

Abstract
Plasmodium falciparum is the etiological agent of malignant malaria and has been shown to exhibit features resembling programmed cell death. This is triggered upon treatment with low micromolar doses of chloroquine or other lysosomotrophic compounds and is associated with leakage of the digestive vacuole contents. In order to exploit this cell death pathway, we developed a high-content screening method to select compounds that can disrupt the parasite vacuole, as measured by the leakage of intravacuolar Ca2+. This assay uses the ImageStream 100, an imaging-capable flow cytometer, to assess the distribution of the fluorescent calcium probe Fluo-4. We obtained two hits from a small library of 25 test compounds, quinacrine and 3′,4′-dichlorobenzamil. The ability of these compounds to permeabilize the digestive vacuole in laboratory strains and clinical isolates was validated by confocal microscopy. The hits could induce programmed cell death features in both chloroquine-sensitive and -resistant laboratory strains. Quinacrine was effective at inhibiting field isolates in a 48-h reinvasion assay regardless of artemisinin clearance status. We therefore present as proof of concept a phenotypic screening method with the potential to provide mechanistic insights to the activity of antimalarial drugs.
INTRODUCTION
Plasmodium falciparum malaria is a global health concern, with 3.3 billion people at risk (1). Currently, the World Health Organization recommends artemisinin combination therapy as the first-line treatment but recent reports suggest emerging resistance against the artemisinins in the form of delayed parasite clearance (2, 3). In light of this, there is a pressing need for new antimalarials, preferably with novel modes of action to avoid cross-resistance. Our laboratory has in recent years uncovered a mode of cell death associated with permeabilization of the digestive vacuole (DV). Treatment with low micromolar concentrations of chloroquine (CQ) induced phenotypes reminiscent of programmed cell death (PCD) in mammalian cells, such as DNA fragmentation, mitochondrial dysfunction, and activation of clan CA cysteine proteases (4, 5). However, CQ was long ago shelved as malaria chemotherapy because of the emergence of resistant mutants. Nevertheless, efforts are being made to resensitize P. falciparum to CQ through the development of chemoreversal agents (6). Interestingly, with the withdrawal of CQ drug pressure, there is evidence that the parasite is undergoing a reversion to CQ susceptibility in Malawi (7).
The canonical mode of action of CQ relies on two key properties. (i) CQ diffuses as a weak base through the erythrocytic and parasitic membranes into the acidic DV, where it becomes diprotonated and trapped within the DV, accumulating to sufficiently high concentrations, and (ii) CQ binds to toxic ferriproporphyrin IX and prevents its polymerization into the more benign hemozoin (8). Our previous work, however, showed that there may be a secondary mechanism through which CQ exerts its antimalarial effects. At sufficiently high concentrations of CQ, the contents of the DV leak into the cytosol, leading to a form of cell death with features different from those of necrosis and which can be averted with certain enzymatic inhibitors. Further tests have indicated that it is not the unique action of CQ that triggers these PCD features, as other unrelated lysosomotrophic compounds such as desipramine (DSP), chlorpromazine (CPZ), and promethazine (PMZ) were able to induce these same phenotypes (5). Given the nonspecificity of these drugs, it is likely that DV permeabilization can be triggered by other, less toxic compounds. We therefore developed a high-content assay to screen for DV-destabilizing compounds that may serve as starting points for drug development.
In this report, we present an assay that uses an imaging-capable flow cytometer, the ImageStream 100, to detect the leakage of DV contents as indicated by Fluo-4 fluorescence. Fluo-4 is a calcium probe that fluoresces green in the presence of Ca2+. The parasite DV is a calcium store, and Ca2+ egress can be used as a proxy for DV permeabilization (9). A schematic representation of the assay work flow is shown in Fig. 1.
FIG 1.
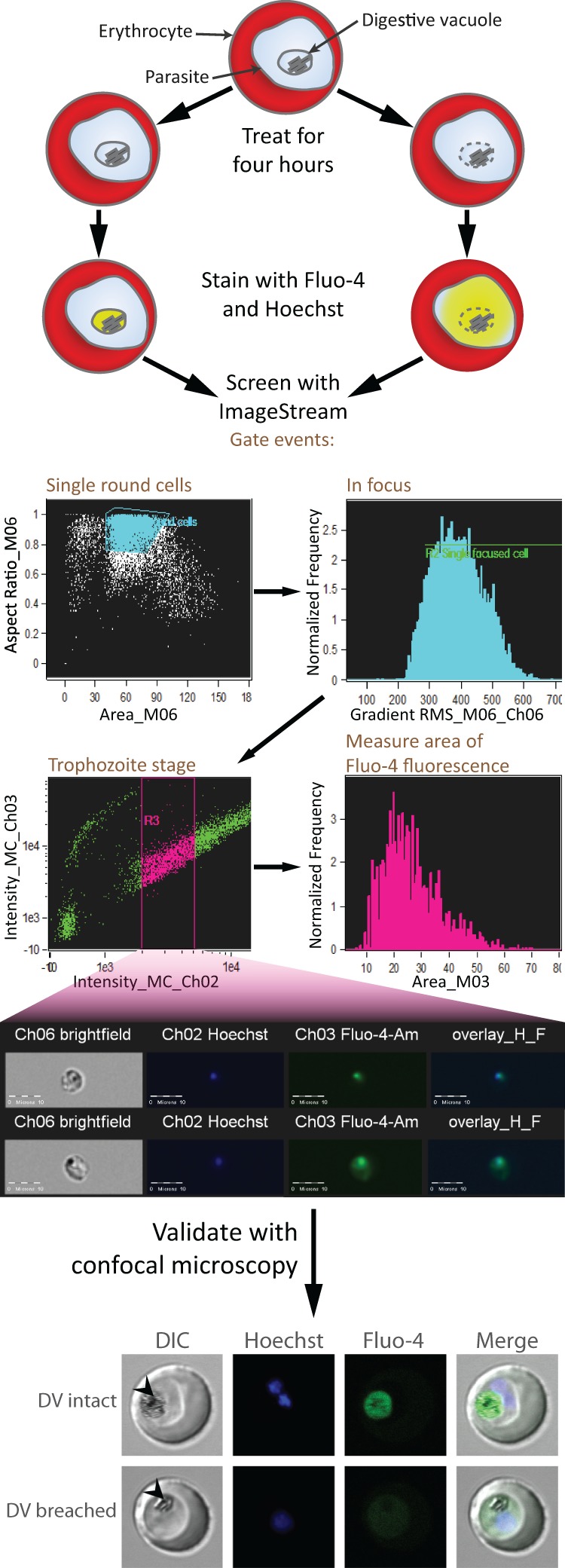
Schematic representation of assay work flow. Erythrocytes infected with trophozoite-stage parasites are treated with the test compounds for 4 h. Following this, the cells are stained with Fluo-4-AM and Hoechst 33342 and analyzed with the ImageStream platform. Hits are validated by confocal microscopy. The confocal images shown are of representative trophozoites with intact or breached DV. Fluo-4 fluorescence localizes to the intact DV, whereas fluorescence is distributed to the parasite cytosol when the DV is permeabilized. Arrowheads indicate the DV. DIC, differential interference contrast.
MATERIALS AND METHODS
Parasite culture and synchronization.
P. falciparum laboratory strains 3D7, 7G8, and K1 (MRA-102, MRA-154, and MRA-159, respectively; MR4, ATCC, Manassas, VA) and Thailand-derived field isolates SMRU 0233, SMRU 0272, SMRU 0270, and SMRU 1116 (Shoklo Malaria Research Unit [SMRU]) were cultured continuously in a malaria culture medium (MCM) consisting of RPMI 1640 (Life Technologies) supplemented with 0.5% (wt/vol) Albumax I (Invitrogen), 0.005% (wt/vol) hypoxanthine, 0.03% (wt/vol) l-glutamate, 0.25% (wt/vol) gentamicin, and human erythrocytes at 2.5% hematocrit. Culture flasks were gassed with 3% CO2, 4% O2, and 93% N2 and incubated at 37°C in the dark. Synchronization of parasite stages was performed by incubation in 5% (wt/vol) d-sorbitol at 37°C for 10 min, after which the cells were washed twice and resuspended in MCM.
Drug preparation and storage.
The library of 25 pharmacologically active compounds (Sigma-Aldrich; Tocris Bioscience) was dissolved in dimethyl sulfoxide (DMSO), phosphate-buffered saline (PBS), or dilute HCl according to the manufacturers' instructions to a stock concentration of 10 mM. CQ, DSP, CPZ, and PMZ (all from Sigma-Aldrich, Dorset, United Kingdom) were prepared by dissolution in PBS. 4-Hydroxytamoxifen (4HT) was dissolved in ethanol. Stock solutions were shielded from light and stored at −20°C. Prior to each experiment, working solutions of 100 or 10 μM were prepared fresh by dilution with PBS. Vehicle controls were prepared with equivalent volumes of PBS.
Assay of IC50.
Ring-stage parasite cultures were diluted to 1.25 to 2% parasitemia at 1.25% hematocrit. Cultures were then incubated with various concentrations of each compound in MCM for 48 h in 96-well plates. Following this, cells were stained with 1 μg/ml Hoechst 33342 for 30 min at 37°C and parasitemia was determined by flow cytometry. Half-maximal inhibitory concentrations (IC50s) were determined in GraphPad Prism 5 by using a variable-slope logistic curve. At least three experiments were performed to obtain the mean IC50s presented.
Parasite treatment and staining for screening.
A 20-μl volume of each drug solution or the vehicle control was added to 180 μl of a trophozoite-stage parasite culture at 2.5% hematocrit with ∼10% parasitemia in flat-bottom 96-well plates. Plates were placed in an incubation chamber, gassed, and incubated at 37°C in the dark for 4 h. Subsequently, parasites were washed twice with MCM and resuspended in a staining solution consisting of 1 μM Fluo-4-AM (Invitrogen) and 1 μg/ml Hoechst 33342 (Invitrogen) in MCM. The cells were then washed twice and resuspended in PBS for confocal imaging or high-content screening.
Confocal imaging.
Wet mounts of stained parasites were visualized at ×100 magnification with an Olympus Fluoview FV1000 (Olympus, Tokyo, Japan) confocal microscope equipped with solid-state and argon ion lasers tuned to 405 and 488 nm, respectively. The following calibrations were used: a 2.0-μs/pixel sampling speed, line Kalman integration, a 488-nm laser (Fluo-4-AM) at 713 V and a transmissivity of 10%, and a 405-nm laser (Hoechst) at 605 V and a transmissivity of 3%. Trophozoite-stage parasites were imaged, and the localization of Fluo-4 fluorescence was determined. For each treatment condition, at least 30 parasitized erythrocytes were enumerated.
ImageStream high-content screening.
Parasitized erythrocytes suspended in PBS to a hematocrit of 5% were assayed with the ImageStream 100 (Amnis, Seattle, WA) fitted with a 40× objective. At least 300 untreated parasites for each single-stain condition were used to create a compensation matrix. During screening, at least 10,000 parasites were acquired for each treatment condition. Gating was performed to select images with single round cells, good focus, and parasites at the trophozoite stage. Round cells were gated as events with aspect ratios (ratios of the minor axis to the major axis) close to 1 and with areas corresponding to single cells as determined by visually inspecting acquired events. Well-focused images were selected by gating on the root mean square of the rate of change of the image intensity profile; a higher rate of change in intensity implies better focus. Aspect ratio, area, and focus were gated on the bright-field channel. Trophozoites were gated on the intensity of Hoechst staining as an indicator of DNA content. Analysis was performed with the IDEAS software (version 4.0).
Assay for PCD features.
Trophozoite-stage parasites were treated with the test compounds for 10 h and then washed twice with MCM. In order to assess mitochondrial depolarization and DNA degradation, the cells were stained with 6 μM JC-1 (Life Technologies) and 0.8 μg/ml Hoechst 33342 dissolved in MCM for 30 min at 37°C. They were then washed twice and resuspended in PBS. Flow cytometry was performed with the BD LSR II Special Order System. JC-1 was excited with a 488-nm laser, and fluorescence was detected with 505LP and 525/50BP filters (396 V) and 570LP and 585/42BP filters (350 V). Hoechst 33342 was excited with a 355-nm laser, and fluorescence was detected with 450LP and 450/50BP filters (380 V). At least 150,000 erythrocytes with 10 to 15% parasitemia were acquired from each sample and analyzed with Dako Summit (version 4.3). JC-1 gating was performed with untreated parasites. Similarly, the sub-G1 population was gated by using the first decile of the Hoechst-stained parasites in the vehicle control, and this gate was unchanged under all of the treatment conditions.
Statistical analyses.
All statistical analyses were performed with SPSS 21. Confocal data were analyzed with Fisher's exact test, comparing DV fluorescence versus cytosolic plus low or no fluorescence. ImageStream data were compared with the vehicle control data by the paired t test. All of the P values reported are two tailed.
Ethics statement.
The blood collection protocol used for in vitro malaria parasite culture was approved by the National University of Singapore Institutional Review Board (NUS IRB; reference code 11-383, approval number NUS-1475). Written informed consent was obtained from all of the participants involved in this study. The clinical isolates used in this study were collected in accordance with the ethical guidelines in the approved protocols (OXTREC reference number 29-09; Center for Clinical Vaccinology and Tropical Medicine, University of Oxford, Oxford, United Kingdom). The use of field isolates for work done at the NUS was in accordance with the NUS IRB (reference code 12-369E).
RESULTS
Validation of high-content screening.
In order to validate the use of the Ca2+ probe Fluo-4 as a proxy for DV permeabilization, synchronous parasites of three laboratory strains were treated with the known lysosome-disrupting compound CQ, DSP, CPZ, PMZ, or 4HT and then stained with Fluo-4-AM and Hoechst. Parasites exhibiting DV-localized, cytosol-localized, or no Fluo-4 fluorescence were enumerated by confocal microscopy (Fig. 1 contains representative confocal images). In 3D7, all five compounds resulted in the redistribution of Fluo-4 fluorescence to the parasite cytosol. 7G8 and K1, however, were more resistant to redistribution by CQ at 3 μM. DSP, CPZ, PMZ, and 4HT resulted in similar DV permeabilization in all three laboratory strains, as indicated by the Fluo-4 redistribution observed (Fig. 2A to C). Using the same treatment and staining regimen, we assayed for the mean area of Fluo-4 fluorescence in 3D7 with the ImageStream 100. CQ, DSP, CPZ, and 4HT induced significantly larger mean areas than the vehicle control (Fig. 2D). However, PMZ-induced relocalization of fluorescence was not detected by the ImageStream platform.
FIG 2.

Trophozoites were treated for 4 h with known lysosome destabilizers, stained with the Ca2+ probe Fluo-4-AM, and subsequently enumerated by confocal microscopy or analyzed with the ImageStream. Panels A, B, and C show the proportions of 3D7, 7G8, and K1 parasites with DV-localized fluorescence (black), cytosolic fluorescence (gray), and low or no fluorescence (white) after drug treatment. At least 30 infected erythrocytes were counted for each condition. Panel D shows the mean area of Fluo-4 fluorescence after treatment of 3D7. The mean areas for all conditions except PMZ were significantly different from the vehicle control (P < 0.05, n = ≥3). Data represent means ± the standard errors of the means. Veh, vehicle control. Concentrations: CPZ, 100 μM; DSP, 200 μM; PMZ, 200 μM; 4HT, 150 μM. When not stated otherwise, the CQ concentration was 3 μM. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
Phenotypic screening of antimalarials for DV permeabilization.
Of the 25 compounds chosen for our present study, 17 were part of the Sigma-Aldrich LOPAC1280 (Library of Pharmacologically Active Compounds) and the National Institute of Neurological Diseases and Stroke drug library and were previously revealed to possess antimalarial activity (10). Of the remaining eight, mefloquine, halofantrine, artemisinin, and sulfadoxine are conventional malaria chemotherapies and were screened to provide insight into their modes of action. Chlorpheniramine, propranolol, ciprofloxacin, and miltefosine are nonclassical antimalarials and have been shown to be ineffective in triggering significant PCD features in P. falciparum (5) and were therefore included as negative controls. Confirmation of the antimalarial activity of our library was done by performing 48-h IC50 assays (Table 1). Although several of our test compounds are known to be fluorescent (11, 12), the library exhibited negligible autofluorescence in our assay (data not shown). In the first round of screening, parasites were treated with the test compounds at 10 μM and assayed with the ImageStream, a flow cytometer with imaging capabilities. Sixteen hits were identified at this stage on the basis of the increase in the mean fluorescence area of Fluo-4 staining (Fig. 3A) and then rescreened at 1 μM, resulting in the selection of three hits, emetine, quinacrine (QC), and 3′,4′-dichlorobenzamil (DCB) (Fig. 3B). Given the small effect size of emetine, we decided to exclude it from further investigations.
TABLE 1.
Mean IC50s of test compounds for 3D7
| Drug name (abbreviation) | IC50 (nM) | Drug name (abbreviation) | IC50 (nM) | Drug name (abbreviation) | IC50 (nM) | ||
|---|---|---|---|---|---|---|---|
| Vincristine (VC) | 5 | Quinacrine (QC) | 56 | 5-(N-Methyl-N-isobutyl)amiloride (A5585) | 4,990 | ||
| Halofantrine (HF) | 5 | Quinine (QUI) | 62 | 5-(N,N-Dimethyl)amiloride hydrochloride (A4562) | 6,592 | ||
| Vinblastine (VB) | 9 | Hexahydro-sila-difenidol hydrochloride (HEXA) | 81 | 4-Hydroxytamoxifen (4HT) | 16,016 | ||
| Mefloquine (MEF) | 11 | 3′,4′-Dichlorobenzamil (DCB) | 90 | Propranolol (PRO) | 19,006 | ||
| Quinidine (QUD) | 25 | BW 284c51 (BIS) | 360 | Chlorpromazine (CPZ) | 20,886 | ||
| Benzamil (BEN) | 27 | Amperoxide (AMP) | 866 | Promethazine (PMZ) | 22,533 | ||
| Emetine (EME) | 28 | S(−)-UH-301 (UH301) | 2,659 | Desipramine (DSP) | 25,630 | ||
| Artemisinin (ART) | 30 | 5-(N-Ethyl-N-isopropyl)amiloride (A3085) | 3,751 | Chlorpheniramine (CPN) | 33,706 | ||
| Chloroquine (CQ) | 46 | SKF 95282 (SKF) | 4,088 | Miltefosine (MTF) | 69,976 | ||
| Cinchonine (CIN) | 48 | Ciprofloxacin (CIP) | 4,402 | Sulfadoxine (SUL) | >105 |
FIG 3.
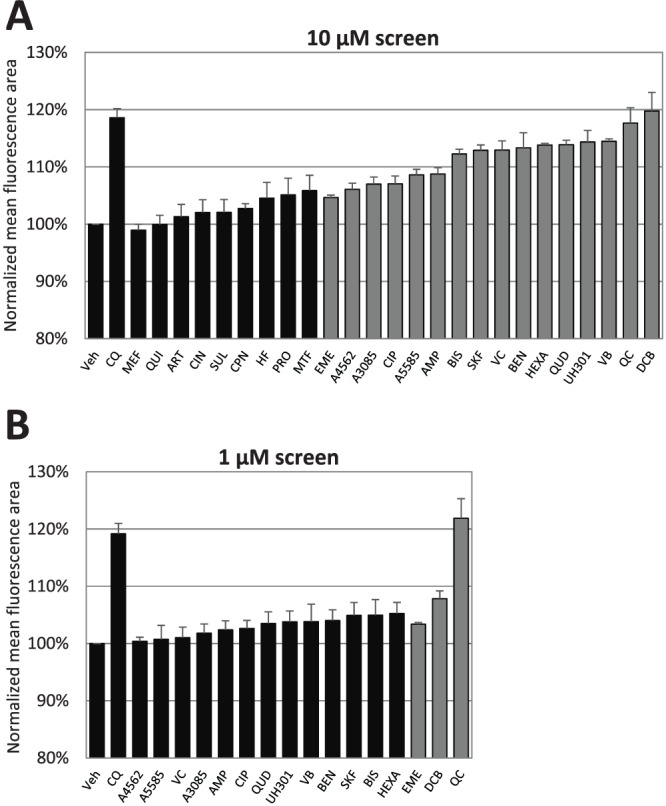
ImageStream screening of a candidate library. 3D7 trophozoites were treated for 4 h and stained with Fluo-4-AM. The mean area of Fluo-4 fluorescence was then assayed with the ImageStream platform. For panel A, candidate compounds were administered at 10 μM and the resulting mean areas were compared with that of the vehicle (Veh) control. For panel B, hits from the 10 μM screening were selected for rescreening at 1 μM. Three hits were identified at this stage: emetine dihydrochloride (EME), DCB, and QC hydrochloride. Compound name abbreviations are provided in Table 1. Gray bars indicate mean areas significantly different from that of the vehicle control (P < 0.05; n = ≥3). CQ at 3 μM was included in both screenings as a positive control. Data represent means ± the standard errors of the means.
Postscreening validation by confocal imaging.
Laboratory strains 3D7, 7G8, and K1 were then treated with the hits QC and DCB and enumerated by confocal microscopy (Fig. 4). Both hits were able to trigger the redistribution of fluorescence in 3D7 at 10 and 1 μM. DCB, however, was unable to induce significant DV permeabilization at ≤1 μM in CQ-resistant strains 7G8 and K1, possibly indicating cross-resistance to DCB. On the other hand, QC retained its potency in these resistant strains. This trend was recapitulated in the IC50 measurements: 7G8 and K1 were far more resistant to DCB than 3D7 was, while QC remained relatively effective in the CQ-resistant strains (Table 2).
FIG 4.
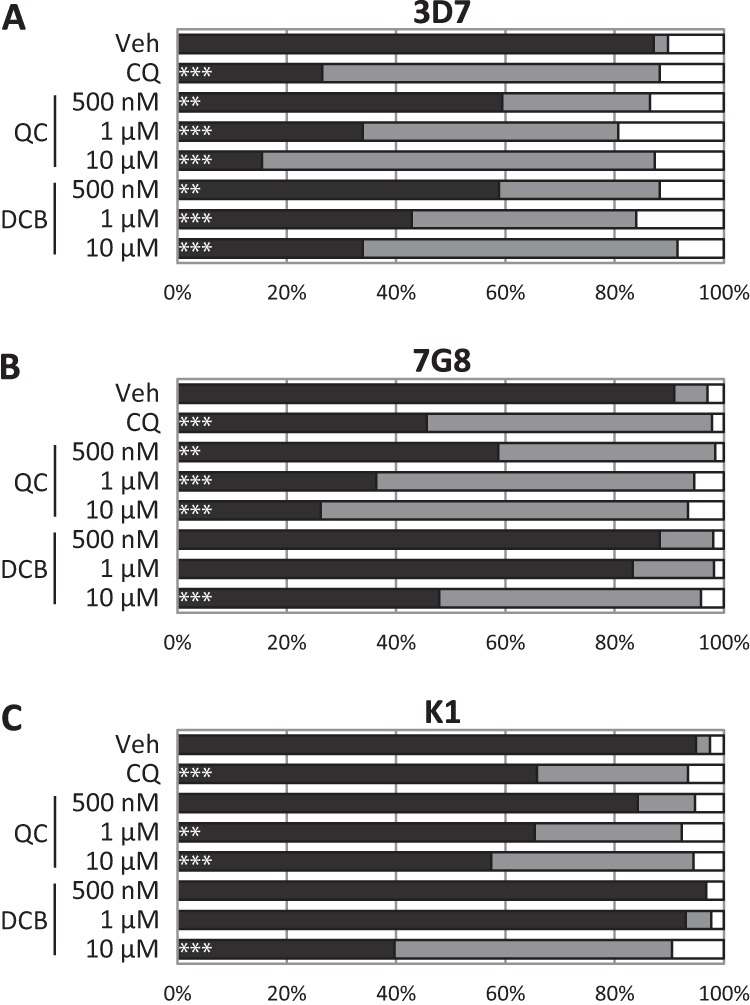
ImageStream hits were validated by confocal microscopy. Drug-treated laboratory strains 3D7, 7G8, and K1 were assessed for DV-localized fluorescence (black), cytosolic fluorescence (gray), or low or no fluorescence (white). In CQ-sensitive strain 3D7, QC and DCB exhibited DV permeabilization efficacy similar to that of CQ. In CQ-resistant strains 7G8 and K1, DCB had reduced potency. At least 30 infected erythrocytes were counted per treatment. ***, P < 0.001; **, P < 0.01; *, P < 0.05. Veh, vehicle.
TABLE 2.
Mean IC50s of hits for laboratory strains and field isolates
| Strain or isolate | Resistance status |
Mean IC50 (nM) |
|||
|---|---|---|---|---|---|
| CQ | Artemisinin | CQ | QC | DCB | |
| Laboratory strains | |||||
| 3D7 | Sensitive | Sensitive | 47 | 56 | 90 |
| 7G8 | Resistant | Sensitive | 292 | 151 | 529 |
| K1 | Resistant | Sensitive | 569 | 100 | 679 |
| Field isolates | |||||
| SMRU 0270 | Resistant | Sensitive | 253 | 64 | 426 |
| SMRU 1116 | Resistant | Sensitive | 391 | 68 | 536 |
| SMRU 0233 | Resistant | Resistant | 139 | 75 | 551 |
| SMRU 0272 | Resistant | Resistant | 158 | 82 | 530 |
Induction of PCD features by QC and DCB.
Given that DV permeabilization is associated with PCD features (4, 5), we believed it would be interesting to investigate whether our two hits can also induce these features. In mammalian cells, apoptotic features include depolarization of the mitochondrial membrane and fragmentation of nuclear DNA, which may be assessed by JC-1 and Hoechst staining, respectively (13, 14). DNA degradation results in a population of cells with DNA content lower even than that of cells at the G1 cell cycle stage, termed the sub-G1 population (14). To this end, the laboratory strains were treated with QC and DCB. A treatment duration of 10 h was used to allow any PCD features to be manifested, after which the cells were assayed for JC-1 and Hoechst staining by flow cytometry. In CQ-sensitive 3D7, CQ, QC, and DCB were similarly effective at inducing mitochondrial membrane potential (ΔΨm) and DNA loss (Fig. 5A). Interestingly, in CQ-resistant strains K1 and 7G8, DCB displayed a greater ability to induce DNA degradation than QC despite having a higher IC50 and being less able to trigger DV disruption in the confocal assays. On the other hand, QC stimulated greater ΔΨm loss than DCB (Fig. 5B and C). An alternative presentation of these data shows that the CQ-resistant parasites were also resistant to CQ induction of PCD features, although 7G8 remained relatively susceptible to ΔΨm loss (Fig. 6A). QC, although unable to stimulate DNA degradation in the CQ-resistant parasites, could trigger depolarization of the mitochondria equivalently in all three strains (Fig. 6B). DCB showed different abilities to trigger PCD features in CQ-sensitive 3D7 versus the CQ-resistant strains while acting similarly on the two resistant strains (Fig. 6C).
FIG 5.

The ability to induce cell death features in P. falciparum was assessed by treating lab strains with various concentrations of the two hits for 10 h. The parasites were then stained with Hoechst 33342 and JC-1 and analyzed by flow cytometry. In CQ-sensitive 3D7, QC and DCB both induced DNA degradation and ΔΨm loss to similar degrees. However, in CQ-resistant strains 7G8 and K1, QC triggered ΔΨm loss at a lower concentration than DCB, while DCB was a better stimulator of DNA degradation than QC. Data represent means ± the standard errors of the means of at least three experiments.
FIG 6.

CQ-resistant strains 7G8 and K1 are generally more resistant to induction of the two cell death features, regardless of the drug tested. QC depolarizes the mitochondria of all three strains to similar extents but does not induce notable DNA fragmentation in CQ-resistant strains. On the other hand, DCB is a weak stimulator of ΔΨm loss but can induce DNA degradation in CQ-resistant strains. Data represent means ± the standard errors of the means of at least three experiments.
Testing of QC and DCB on field isolates.
The end goal of this screening method is to identify lead compounds that are effective in the real world. Therefore, QC and DCB were tested on two artemisinin-sensitive and delayed-clearance clinical field isolates from Thailand. Drug susceptibility assays showed that the efficacy of QC and DCB was similar to that against the laboratory strains (Table 2), regardless of artemisinin clearance status. In the confocal counts, QC consistently outperformed CQ in stimulating the redistribution of fluorescence across all of the strains (Fig. 7, comparing CQ and QC at 500 nM, P ≤ 0.001 for all). The efficacies of DCB against the field isolates and the CQ-resistant laboratory strains were similar (Fig. 2B and C and 7).
FIG 7.
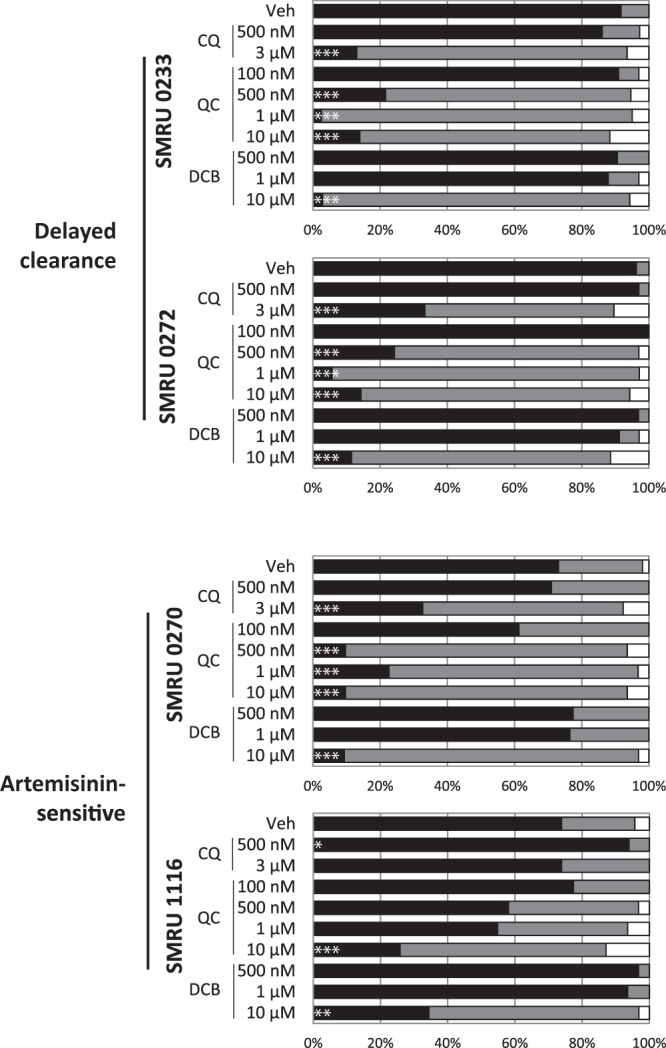
The DV-destabilizing effects of QC and DCB are recapitulated in field isolates. Two artemisinin-sensitive and two delayed-clearance isolates were treated with QC and DCB for 4 h and then stained with Fluo-4-AM. Confocal microscopy was performed to assess the percentages of parasites with DV-localized fluorescence (black), cytosolic fluorescence (gray), and low or no fluorescence (white). QC retains a potent DV-permeabilizing effect in both the delayed-clearance isolates and one of the artemisinin-sensitive isolates. At least 30 infected erythrocytes were counted per treatment. ***, P < 0.001; **, P < 0.01; *, P < 0.05. Veh, vehicle.
Strictly standardized mean difference.
To obtain a statistical assessment of our assay, we retrospectively calculated the strictly standardized mean difference (SSMD) score by comparing the normalized mean fluorescence areas of the vehicle control and the CQ control in the 10 and 1 μM screenings. The SSMD score allows the quantification of effect size by using paired data (15), unlike the commonly used z factor. Our sample size was 16 pairs of vehicle and 3 μM CQ controls. Using these data, the calculated SSMD score was 2.62, which is considered to be superior resolution (15).
DISCUSSION
Given the association of DV permeabilization with PCD features (4, 5) and the centrality of the DV to the erythrocytic stage of P. falciparum, it was a natural progression to the search for novel compounds that compromise the DV. Despite the popularity of molecular-target-based high-throughput screening methods, these assays tend to result in few leads; this is presumably due to the ease with which mutations conferring resistance are obtained and physical barriers to the drug targets, among other factors (16). A phenotypic screening method, on the other hand, may produce fewer false-positive results but not clearly delineate the mode of action. At a time when resistance to front-line chemotherapies appears to be emerging, we may not be able to afford the luxury of searching for compounds with well-defined targets. Fortuitously, a confluence of technology and elucidation of parasite biology has allowed us to take advantage of the ImageStream's imaging capabilities to screen for compounds that destabilize the parasite DV; this approach has also recently been used to screen for antimicrobial peptides with activity against P. falciparum (17).
7G8 and K1 are CQ resistant, and their resistance is attributed to mutations in the P. falciparum crt (chloroquine resistance transporter) gene (18, 19). These mutations facilitate the efflux of CQ from the DV, resulting in lower intravacuolar concentrations of CQ (20–23). This would explain the lowered ability of CQ to induce Ca2+ redistribution in 7G8 and K1 (Fig. 2A to C). CQ resistance might also presumably provide general protection against lysosomotrophic compounds. Our results show that CQ resistance status does not influence the redistribution of DV contents triggered by the non-CQ-related drugs DSP, CPZ, PMZ, and 4HT at the concentrations tested. However, Fluo-4 redistribution was not observed in 7G8 and K1 with DCB treatment up to 1 μM (Fig. 4), hinting that the action of DCB is modulated by CQ resistance despite different origins of resistance in 7G8 and K1. Interestingly, of the clinical isolates, SMRU 1116 appeared to be resistant to DV destabilization regardless of the compound tested (Fig. 7). We are unable to explain this phenotype. Some possibilities might include variant pfmdr and pfmrp, both of which have been suggested to play roles in drug resistance (24–27).
Of the two hits from our assay, DCB appears to be far less potent in terms of DV permeabilization and inhibition of parasite reinvasion. DCB is a Na+/Ca2+ exchanger inhibitor (28), and this highlights a limitation of our assay. Given that our screening method relies on the Ca2+ probe Fluo-4-AM, any compound that interferes with the Ca2+ uptake of the DV may result in a hit, as the net flux could be tilted toward an increase in cytosolic Ca2+. Strictly speaking, therefore, our assay identifies not DV-permeabilizing compounds but rather compounds that affect the distribution of Ca2+. This could confound our objective of identifying compounds that disrupt the DV. Nevertheless, Ca2+ dysfunction would still be detrimental to parasite development, as blocking of Ca2+ oscillations has been shown to be lethal to the parasite (29, 30). Despite this, we believe that DCB does indeed destabilize the DV membrane, as postscreening validation showed that DCB induces phenotypes reminiscent of PCD (Fig. 5 and 6), consistent with our previous work on DV disruption (5). A note of interest is that CQ resistance appears to be cross-protective against DCB. The physiological function of the P. falciparum CRT (PfCRT) protein has yet to be elucidated, but it is suspected to be involved in Ca2+ flux (31). Verapamil, a Ca2+ channel blocker, is believed to bind to mutant forms of PfCRT that confer resistance to CQ, thus blocking CQ efflux from the DV (32). Given that DCB is also an inhibitor of Ca2+ transport, it may bind preferentially to CQ-sensitive PfCRT instead.
The second hit, QC, is a more promising candidate, with its ability to induce Ca2+ redistribution at submicromolar levels (Fig. 7) and low IC50s (Table 2) in field isolates. QC was the antimalarial drug of choice prior to CQ and is structurally similar to CQ, save for an additional benzene ring (33). Both CQ and QC are known to bind to heme and inhibit its degradation and detoxification (34). Interestingly, although QC is an analog of CQ, resistance to CQ did not offer the field isolates much protection against QC. It has been postulated that benzene moieties may impart chemoreversal properties, that is, a resensitization of resistant parasites to CQ, by blocking the export of CQ by mutant PfCRT (6, 35). The additional aromatic ring may serve to partially reverse resistance and prevent the efflux of QC. Interestingly, Sanchez and colleagues previously reported a remarkable effect of QC on CQ uptake: at a low concentration of QC, accumulation of radiolabeled CQ is boosted in a CQ-resistant P. falciparum strain but this effect is rapidly lost as the QC concentration increases (21). This supports our model of QC as both a chemoreversal agent and a lysosomotrophic detergent. At low concentrations of QC, efflux of CQ by mutant PfCRT is inhibited. However, at higher concentrations, the DV is permeabilized, resulting in CQ leakage out of the DV. The efficacy of QC in disrupting the DV of field isolates may also serve as a proof of concept for the development of CQ analogs to target the DV. It has been previously reported that certain regimens of QC therapy can raise blood QC levels beyond 100 nM for days (36). Although our confocal assessments showed a lack of DV instability at 100 nM, our 4-h assay could have been too short to allow any observable phenotypic changes. DV compromise could still possibly be a mechanism of QC killing with a longer duration of exposure.
Despite screening only a small library of compounds, our assay has uncovered a novel mode of killing by a previously known antimalarial drug, QC, and possibly a new class of DV-destabilizing agents in DCB. Although the current setup allows the screening of only a small library because of the single-tube reading format of the ImageStream 100, newer models such as the ImageStreamX Mark II are able to assay in a 96-well plate format and at better resolutions, potentially increasing the throughput of this assay. Further work beyond this screening assay could include testing for drug-induced hemolysis. There are conflicting reports of the ability of one of our hits, QC, to induce hemolysis in glucose-6-phosphate dehydrogenase-deficient patients (37), highlighting the need to characterize potential drugs before further development. Confounding this further, metabolic acidosis is a hallmark of severe malaria (38) and may modify the drug response. Drug-induced hemolysis in patients with malaria may worsen any parasite-induced anemia and potentially be fatal. Therefore, as a preliminary test, an ex vivo assay for hemolysis that allows pH adjustment may be performed (39). Our assay could perhaps also be used to search for transmission-blocking compounds by screening another DV-dependent stage, the gametocyte. However, further characterization of the gametocyte would first have to be performed to assess its amenability to this screening platform.
ACKNOWLEDGMENTS
We thank the patients and staff of the SMRU for their contributions to this study.
SMRU is funded by the Wellcome Trust of Great Britain, as part of the Oxford Tropical Medicine Research Program of Wellcome Trust—Mahidol University. We are also grateful for the following strains obtained through MR4's BEI Resources Repository, NIAID, NIH: P. falciparum 3D7, MRA-102, deposited by D. J. Carucci; P. falciparum 7G8, MRA-154, deposited by D. E. Kyle; and P. falciparum K1, MRA-159, deposited by D. E. Kyle. This study was supported by generous grants from the National Medical Research Council, Singapore (NMRC/1310/2011 and NMRC/EDG/1038/2011).
Footnotes
Published ahead of print 11 November 2013
REFERENCES
- 1.World Health Organization 2012. World malaria report 2012 World Health Organization, Geneva, Switzerland: http://www.who.int/malaria/publications/world_malaria_report_2012/report/en/index.html [Google Scholar]
- 2.Mita T, Tanabe K. 2012. Evolution of Plasmodium falciparum drug resistance: implications for the development and containment of artemisinin resistance. Jpn. J. Infect. Dis. 65:465–475. 10.7883/yoken.65.465 [DOI] [PubMed] [Google Scholar]
- 3.Takala-Harrison S, Clark TG, Jacob CG, Cummings MP, Miotto O, Dondorp AM, Fukuda MM, Nosten F, Noedl H, Imwong M, Bethell D, Se Y, Lon C, Tyner SD, Saunders DL, Socheat D, Ariey F, Phyo AP, Starzengruber P, Fuehrer H-P, Swoboda P, Stepniewska K, Flegg J, Arze C, Cerqueira GC, Silva JC, Ricklefs SM, Porcella SF, Stephens RM, Adams M, Kenefic LJ, Campino S, Auburn S, MacInnis B, Kwiatkowski DP, Su X, White NJ, Ringwald P, Plowe CV. 2013. Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in Southeast Asia. Proc. Natl. Acad. Sci. U. S. A. 110:240–245. 10.1073/pnas.1211205110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ch'ng J-H, Kotturi SR, Chong AG-L, Lear MJ, Tan KS-W. 2010. A programmed cell death pathway in the malaria parasite Plasmodium falciparum has general features of mammalian apoptosis but is mediated by clan CA cysteine proteases. Cell Death Dis. 1:e26. 10.1038/cddis.2010.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ch'ng J-H, Liew K, Goh AS-P, Sidhartha E, Tan KS-W. 2011. Drug-induced permeabilization of parasite's digestive vacuole is a key trigger of programmed cell death in Plasmodium falciparum. Cell Death Dis. 2:e216. 10.1038/cddis.2011.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ch'ng J-H, Mok S, Bozdech Z, Lear MJ, Boudhar A, Russell B, Nosten F, Tan KS-W. 2013. A whole cell pathway screen reveals seven novel chemosensitizers to combat chloroquine resistant malaria. Sci. Rep. 3:1734. 10.1038/srep01734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laufer MK, Thesing PC, Dzinjalamala FK, Nyirenda OM, Masonga R, Laurens MB, Stokes-Riner A, Taylor TE, Plowe CV. 2012. A longitudinal trial comparing chloroquine as monotherapy or in combination with artesunate, azithromycin or atovaquone-proguanil to treat malaria. PLoS One 7:e42284. 10.1371/journal.pone.0042284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ginsburg H, Ward S, Bray P. 1999. An integrated model of chloroquine action. Parasitol. Today 15:357–360. 10.1016/S0169-4758(99)01502-1 [DOI] [PubMed] [Google Scholar]
- 9.Rohrbach P, Friedrich O, Hentschel J, Plattner H, Fink RHA, Lanzer M. 2005. Quantitative calcium measurements in subcellular compartments of Plasmodium falciparum-infected erythrocytes. J. Biol. Chem. 280:27960–27969. 10.1074/jbc.M500777200 [DOI] [PubMed] [Google Scholar]
- 10.Lucumi E, Darling C, Jo H, Napper AD, Chandramohanadas R, Fisher N, Shone AE, Jing H, Ward SA, Biagini GA, DeGrado WF, Diamond SL, Greenbaum DC. 2010. Discovery of potent small-molecule inhibitors of multidrug-resistant Plasmodium falciparum using a novel miniaturized high-throughput luciferase-based assay. Antimicrob. Agents Chemother. 54:3597–3604. 10.1128/AAC.00431-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bohórquez EB, Chua M, Meshnick SR. 2012. Quinine localizes to a non-acidic compartment within the food vacuole of the malaria parasite Plasmodium falciparum. Malar. J. 11:350. 10.1186/1475-2875-11-350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peti-Peterdi J, Fintha A, Fuson AL, Tousson A, Chow RH. 2004. Real-time imaging of renin release in vitro. Am. J. Physiol. Renal Physiol. 287:F329–F335. 10.1152/ajprenal.00420.2003 [DOI] [PubMed] [Google Scholar]
- 13.Vermes I, Haanen C, Reutelingsperger C. 2000. Flow cytometry of apoptotic cell death. J. Immunol. Methods 243:167–190. 10.1016/S0022-1759(00)00233-7 [DOI] [PubMed] [Google Scholar]
- 14.Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P, Traganos F. 1992. Features of apoptotic cells measured by flow cytometry. Cytometry 13:795–808. 10.1002/cyto.990130802 [DOI] [PubMed] [Google Scholar]
- 15.Zhang X. 2008. Genome-wide screens for effective siRNAs through assessing the size of siRNA effects. BMC Res. Notes 1:33. 10.1186/1756-0500-1-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. 2007. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6:29–40. 10.1038/nrd2201 [DOI] [PubMed] [Google Scholar]
- 17.Love MS, Millholland MG, Mishra S, Kulkarni S, Freeman KB, Pan W, Kavash RW, Costanzo MJ, Jo H, Daly TM, Williams DR, Kowalska MA, Bergman LW, Poncz M, DeGrado WF, Sinnis P, Scott RW, Greenbaum DC. 2012. Platelet factor 4 activity against P. falciparum and its translation to nonpeptidic mimics as antimalarials. Cell Host Microbe 12:815–823. 10.1016/j.chom.2012.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LMB, bir Singh Sidhu A, Naudé B, Deitsch KW, Su X, Wootton JC, Roepe PD, Wellems TE. 2000. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell 6:861–871. 10.1016/S1097-2765(05)00077-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen N, Kyle DE, Pasay C, Fowler EV, Baker J, Peters JM, Cheng Q. 2003. pfcrt allelic types with two novel amino acid mutations in chloroquine-resistant Plasmodium falciparum isolates from the Philippines. Antimicrob. Agents Chemother. 47:3500–3505. 10.1128/AAC.47.11.3500-3505.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sidhu ABS, Verdier-Pinard D, Fidock DA. 2002. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science 298:210–213. 10.1126/science.1074045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanchez CP, McLean JE, Stein W, Lanzer M. 2004. Evidence for a substrate specific and inhibitable drug efflux system in chloroquine resistant Plasmodium falciparum strains. Biochemistry 43:16365–16373. 10.1021/bi048241x [DOI] [PubMed] [Google Scholar]
- 22.Sanchez CP, Stein W, Lanzer M. 2003. Trans stimulation provides evidence for a drug efflux carrier as the mechanism of chloroquine resistance in Plasmodium falciparum. Biochemistry 42:9383–9394. 10.1021/bi034269h [DOI] [PubMed] [Google Scholar]
- 23.Paguio MF, Cabrera M, Roepe PD. 2009. Chloroquine transport in Plasmodium falciparum. 2. Analysis of PfCRT-mediated drug transport using proteoliposomes and a fluorescent chloroquine probe. Biochemistry 48:9482–9491. 10.1021/bi901035j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Price RN, Cassar C, Brockman A, Duraisingh M, Vugt M van, White NJ, Nosten F, Krishna S. 1999. The pfmdr1 gene is associated with a multidrug-resistant phenotype in Plasmodium falciparum from the western border of Thailand. Antimicrob. Agents Chemother. 43:2943–2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim P, Alker AP, Khim N, Shah NK, Incardona S, Doung S, Yi P, Bouth DM, Bouchier C, Puijalon OM, Meshnick SR, Wongsrichanalai C, Fandeur T, Bras JL, Ringwald P, Ariey F. 2009. Pfmdr1 copy number and arteminisin derivatives combination therapy failure in falciparum malaria in Cambodia. Malar. J. 8:11. 10.1186/1475-2875-8-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veiga MI, Ferreira PE, Jörnhagen Malmberg LM, Kone A, Schmidt BA, Petzold M, Björkman A, Nosten F, Gil JP. 2011. Novel polymorphisms in Plasmodium falciparum ABC transporter genes are associated with major ACT antimalarial drug resistance. PLoS One 6:e20212. 10.1371/journal.pone.0020212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pirahmadi S, Zakeri S, Afsharpad M, Djadid ND. 2013. Mutation analysis in pfmdr1 and pfmrp1 as potential candidate genes for artemisinin resistance in Plasmodium falciparum clinical isolates 4 years after implementation of artemisinin combination therapy in Iran. Infect. Genet. Evol. 14:327–334. 10.1016/j.meegid.2012.12.014 [DOI] [PubMed] [Google Scholar]
- 28.Oda T, Kume T, Izumi Y, Ishihara K, Sugmimoto H, Akaike A. 2011. Na+/Ca2+ exchanger inhibitors inhibit neurite outgrowth in PC12 cells. J. Pharmacol. Sci. 116:128–131. 10.1254/jphs.11011SC [DOI] [PubMed] [Google Scholar]
- 29.Enomoto M, Kawazu S, Kawai S, Furuyama W, Ikegami T, Watanabe J, Mikoshiba K. 2012. Blockage of spontaneous Ca2+ oscillation causes cell death in intraerythrocitic Plasmodium falciparum. PLoS One 7:e39499. 10.1371/journal.pone.0039499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adovelande J, Bastide B, Deleze J, Schrevel J. 1993. Cytosolic free calcium in Plasmodium falciparum-infected erythrocytes and the effect of verapamil: a cytofluorometric study. Exp. Parasitol. 76:247–258. 10.1006/expr.1993.1030 [DOI] [PubMed] [Google Scholar]
- 31.Biagini GA, Fidock DA, Bray PG, Ward SA. 2005. Mutations conferring drug resistance in malaria parasite drug transporters Pgh1 and PfCRT do not affect steady-state vacuolar Ca2+. Antimicrob. Agents Chemother. 49:4807–4808. 10.1128/AAC.49.11.4807-4808.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin SK, Oduola AMJ, Milhous WK. 1987. Reversal of chloroquine resistance in Plasmodium falciparum by verapamil. Science 235:899–901. 10.1126/science.3544220 [DOI] [PubMed] [Google Scholar]
- 33.Tanenbaum LTD. 1980. Antimalarial agents: chloroquine, hydroxychloroquine, and quinacrine. Arch. Dermatol. 116:587–591. 10.1001/archderm.1980.01640290097026 [DOI] [PubMed] [Google Scholar]
- 34.Loria P, Miller S, Foley M, Tilley L. 1999. Inhibition of the peroxidative degradation of haem as the basis of action of chloroquine and other quinoline antimalarials. Biochem. J. 339:363–370. 10.1042/0264-6021:3390363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhattacharjee AK, Kyle DE, Vennerstrom JL, Milhous WK. 2002. A 3D QSAR pharmacophore model and quantum chemical structure-activity analysis of chloroquine (CQ)-resistance reversal. J. Chem. Inf. Comput. Sci. 42:1212–1220. 10.1021/ci0200265 [DOI] [PubMed] [Google Scholar]
- 36.Shannon JA, Earle DP, Brodie BB, Taggart JV, Berliner RW. 1944. The pharmacological basis for the rational use of atabrine in the treatment of malaria. J. Pharmacol. Exp. Ther. 81:307–330 http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.32.2459&rep=rep1&type=pdf [Google Scholar]
- 37.Youngster DI, Arcavi L, Schechmaster R, Akayzen Y, Popliski H, Shimonov J, Beig S, Berkovitch M. 2010. Medications and glucose-6-phosphate dehydrogenase deficiency. Drug Saf. 33:713–726. 10.2165/11536520-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 38.Maitland K, Newton CRJC. 2005. Acidosis of severe falciparum malaria: heading for a shock? Trends Parasitol. 21:11–16. 10.1016/j.pt.2004.10.010 [DOI] [PubMed] [Google Scholar]
- 39.Evans BC, Nelson CE, Yu SS, Beavers KR, Kim AJ, Li H, Nelson HM, Giorgio TD, Duvall CL. 2013. Ex vivo red blood cell hemolysis assay for the evaluation of pH-responsive endosomolytic agents for cytosolic delivery of biomacromolecular drugs. J. Vis. Exp. 73:e50166. 10.3791/50166 [DOI] [PMC free article] [PubMed] [Google Scholar]