. Mol. Med. 80:233–242. 10.1007/s00109-001-0300-8 [DOI] [PubMed] [Google Scholar]
- 18.Kaptein SJ, Efferth T, Leis M, Rechter S, Auerochs S, Kalmer M, Bruggeman CA, Vink C, Stamminger T, Marschall M. 2006. The anti-malaria drug artesunate inhibits replication of cytomegalovirus in vitro and in vivo. Antiviral Res. 69:60–69. 10.1016/j.antiviral.2005.10.003 [DOI] [PubMed] [Google Scholar]
- 19.Shapira MY, Resnick IB, Chou S, Neumann AU, Lurain NS, Stamminger T, Caplan O, Saleh N, Efferth T, Marschall M, Wolf DG. 2008. Artesunate as a potent antiviral agent in a patient with late drug-resistant cytomegalovirus infection after hematopoietic stem cell transplantation. Clin. Infect. Dis. 46:1455–1457. 10.1086/587106 [DOI] [PubMed] [Google Scholar]
- 20.Wolf DG, Shimoni A, Resnick IB, Stamminger T, Neumann AU, Chou S, Efferth T, Caplan O, Rose J, Nagler A, Marschall M. 2011. Human cytomegalovirus kinetics following institution of artesunate after hematopoietic stem cell transplantation. Antiviral Res. 90:183–186. 10.1016/j.antiviral.2011.03.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lau PK, Woods ML, Ratanjee SK, John GT. 2011. Artesunate is ineffective in controlling valganciclovir-resistant cytomegalovirus infection. Clin. Infect. Dis. 52:279. 10.1093/cid/ciq050 [DOI] [PubMed] [Google Scholar]
- 22.Sellar RS, Ward KN, Thomson KJ, Peggs KS. 2012. Evidence for clinical activity of artesunate in multidrug-resistant herpes simplex infection following HSCT. Bone Marrow Transplant. 47:1482–1483. 10.1038/bmt.2012.46 [DOI] [PubMed] [Google Scholar]
- 23.Hakacova N, Klingel K, Kandolf R, Engdahl E, Fogdell-Hahn A, Higgins T. 2013. First therapeutic use of artesunate in treatment of human herpesvirus 6B myocarditis in a child. J. Clin. Virol. 57:157–160. 10.1016/j.jcv.2013.02.005 [DOI] [PubMed] [Google Scholar]
- 24.Auerochs S, Korn K, Marschall M. 2011. A reporter system for Epstein-Barr virus (EBV) lytic replication: anti-EBV activity of the broad anti-herpesviral drug artesunate. J. Virol. Methods 173:334–339. 10.1016/j.jviromet.2011.03.005 [DOI] [PubMed] [Google Scholar]
- 25.Milbradt J, Auerochs S, Korn K, Marschall M. 2009. Sensitivity of human herpesvirus 6 and other human herpesviruses to the broad-spectrum antiinfective drug artesunate. J. Clin. Virol. 46:24–28. 10.1016/j.jcv.2009.05.017 [DOI] [PubMed] [Google Scholar]
- 26.Romero MR, Efferth T, Serrano MA, Castano B, Macias RI, Briz O, Marin JJ. 2005. Effect of artemisinin/artesunate as inhibitors of hepatitis B virus production in an “in vitro” replicative system. Antiviral Res. 68:75–83. 10.1016/j.antiviral.2005.07.005 [DOI] [PubMed] [Google Scholar]
- 27.Efferth T, Romero MR, Wolf DG, Stamminger T, Marin JJ, Marschall M. 2008. The antiviral activities of artemisinin and artesunate. Clin. Infect. Dis. 47:804–811. 10.1086/591195 [DOI] [PubMed] [Google Scholar]
- 28.Romero MR, Serrano MA, Vallejo M, Efferth T, Alvarez M, Marin JJ. 2006. Antiviral effect of artemisinin from Artemisia annua against a model member of the Flaviviridae family, the bovine viral diarrhoea virus (BVDV). Planta Med. 72:1169–1174. 10.1055/s-2006-947198 [DOI] [PubMed] [Google Scholar]
- 29.Morris CA, Duparc S, Borghini-Fuhrer I, Jung D, Shin CS, Fleckenstein L. 2011. Review of the clinical pharmacokinetics of artesunate and its active metabolite dihydroartemisinin following intravenous, intramuscular, oral or rectal administration. Malaria J. 10:263. 10.1186/1475-2875-10-263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olsen GH, Hirsch HH, Rinaldo CH. 2009. Functional analysis of polyomavirus BK non-coding control region quasispecies from kidney transplant recipients. J. Med. Virol. 81:1959–1967. 10.1002/jmv.21605 [DOI] [PubMed] [Google Scholar]
- 31.Rinaldo CH, Gosert R, Bernhoff E, Finstad S, Hirsch HH. 2010. 1-O-Hexadecyloxypropyl cidofovir (CMX001) effectively inhibits polyomavirus BK replication in primary human renal tubular epithelial cells. Antimicrob. Agents Chemother. 54:4714–4722. 10.1128/AAC.00974-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma BN, Li R, Bernhoff E, Gutteberg TJ, Rinaldo CH. 2011. Fluoroquinolones inhibit human polyomavirus BK (BKV) replication in primary human kidney cells. Antiviral Res. 92:115–123. 10.1016/j.antiviral.2011.07.012 [DOI] [PubMed] [Google Scholar]
- 33.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 34.Hirsch HH, Mohaupt M, Klimkait T. 2001. Prospective monitoring of BK virus load after discontinuing sirolimus treatment in a renal transplant patient with BK virus nephropathy. J. Infect. Dis. 184:1494–1496. 10.1086/324425 [DOI] [PubMed] [Google Scholar]
- 35.Randhawa PS, Vats A, Zygmunt D, Swalsky P, Scantlebury V, Shapiro R, Finkelstein S. 2002. Quantitation of viral DNA in renal allograft tissue from patients with BK virus nephropathy. Transplantation 74:485–488. 10.1097/00007890-200208270-00009 [DOI] [PubMed] [Google Scholar]
- 36.Rinaldo CH, Myhre MR, Alstad H, Nilssen O, Traavik T. 2003. Human polyomavirus BK (BKV) transiently transforms and persistently infects cultured osteosarcoma cells. Virus Res. 93:181–187. 10.1016/S0168-1702(03)00096-0 [DOI] [PubMed] [Google Scholar]
- 37.Hey AW, Johnsen JI, Johansen B, Traavik T. 1994. A two fusion partner system for raising antibodies against small immunogens expressed in bacteria. J. Immunol. Methods 173:149–156. 10.1016/0022-1759(94)90294-1 [DOI] [PubMed] [Google Scholar]
- 38.Rinaldo CH, Traavik T, Hey A. 1998. The agnogene of the human polyomavirus BK is expressed. J. Virol. 72:6233–6236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riss TL, Moravec RA. 2004. Use of multiple assay endpoints to investigate the effects of incubation time, dose of toxin, and plating density in cell-based cytotoxicity assays. Assay Drug Dev. Technol. 2:51–62. 10.1089/154065804322966315 [DOI] [PubMed] [Google Scholar]
- 40.Cuesta I, Nunez-Ramirez R, Scheres SH, Gai D, Chen XS, Fanning E, Carazo JM. 2010. Conformational rearrangements of SV40 large T antigen during early replication events. J. Mol. Biol. 397:1276–1286. 10.1016/j.jmb.2010.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.An P, Saenz Robles MT, Pipas JM. 2012. Large T antigens of polyomaviruses: amazing molecular machines. Annu. Rev. Microbiol. 66:213–236. 10.1146/annurev-micro-092611-150154 [DOI] [PubMed] [Google Scholar]
- 42.Cole CN. 1996. Polyomavirinae: the viruses and their replication, p 1997–2025 In Fields BN, Knipe DM, Howley PM. (ed), Fields virology, 3rd ed. Lippincott-Raven Press, Philadelphia, PA [Google Scholar]
- 43.Jiang Z, Chai J, Chuang HH, Li S, Wang T, Cheng Y, Chen W, Zhou D. 2012. Artesunate induces G0/G1 cell cycle arrest and iron-mediated mitochondrial apoptosis in A431 human epidermoid carcinoma cells. Anticancer Drugs 23:606–613. 10.1097/CAD.0b013e328350e8ac [DOI] [PubMed] [Google Scholar]
- 44.Finaurini S, Basilico N, Corbett Y, D'Alessandro S, Parapini S, Olliaro P, Haynes RK, Taramelli D. 2012. Dihydroartemisinin inhibits the human erythroid cell differentiation by altering the cell cycle. Toxicology 300:57–66. 10.1016/j.tox.2012.05.024 [DOI] [PubMed] [Google Scholar]
- 45.Li S, Xue F, Cheng Z, Yang X, Wang S, Geng F, Pan L. 2009. Effect of artesunate on inhibiting proliferation and inducing apoptosis of SP2/0 myeloma cells through affecting NFkappaB p65. Int. J. Hematol. 90:513–521. 10.1007/s12185-009-0409-z [DOI] [PubMed] [Google Scholar]
- 46.Xu Q, Li ZX, Peng HQ, Sun ZW, Cheng RL, Ye ZM, Li WX. 2011. Artesunate inhibits growth and induces apoptosis in human osteosarcoma HOS cell line in vitro and in vivo. J. Zhejiang Univ. Sci. B 12:247–255. 10.1631/jzus.B1000373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang M, Zhao L, Gamez M, Imperiale MJ. 2012. Roles of ATM and ATR-mediated DNA damage responses during lytic BK polyomavirus infection. PLoS Pathog. 8:e1002898. 10.1371/journal.ppat.1002898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chou S, Marousek G, Auerochs S, Stamminger T, Milbradt J, Marschall M. 2011. The unique antiviral activity of artesunate is broadly effective against human cytomegaloviruses including therapy-resistant mutants. Antiviral Res. 92:364–368. 10.1016/j.antiviral.2011.07.018 [DOI] [PubMed] [Google Scholar]
- 49.Bethell D, Se Y, Lon C, Tyner S, Saunders D, Sriwichai S, Darapiseth S, Teja-Isavadharm P, Khemawoot P, Schaecher K, Ruttvisutinunt W, Lin J, Kuntawungin W, Gosi P, Timmermans A, Smith B, Socheat D, Fukuda MM. 2011. Artesunate dose escalation for the treatment of uncomplicated malaria in a region of reported artemisinin resistance: a randomized clinical trial. PLoS One 6:e19283. 10.1371/journal.pone.0019283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Efferth T, Kaina B. 2010. Toxicity of the antimalarial artemisinin and its derivatives. Crit. Rev. Toxicol. 40:405–421. 10.3109/10408441003610571 [DOI] [PubMed] [Google Scholar]
- 51.Schnepf N, Corvo J, Pors MJ, Mazeron MC. 2011. Antiviral activity of ganciclovir and artesunate towards human cytomegalovirus in astrocytoma cells. Antiviral Res. 89:186–188. 10.1016/j.antiviral.2010.12.002 [DOI] [PubMed] [Google Scholar]
- 52.Zoller T, Junghanss T, Kapaun A, Gjorup I, Richter J, Hugo-Persson M, Morch K, Foroutan B, Suttorp N, Yurek S, Flick H. 2011. Intravenous artesunate for severe malaria in travelers, Europe. Emerg. Infect. Dis. 17:771–777. 10.3201/eid1705.101229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Q, Cantilena LR, Leary KJ, Saviolakis GA, Miller RS, Melendez V, Weina PJ. 2009. Pharmacokinetic profiles of artesunate after single intravenous doses at 0.5, 1, 2, 4, and 8 mg/kg in healthy volunteers: a phase I study. Am. J. Trop. Med. Hyg. 81:615–621. 10.4269/ajtmh.2009.09-0150 [DOI] [PubMed] [Google Scholar]
- 54.Miller RS, Li Q, Cantilena LR, Leary KJ, Saviolakis GA, Melendez V, Smith B, Weina PJ. 2012. Pharmacokinetic profiles of artesunate following multiple intravenous doses of 2, 4, and 8 mg/kg in healthy volunteers: phase 1b study. Malaria J. 11:255. 10.1186/1475-2875-11-255 [DOI] [PMC free article] [PubMed] [Google Scholar]