

Abstract
The stress-inducible molecules GADD45β and GADD45γ have been implicated in regulating IFNγ production in CD4 T cells. However, how GADD45 proteins function has been controversial. MEKK4 is a MAP kinase kinase kinase that interacts with GADD45 in vitro. Here we generated MEKK4-deficient mice to define the function and regulation of this pathway. CD4 T cells from MEKK4−/− mice have reduced p38 activity and defective IFNγ synthesis. Expression of GADD45β or GADD45γ promotes IFNγ production in MEKK4+/+ T cells, but not in MEKK4−/− cells or in cells treated with a p38 inhibitor. Thus, MEKK4 mediates the action of GADD45β and GADD45γ on p38 activation and IFNγ production. During Th1 differentiation, the GADD45β/GADD45γ/MEKK4 pathway appears to integrate upstream signals transduced by both T cell receptor and IL12/STAT4, leading to augmented IFNγ production in a process independent of STAT4.
Keywords: GADD45, interferon, p38, STAT4, T cell differentiation
Introduction
Stress-activated MAP kinase (MAPK), including p38 and JNK, regulate a variety of intracellular processes in response to environmental stresses, growth factors, cytokines, and other stimuli. p38 and JNK are activated via signaling cascades involving a MAPK kinase (MAPKK) that is responsible for phosphorylation of the appropriate MAPK, and a MAPK kinase kinase (MAPKKK or MAP3K) that phosphorylates and activates MAPKK (Davis, 2000; Kyriakis and Avruch, 2001). Recent studies have suggested a new mode of p38/JNK activation, which involves an interaction between GADD45 proteins and a MAP3K, MEKK4 (MTK1 in human) (Takekawa et al, 1997; Takekawa and Saito, 1998). GADD45 proteins include three closely related members (α, β, and γ) that are transcriptionally induced by environmental stress. These proteins bind MEKK4 and augment its activity. Overexpression of each individual GADD45 protein by transient transfection activates p38/JNK and causes apoptosis, which can be partially suppressed by coexpression of a dominant inhibitory MEKK4 protein. These studies suggest that GADD45 may mediate activation of the p38/JNK pathway, through MEKK4, in response to environmental stress (Takekawa and Saito, 1998).
The function of GADD45 proteins in regulating stress-activated MAPK is starting to be explored in the immune system, with the use of T helper type 1 (Th1) cells as a model system. Th1 cells produce a signature cytokine, IFNγ, in response to antigen receptor challenge or combined stimulation by IL12 and IL18 (Yang et al, 1999). IL12/IL18 stimulation induces high-level expression of GADD45β and GADD45γ. Retroviral overexpression of GADD45β leads to sustained p38 activation and enhanced IFNγ production in IL12/IL18-stimulated Th1 cells (Yang et al, 2001). A study from our group has found that GADD45γ is strongly induced during Th1 cell differentiation (Lu et al, 2001). Th1 effector cells deficient in GADD45γ show reduced IFNγ production and p38/JNK activation in response to T cell receptor (TCR) stimulation. In vivo, GADD45γ−/− mice exhibit suppressed contact hypersensitivity, indicative of the significance of this protein in regulating immune function (Lu et al, 2001). Recently, we have generated GADD45β−/− mice and demonstrated that GADD45β is required for MAPK activation and signaling of both antigen receptor and inflammatory cytokines in T cells (Lu et al, 2004). GADD45 proteins also appear to regulate p38/JNK outside the immune system. GADD45α function was shown to be critical for the maintenance of sustained p38/JNK activities in UV-irradiated keratinocytes, and thus represents a key component protecting the skin against UV-induced tumors (Hildesheim et al, 2002).
However, results from several other studies conflict with the notion that GADD45 proteins activate p38/JNK through MEKK4. Notably, JNK and p38 activation by genotoxic stress occurs significantly earlier than the transcriptional induction of all three GADD45 members, arguing against a role for GADD45 in activating p38/JNK (Shaulian and Karin, 1999; Wang et al, 1999). Transient overexpression of GADD45α does not cause JNK or p38 activation in several cell lines, including HeLa, NIH3T3, and COS-7 (Wang et al, 1999). In contrast, overexpression of GADD45β in 3DO T cells effectively inhibits JNK activation induced by TNFα and hydrogen peroxide, whereas antisense GADD45β enhances it (De Smaele et al, 2001). Furthermore, a recent study has found that GADD45α binds p38 directly, and is capable of mediating H-Ras-induced activation of p38, but not JNK or ERK, in a pathway apparently independent of MEKK4 (Bulavin et al, 2003). The reason for the discrepancy between these studies is not clear, but is likely to reflect the multiple functions of GADD45 proteins in regulating intracellular processes. GADD45 proteins are known to interact with a number of nuclear proteins such as proliferating cell nuclear antigen (PCNA), p21, cdc2, and core histone protein (Smith et al, 1994; Kearsey et al, 1995; Carrier et al, 1999; Zhan et al, 1999), in addition to activating MEKK4 (Takekawa and Saito, 1998). Conceivably, in different cell types and/or stimulation conditions, GADD45 proteins could preferentially interact with one or several particular pathways, leading to the differential regulation of p38/JNK activities and other downstream events through distinct mechanisms.
Despite recent advances in our understanding of GADD45 and MEKK4, a critical issue to be addressed is whether GADD45 and MEKK4 are in the same genetic pathway in vivo and whether this pathway is physiologically important. Previous work using dominant-negative MEKK4 is suggestive but has obvious limitations inherent with the use of the dominative-negative approach (Takekawa et al, 1997; Takekawa and Saito, 1998; Yang et al, 2001); for example, the overexpressed MEKK4 could interfere with other pathways nonspecifically by interacting with upstream adaptors or downstream kinases not physiologically involved with MEKK4, thus making the interpretation of the results difficult. To elucidate the relationship between GADD45 and MEKK4 and the functional significance of the MEKK4-dependent pathway, we created MEKK4-deficient mice. We present here the first genetic evidence that GADD45 proteins and MEKK4 are in the same pathway in CD4 T cells. Furthermore, our study has provided new insights into the regulation and function of this important pathway during Th1 cell differentiation. Our results suggest that this pathway integrates signals from TCR and IL12/STAT4 in developing Th1 cells and promotes STAT4-independent production of IFNγ.
Results
Generation of MEKK4-deficient mice
We created MEKK4-deficient mice by homologous recombination in embryonic stem (ES) cells. We designed a targeting vector in which a lox recombination site was inserted upstream of exon 3 in the MEKK4 gene, and a neomycin cassette (flanked by two lox sites) downstream of exon 3 (Figure 1A). Correctly targeted ES cells were screened and verified by Southern blotting, and then used for blastocyst injection. Offspring from germline chimeras were crossed with splicer female mice expressing a transgene pTet-Cre (Koni et al, 2001), which caused complete deletion of the neomycin cassette and exon 3 (Figure 1B). This exon encodes 452 aa (out of 1597) of MEKK4 protein sequence and contains the entire GADD45 binding domain. Furthermore, we anticipated that this targeting strategy would result in a frame shift and premature termination of the coding sequence downstream of exon 3 in the MEKK4 protein, including the kinase domain. Western blotting using two antibodies against the N-and C-termini of MEKK4 could not detect the expression of MEKK4 protein in MEKK4−/− T cells (Figure 1C), confirming the null mutation of MEKK4 in MEKK4−/− mice.
Figure 1.
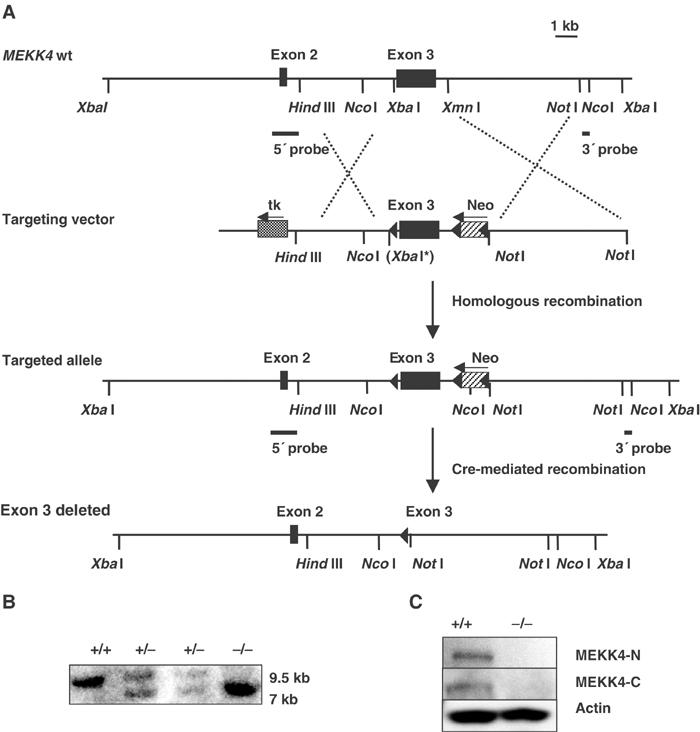
Generation of MEKK4 mutant mice. (A) Targeting strategy. The targeting vector to replace the endogenous MEKK4 exon 3 sequence contains a lox site (indicated by the filled arrow) inserted upstream of MEKK4 exon 3, and a neomycin cassette (neo) flanked by two lox sites downstream of exon 3. Cre-mediated recombination in vivo resulted in the complete deletion of MEKK4 exon 3. XbaI* indicates that the restriction site was destroyed. (B) Southern blot analysis of genomic DNA from MEKK4+/+, +/− and −/− mice. DNA was digested with NcoI and hybridized to the 3′ probe. The wild-type allele is 9.5 kb in length, and the mutant allele (with exon 3 deleted) is 7 kb. (C) Western blot analysis of activated CD4 T cells from MEKK4 +/+ and −/− mice. Two polyclonal antibodies that recognize the N-terminus (upper panel) and C-terminus (middle panel) of MTK1/MEKK4 coding sequence were used.
MEKK4 mediates IFNγ production in differentiating Th1 cells
In the progeny from MEKK4+/− crosses, MEKK4−/− mice were born with a decreased Mendelian ratio (out of 280 mice analyzed, 30, 54.6, and 15.4% were MEKK4+/+, +/−, and −/−, respectively), indicating that some of them had died during the embryonic development (to be described elsewhere). The MEKK4−/− mice that were born live exhibited a slightly reduced body size at a young age, but grew to adulthood with no other obvious defects. FACS analysis of the expression of lymphoid markers (including CD4, CD8, CD3, B220, and CD19) in the thymus, spleen, and lymph nodes of MEKK4−/− mice did not reveal major abnormalities in the development of the immune system. Also, MEKK4+/+ and −/− T cells had comparable levels of CD25, CD44, CD62L, and CD69 expression, suggesting that the homeostatic status of peripheral T cells is normal in the mutant mice (data not shown). To analyze the effect of MEKK4 deficiency on T cell activation, we stimulated CD4 T cells with anti-CD3 and/or anti-CD28, and measured their proliferation and cytokine production. We observed a similar degree of thymidine incorporation and IL2 production in MEKK4+/+ and −/− T cells (data not shown). These results suggest that TCR signaling appears to be largely intact in MEKK4−/− primary T cells.
p38 MAPK is known to have an important role in mediating IL12-driven IFNγ production in developing CD4 T cells (Rincon et al, 1998; Lu et al, 1999; Zhang and Kaplan, 2000). In light of a potential role for MEKK4 in regulating p38 activation, we examined whether IFNγ synthesis was affected by MEKK4 deficiency in developing T cells. CD4 T cells were activated by anti-CD3 and anti-CD28 in the presence of IL12 and anti-IL4 (Th1 conditions). After 4 days, the culture supernatant was harvested and IFNγ was measured by ELISA. As expected, IL12 induced a dose-dependent increase in IFNγ production in MEKK4+/+ T cells. MEKK4−/− T cells showed an approximately three-fold reduction in the IFNγ produced (Figure 2A). The reduction in IFNγ levels was detectable at different times, ranging from 3 to 6 days after primary stimulation (Figure 2B). The decreased IFNγ production in MEKK4−/− T cells was not due to differences in the cell number, as MEKK4−/− T cells showed similar or slightly increased cell number at the end of the culture period (data not shown). We also measured the expression of IFNγ mRNA by real-time PCR analysis of RNA harvested after 3 and 6 days of T cell stimulation under Th1 conditions. IFNγ mRNA was significantly reduced in MEKK4−/− T cells compared with MEKK4+/+ T cells, suggesting that the decrease in IFNγ secretion was a result of reduced expression of IFNγ mRNA by MEKK4−/− T cells (Figure 2C).
Figure 2.
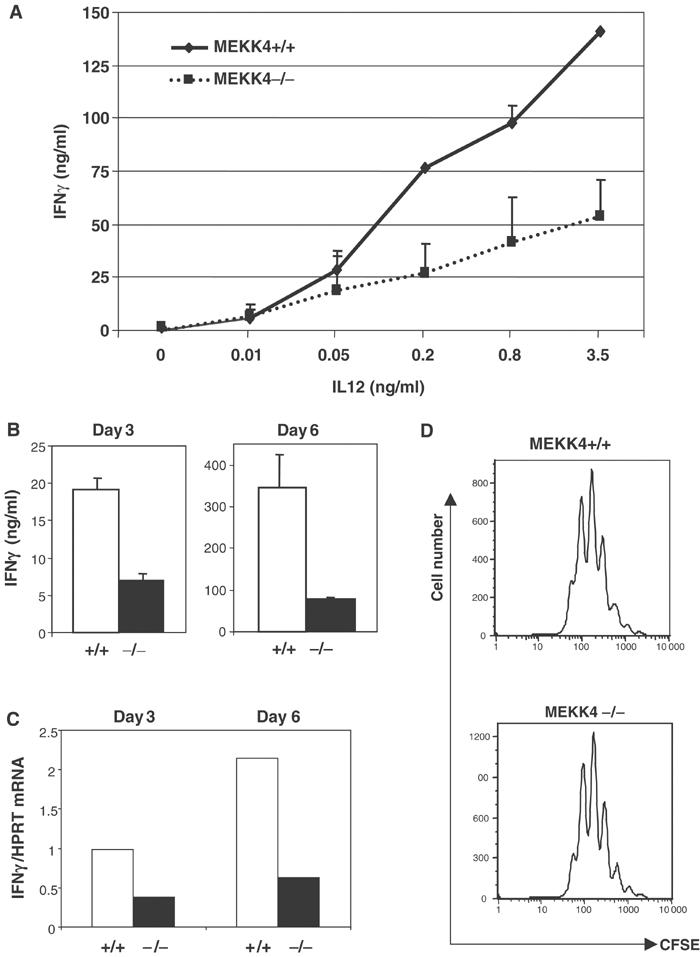
IFNγ synthesis is reduced in differentiating Th1 cells from MEKK4−/− mice. (A) CD4 T cells were activated by anti-CD3, anti-CD28, anti-IL4, and different doses of IL12 for 4 days, and IFNγ levels were measured by ELISA. (B) CD4 T cells were activated as in (A) in the presence of 2 ng/ml IL12, and IFNγ levels were measured by ELISA after 3 and 6 days of stimulation. (C) Real-time PCR analysis of IFNγ mRNA was performed using RNA isolated from cells after 3 and 6 days of Th1 differentiation. The amount of IFNγ mRNA was normalized to that of HPRT mRNA. (D) CFSE-labeled CD4 T cells were activated under Th1 conditions for 3 days, followed by FACS analysis.
In addition, we examined whether MEKK4 contributes to other functions of IL12 in primary T cells, such as potentiation of T cell proliferation (Trinchieri, 1995). IL12 stimulated a similar degree of T cell proliferation in MEKK4+/+ and −/− CD4 T cells, as measured by CFSE labeling analysis (Figure 2D). These results suggest that MEKK4 regulates IFNγ production but not proliferation in developing Th1 cells.
MEKK4 is important for both TCR and cytokine-induced IFNγ production in Th1 effector cells
Next, we evaluated the function of MEKK4 in effector cytokine production. We stimulated naive CD4 T cells from MEKK4+/+ and −/− mice under Th1 polarizing conditions. After 5–6 days of culture, live T cells were harvested, washed, and restimulated with plate-bound anti-CD3. We found that, compared with wild-type cells, MEKK4−/− Th1 cells produced significantly reduced levels of IFNγ, representing an approximately 70% reduction (Figure 3A).
Figure 3.
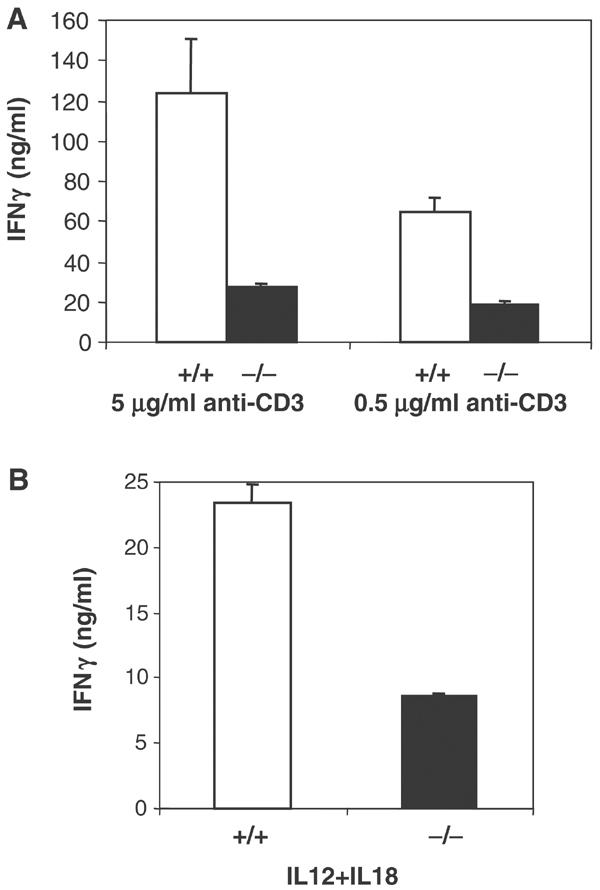
IFNγ production is decreased in MEKK4−/− Th1 effector cells. CD4 T cells were activated under Th1 conditions for 6 days. Live cells were purified by Ficoll centrifugation and restimulated with 5 μg/ml anti-CD3, 0.5 μg/ml anti-CD3 (A) or 2 ng/ml IL12 and 20 ng/ml IL18 (B). IFNγ production was measured by ELISA.
Th1 effector cells also produce IFNγ in response to combined IL12 and IL18 stimulation, in a pathway independent of TCR signaling (Yang et al, 1999, 2001). We therefore stimulated Th1 effector cells with IL12 and IL18, and measured IFNγ by ELISA. Similar to what we found using anti-CD3 stimulation, MEKK4−/− Th1 cells showed a defect in IFNγ production in response to IL12 and IL18 (Figure 3B). This is consistent with previous findings by Yang et al (2001) that a dominant-negative MEKK4 inhibits IL12/IL18-induced IFNγ in Th1 cells. Together, these results indicate that MEKK4 plays an important role in effector cytokine production in response to both TCR and IL12/1L18 stimulation.
MEKK4 promotes p38 MAPK activation in CD4 T cells
We next examined which intracellular signaling pathway was affected in MEKK4−/− T cells. The most likely candidate was that of p38 MAPK, which is known to be an important positive regulator of IFNγ and Th1 cell differentiation (Rincon et al, 1998; Lu et al, 1999; Zhang and Kaplan, 2000). A dominant-negative MEKK4 has been shown to inhibit p38 activation in several cell lines, although no genetic evidence has been provided (Takekawa et al, 1997; Takekawa and Saito, 1998). To measure the activity of p38, we used an antibody that specifically recognizes the activated form of the kinase. Th1 cells were either left untreated, or treated with anti-CD3 or IL12/IL18, then harvested for protein extraction and Western blot analysis. As shown in Figures 4A and B, there was a low level of constitutive p38 activity in wild-type Th1 cells before stimulation. This basal activity was barely detectable in MEKK4−/− cells. In response to anti-CD3 or IL12/IL18 stimulation, p38 activity was rapidly upregulated in wild-type T cells, then gradually abated. The early phase of p38 activation immediately after stimulation was slightly reduced in MEKK4−/− T cells. In contrast, p38 activities were more dramatically reduced in MEKK4−/− T cells at later time points (Figures 4A and B). Thus, MEKK4 plays an essential role in the sustained p38 activation in T cells. With a similar approach, we measured JNK activity and found that it was minimally affected by MEKK4 deficiency (Figure 4C). These results demonstrate that MEKK4 contributes to the regulation of p38, but not JNK, in T cells.
Figure 4.
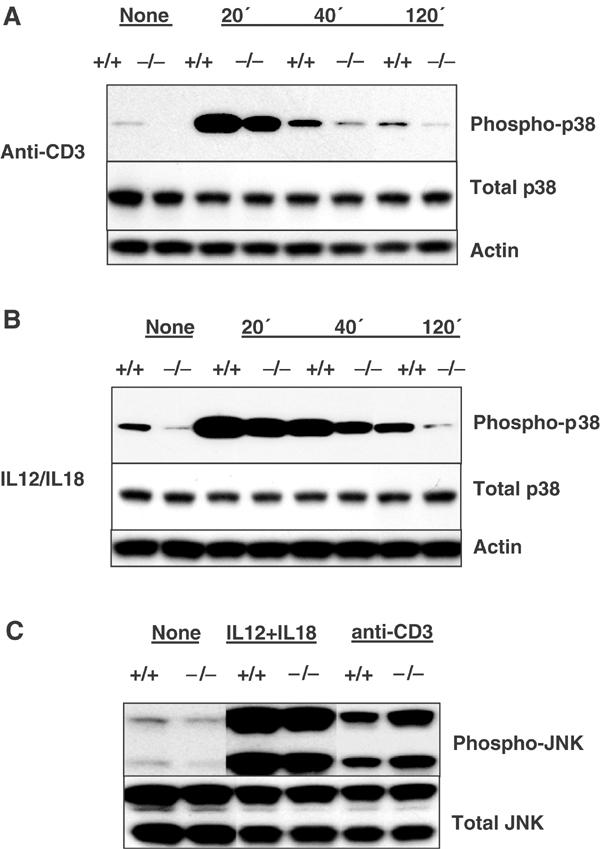
MEKK4−/− Th1 cells exhibit defective p38 activation. CD4 T cells were activated under Th1 conditions for 6 days. Live cells were left untreated, or stimulated with 5 μg/ml anti-CD3 or 2 ng/ml IL12 and 20 ng/ml IL18. Protein extracts were isolated and analyzed by Western blot using antibodies recognizing phospho-p38, total p38, actin, phospho-JNK, and total JNK. (A) p38 activities after anti-CD3 stimulation. (B) p38 activities after IL12/IL18 stimulation. (C) JNK activities after 40 min stimulation by anti-CD3 or IL12/IL18.
MEKK4 and p38 are downstream of GADD45β/GADD45γ in CD4 T cells
We noticed that MEKK4−/− Th1 effector cells exhibited defects similar to those of GADD45γ−/− and GADD45β−/− mice, namely, the reduction in IFNγ production and p38 activation (Lu et al, 2001, 2004). Therefore, MEKK4 may be a component of the same signaling pathway as GADD45 proteins in T cells. In order to determine the direct relationship between GADD45β/GADD45γ and MEKK4, we overexpressed GADD45β and GADD45γ in MEKK4+/+ and −/− T cells using GFP-tagged bicistronic retroviral vectors (Yang et al, 2001). We did not analyze the effect of GADD45α here, as this gene is expressed at low levels in peripheral T cells and does not undergo significant regulation upon T cell activation (Lu et al, 2001). Naive CD4 T cells were stimulated under Th1 conditions for 20 h and infected with GFP-RV, GADD45β-RV, or GADD45γ-RV. To measure the expression of IFNγ at earlier time points in primary cells, we used an intracellular staining method as described by Afkarian et al (2002). When gated on GFP-positive cells, overexpression of GADD45β or GADD45γ in wild-type cells resulted in an approximately 100% increase in the number of IFNγ expressing cells, compared with control virus (GFP-RV)-infected cells (Figure 5A). There was no significant difference in IFNγ levels in cells gated on the GFP-negative population (data not shown). In accordance with our earlier data, the number of IFNγ expressing cells was 60% lower in the MEKK4−/− control group than in the GFP-RV transduced wild-type cells. Significantly, the increase in IFNγ expression following GADD45β or GADD45γ transduction in the wild-type cells was completely abrogated in MEKK4−/− T cells. To verify the intracellular staining result, we measured IFNγ secretion by ELISA from virally infected cell supernatants. This approach was feasible as we could achieve a relatively high (more than 50%) transduction efficiency. Consequently, we were able to detect the effect of GADD45 overexpression in a mixed cell population that included GFP-negative, nontransduced cells. As shown in Figure 5B, retroviral expression of GADD45β or GADD45γ induced a three-fold increase in IFNγ levels in MEKK4+/+ cells after 4 days, and this effect was entirely lost in MEKK4−/− cells. These results indicate that MEKK4 is required for GADD45β/GADD4γ-induced IFNγ production in primary CD4 T cells.
Figure 5.
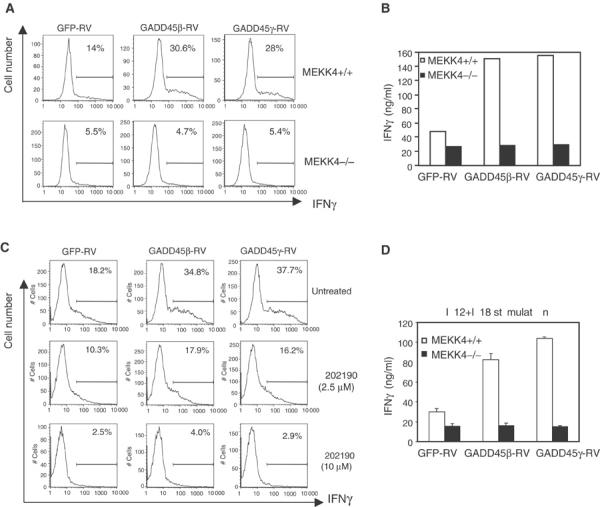
MEKK4 and p38 are required for GADD45β and GADD45γ-induced IFNγ production. (A) IFNγ levels were analyzed by intracellular staining 2 days after retroviral transduction of MEKK4+/+ and −/− T cells. (B) IFNγ levels were measured by ELISA 4 days after retroviral transduction. (C) CD4 T cells were treated with SB202190 at the time of viral transduction. After 2 days, IFNγ levels were analyzed by intracellular staining. (D) At 4 days after viral transduction, GFP-positive cells were purified by FACS and restimulated with 2 ng/ml IL12 and 20 ng/ml IL18. IFNγ was measured by ELISA.
As MEKK4−/− T cells showed reduced p38 activity, we tested whether decreasing p38 activity alone had similar effects as MEKK4 deficiency. We added a specific p38 inhibitor, SB202190, to wild-type cells infected with three types of viral vectors, and measured IFNγ production by intracellular staining. As shown in Figure 5C, SB202190 treatment of wild-type cells reduced the production of IFNγ in a dose-dependent manner. Addition of 10 μM SB202190 eliminated the effect of GADD45β/GADD45γ on IFNγ production, which was very similar to MEKK4 deficiency. Thus, the reduced p38 activation in MEKK4−/− cells may account for the defective IFNγ induction downstream of GADD45β and GADD45γ.
We further evaluated whether IFNγ production in Th1 effector cells was also regulated by the GADD45β/GADD45γ/MEKK4 pathway. At 4 days after retroviral infection, GFP-expressing cells were sorted by FACS, and restimulated with anti-CD3 or IL12/IL18. There were no strong effects of GADD45β or GADD45γ overexpression on IFNγ production when the wild-type cells were stimulated with anti-CD3 (data not shown). Thus, we were unable to determine whether MEKK4 is required for the function of GADD45 under such conditions. In contrast, when wild-type Th1 cells were stimulated with IL12/IL18, GADD45β or GADD45γ overexpression led to significantly enhanced production of IFNγ. No such effect was observed in MEKK4−/− cells (Figure 5D).
Taken together, all these results support the thesis that GADD45β/GADD45γ, MEKK4, and p38 are in the same genetic pathway that augments IFNγ production in both primary and effector CD4 T cells.
GADD45/MEKK4 induces STAT4-independent IFNγ production
STAT4 is a major intracellular pathway mediating the effect of IL12 in CD4 T cells (Kaplan et al, 1996; Thierfelder et al, 1996; Usui et al, 2003). Previous studies demonstrated that GADD45β and GADD45γ induce serine 721 phosphorylation of STAT4 through the MKK6/p38 pathway. Such a phosphorylation event is critical for the function of STAT4 in Th1 cell differentiation and IFNγ synthesis (Visconti et al, 2000; Morinobu et al, 2002). Using MEKK4−/− mice that have a complete abrogation of the effect of GADD45β and GADD45γ on IFNγ production, we explored the interaction between the MEKK4/p38 and STAT4 pathways. CD4 T cells were activated by anti-CD3 and anti-CD28 for 3 days, extensively washed, and stimulated with 10 ng/ml IL12. As shown in Figure 6A, IL12 stimulated a similar degree of serine phosphorylation of STAT4 in MEKK4+/+ and −/− cells. As a control, tyrosine phosphorylation of STAT4 was measured, which also showed no difference between MEKK4+/+ and −/− cells. Hence, STAT4 phosphorylation does not require MEKK4 function.
Figure 6.
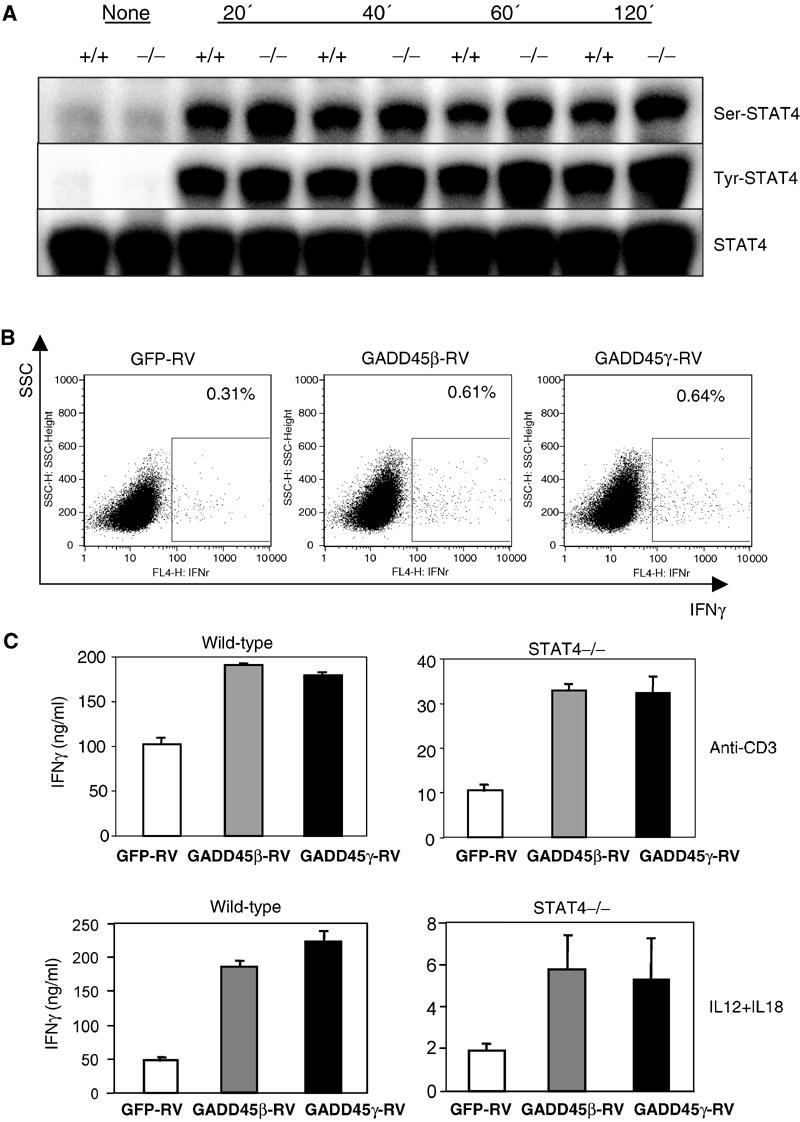
GADD45/MEKK4 induces STAT4-independent IFNγ production. (A) CD4 T cells were activated by anti-CD3 and anti-CD28 for 3 days. After washing and resting for 4 h, they were stimulated with 10 ng/ml IL12, and protein extracts were isolated at the times indicated. Western blot analysis was performed using antibodies against serine phosphorylated, tyrosine phosphorylated, and total STAT4. (B) STAT4−/− CD4 T cells were transduced with retroviruses, and analyzed by intracellular staining for IFNγ levels after 2 days. (C) STAT4+/+ and −/− CD4 T cells were transduced with retroviruses, and GFP-positive cells purified after 4 days were stimulated with 5 μg/ml anti-CD3 or 2 ng/ml IL12 and 20 ng/ml IL18. IFNγ was measured by ELISA.
We then tested whether the ability of the GADD45/MEKK4 pathway to induce IFNγ is dependent on STAT4 by expressing GADD45β or GADD45γ in STAT4−/− CD4 T cells. As in the previous experiment, we analyzed IFNγ production in primary T cells by intracellular staining 2 days after viral transduction. A moderately increased number of STAT4−/− CD4 T cells expressed IFNγ after infection with GADD45β-RV or GADD45γ-RV virus, compared with control GFP-RV virus. IFNγ levels in effector cells were measured by ELISA after restimulating the sorted, GFP-positive population. As shown in Figure 6C, when STAT4−/− Th1 cells were restimulated with anti-CD3, control cells expressing GFP alone synthesized 10 ng/ml IFNγ, while GADD45β or GADD45γ-expressing cells produced 30 ng/ml IFNγ. Upon combined IL12 and IL18 stimulation, control cells produced 2 ng/ml IFNγ, while GADD45β or γ-expressing cells produced 5–6 ng/ml IFNγ. Thus, GADD45β or GADD45γ overexpression yields STAT4-independent IFNγ production.
Expression of GADD45β and GADD45γ is differentially regulated by STAT4
The experiment shown above indicates that STAT4 is not required downstream of the GADD45/MEKK4 pathway. However, as GADD45β and GADD45γ are induced after T cell activation, especially under Th1 conditions (Lu et al, 2001), it is possible that STAT4 plays a role upstream of this pathway. We therefore assessed the requirement for STAT4 in the induction of GADD45β and GADD45γ. We activated wild-type and STAT4−/− CD4 T cells with anti-CD3 and anti-CD28 under Th1 (IL12 and anti-IL4) or neutralized (anti-IL12 and anti-IL4) conditions. Expression of GADD45β and GADD45γ mRNA was analyzed by Northern blot after 60 h. We found that GADD45β and GADD45γ induction displayed differential regulation by STAT4. As shown in Figure 7A, the expression of GADD45β was independent of exogenous IL12 stimulation, and only slightly reduced in STAT4−/− T cells under Th1 conditions, suggesting that its induction is largely driven by TCR activation. In contrast, GADD45γ induction required IL12/STAT4: the GADD45γ level was significantly higher in IL12-treated cultures, and this increase was blocked in STAT4−/− cells. This result indicates that GADD45γ induction requires STAT4 signaling, while GADD45β expression is largely STAT4 independent.
Figure 7.
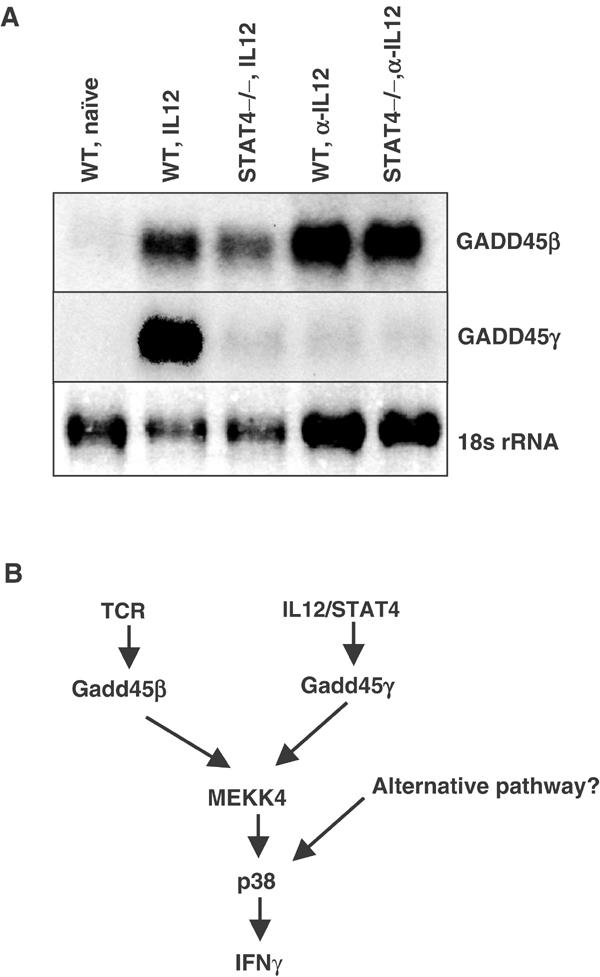
Expression of GADD45β and GADD45γ is differentially regulated by STAT4. (A) CD4 T cells from wild-type and STAT4−/− were activated by anti-CD3 and anti-CD28 under Th1 conditions (2 ng/ml IL12 and 10 μg/ml anti-IL4) or neutralized conditions (anti-IL12 and anti-IL4) for 60 h. Lane 1: WT naïve T cells. Lane 2: WT Th1 cells. Lane 3: STAT4−/− Th1 cells. Lane 4: WT cells activated under neutralized conditions. Lane 5: STAT4−/− cells activated under neutralized conditions. Note the loading difference between Th1 and neutralized conditions. (B) A diagram for the regulation of the GADD45/MEKK4 pathway in CD4 T cells.
Discussion
Previous studies have produced conflicting results concerning the relationship between GADD45 proteins and the MEKK4-mediated MAPK pathway. Using Th1 cells as a model system, we have provided the first definitive evidence that GADD45β/GADD45γ and MEKK4 are in the same genetic pathway. Our conclusion is based on the following observations. First, MEKK4−/− mice had phenotypes similar to those in the GADD45γ−/− and GADD45β−/− mice (Lu et al, 2001, 2004). CD4 T cells from these genetically modified mice expressed reduced levels of effector cytokine IFNγ accompanied by compromised activation of p38 MAPK. Second, by retroviral transduction, we demonstrated that while GADD45β and GADD45γ can drive the expression of IFNγ in MEKK4+/+ cells, this ability is completely lost in MEKK4−/− cells, indicating that MEKK4 is downstream of GADD45β and GADD45γ. Furthermore, a specific p38 inhibitor can block the IFNγ-promoting effect of GADD45β and GADD45γ. Thus, the defective p38 activation in MEKK4−/− cells likely accounts for the inability of these cells to support GADD45 protein-induced IFNγ production.
It is important to note that MEKK4−/− T cells still had significant levels of p38 activity immediately after acute stimulation, suggesting the presence of an alternative pathway(s) for activating p38 in CD4 T cells. This correlates with partial inhibition of IFNγ in MEKK4−/− cells, as p38 inhibitor further reduced IL12-induced IFNγ production in MEKK4−/− cells (data not shown). MEKK4−/− T cells showed a more dramatic reduction of p38 activity at delayed time points, indicating that the major function of MEKK4 is in the maintenance of sustained p38 activation. This most likely reflects the fact that the upstream regulators of MEKK4, GADD45β and GADD45γ, require transcriptional induction. Such phenomena have been observed in other systems. For example, TGFβ treatment of pancreatic cells and hepatocytes leads to delayed activation of p38, which is mediated by Smad-dependent expression of GADD45β (Takekawa et al, 2002; Yoo et al, 2003). Similarly, GADD45α is induced in keratinocytes exposed to UV radiation and plays an indispensable role in the late but not early phases of p38/JNK MAPK activation (Hildesheim et al, 2002). An alternative explanation is that GADD45/MEKK4 may inhibit the expression of p38 phosphatases. We have analyzed the mRNA levels by RT-PCR of three prominent p38 phosphatases, MKP1 (DUSP1), MKP5 (DUSP10), and M3/6 (DUSP8) (Camps et al, 2000; Theodosiou and Ashworth, 2002), in the MEKK4+/+ and −/− T cells. M3/6 was undetectable in either cell type, while MKP1 and MKP5 were expressed at similar levels between MEKK4+/+ and −/− cells (data not shown). Therefore, it seems unlikely that MEKK4 regulates the expression of these phosphatases.
Interestingly, MEKK4−/− T cells show a different phenotype than GADD45β−/− and GADD45γ−/− cells with regard to JNK activation. While GADD45β−/− or GADD45γ−/− Th1 cells display reduced activation of JNK (Lu et al, 2001, 2004), JNK activity is apparently normal in MEKK4−/− T cells. The simplest explanation is that GADD45γ may regulate JNK through another related MAP3K, or more likely, an indirect mechanism. As GADD45 proteins can affect chromatin structure and interact with multiple nuclear proteins and transcription factors (Smith et al, 1994; Kearsey et al, 1995; Carrier et al, 1999; Zhan et al, 1999; Yi et al, 2000), they play an important role in affecting cell cycle progression and possibly gene expression, which in turn could lead to altered JNK activity (and other biochemical and physiological changes). Such an activity of GADD45 is unlikely to be mediated by MEKK4. The MEKK4-independent function of GADD45 offers a potential mechanism for the effect of GADD45β in inhibiting JNK activity (De Smaele et al, 2001).
Another new conclusion to arise from this study is that the MEKK4 pathway plays an important role in inducing STAT4-independent production of IFNγ. We have found that serine and tyrosine phosphorylation of STAT4 is unaffected by MEKK4 deficiency, and that overexpression of GADD45 induces IFNγ production in STAT4−/− cells. Thus, GADD45/MEKK4 represents a new pathway that may mediate STAT4-independent development of Th1 cells (Kaplan et al, 1998). Future work to identify the downstream molecules targeted by this pathway will provide novel insights into our understanding of how Th1 cells develop in a STAT4-independent manner. Candidate downstream targets of p38 regulation in T cells include transcription factors of the ATF and NFAT families (Zhang and Kaplan, 2000; Wu et al, 2003). Recently, p38-dependent phosphorylation of histone H3 has been shown to promote the recruitment of NF-κB, leading to the activation of certain cytokine genes (Saccani et al, 2002). As Th1 cell development is associated with remodeling of the chromatin structure of the IFNγ locus (Avni et al, 2002; Fields et al, 2002), p38 may also play a significant role in this epigenetic process.
We have noted that our conclusion is in apparent conflict with earlier work indicating that serine 721 phosphorylation of STAT4 mediated by p38 is critical for IFNγ production and Th1 cell development (Visconti et al, 2000; Morinobu et al, 2002). However, as there is residual p38 activity in MEKK4−/− T cells, it is possible that a GADD45/MEKK4-independent pathway(s) is sufficient serine phosphorylation of STAT4. One potential candidate is Rac2, a small G protein specifically expressed in Th1 cells that has the capacity to activate p38 and induce IFNγ production (Li et al, 2000). It will be interesting to examine whether the function of Rac2 in this process requires the phosphorylation of STAT4.
In light of the STAT4-independent IFNγ production by GADD45/MEKK4, it becomes all the more intriguing to find that the expression of GADD45β and GADD45γ is differentially regulated by STAT4. The GADD45β level is rapidly increased upon TCR stimulation (Lu et al, 2004), and this induction is largely independent of IL12/STAT4 signaling. In contrast, IL12/STAT4 signaling plays an essential role in the induction of GADD45γ expression. Thus, despite the fact that upon overexpression, both GADD45β and GADD45γ can augment IFNγ expression, their function in vivo may be distinct. It is likely that GADD45β mediates an early phase of IFNγ production induced by TCR, while GADD45γ plays a role in the late stage of IFNγ synthesis downstream of IL12/STAT4. This proposal is consistent with a two-step T cell differentiation model (Avni and Rao, 2000; Avni et al, 2002; Fields et al, 2002): First, TCR stimulation of naive T cells initiates chromatin remodeling and transcription of IFNγ (and Th2 cytokine IL4), independent of the cytokine milieu. Second, IL12/STAT4 signals drive the expansion of uncommitted cells and lead to the development of Th1 effector cells. As MEKK4 is downstream of both GADD45β and GADD45γ, it appears to play an important role in integrating these signals during Th1 cell differentiation (Figure 7B).
In the current study, we found that Th1 effector cells from MEKK4−/− mice exhibit a defective response to both TCR and cytokine (IL12/IL18) stimulation. This finding differs from the results reported by Yang et al (2001), which suggests that GADD45β and MEKK4 mediate only IL12/IL18-induced, but not TCR-induced IFNγ production. Indeed, we also found that GADD45 overexpression had no strong effect on IFNγ levels in effector cells restimulated with anti-CD3, which could lead to the conclusion that GADD45 proteins play no role in the TCR signaling. However, under such conditions endogenous GADD45 proteins are strongly upregulated (Lu et al, 2004) and could obscure the effect of exogenously expressed GADD45 proteins. Consistent with this notion, retroviral expression of GADD45 proteins in STAT4−/− T cells, in which expression of endogenous GADD45 proteins is reduced, resulted in significantly increased IFNγ production after TCR stimulation. Importantly, when the endogenous components of the GADD45/MEKK4 pathway are disrupted, as in GADD45β−/−, GADD45γ−/−, and MEKK4−/− mice (Lu et al, 2001, 2004), Th1 effector cells have impaired responses to both TCR stimulation and IL12/IL18, supporting that this pathway is downstream of both TCR and IL12/IL18 in mediating effector cytokine expression.
Materials and methods
Generation of MEKK4−/− mice
The mouse MEKK4 gene was isolated from a 129/SvE mouse genomic library arrayed in high-density membranes (Research Genetics), using a human MEKK4/MTK1 cDNA probe. The targeting vector contained, in order, a 4.6-kb HindIII–XbaI fragment 5′ to MEKK4 exon 3, a lox recombination site, a 2.4-kb XbaI–XmnI fragment containing MEKK4 exon 3, a neomycin cassette flanked by two lox sites, and a 5.5-kb XmnI–NotI fragment 3′ to exon 3 (Figure 1A). The Easy-Flox vector (a gift from Dr K Rajewsky) was used as the backbone to construct the targeting vector. The targeting vector was electroporated into TC1 ES cells, and drug-resistant clones were initially screened with Southern blot analysis of XbaI-digested DNA using a probe that is 5′ to the MEKK4 genomic sequence contained in the targeting vector (data not shown). The candidate ES cell clones were expanded and verified by Southern blot analysis of NcoI-digested DNA using a probe that is 3′ to the genomic sequence contained in the targeting vector (Figure 1B). Two independent targeted ES cell clones were injected into C57BL/6 blastocysts. Chimeras were bred with splicer female mice on a C57BL/6 background (Koni et al, 2001) for germline transmission and the Cre-mediated in vivo deletion of MEKK4 exon 3 and the neomycin cassette. Age- and sex-matched MEKK4+/+ and −/− mice on a mixed C57BL/6 × 129SvE background were used in this study.
Mouse strains
C57BL/6 and BALB/C mice were purchased from the National Cancer Institute, and STAT4−/− mice from The Jackson Laboratory.
T cell culture
CD4 T cells were isolated from spleens and lymph nodes of 6–10-week-old mice. Briefly, the CD4 T cells were first enriched by immunomagnetic negative selection using antibodies against CD8, MHC class II, and NK1.1, and magnetic beads conjugated with goat anti-mouse and anti-rat Ig (Polysciences, Inc.). Naive CD62LhighCD44lowCD4+ T cells were further purified by FACS sorting. For Th1 cell differentiation, the purified CD4 T cells were stimulated with 5 μg/ml anti-CD3 (145-2C11), 2 μg/ml anti-CD28 (37.5.1), 30 U/ml human IL2, 2 ng/ml IL12 (a gift from Wyeth Research), and 10 μg/ml anti-IL4 (11B11) in the presence of irradiated splenocytes. After 5–6 days of T cell differentiation, live effector cells were obtained by Ficoll centrifugation and restimulated with plate-bound anti-CD3 or combined IL12 (2 ng/ml) and IL18 (20 ng/ml). CFSE labeling was performed as described elsewhere (Lyons, 2000).
Retroviral constructs and transduction
The original retroviral vector (GFP-RV) was kindly provided by Dr K Murphy (Washington University, St Louis, MO). GADD45β and GADD45γ were amplified by RT–PCR and subcloned as described in Yang et al (2001). Phoenix-Eco packaging cell line was transfected using Lipofectamine 2000 (Invitrogen), and the resulting retrovirus was harvested 48 and 72 h later. Primary T cells were stimulated under Th1 conditions for 20 h before being infected with retroviral supernatants. At day 4–5, GFP-positive populations were sorted and restimulated.
Intracellular staining
Monensin (GolgiStop from Pharmingen) was added in the final 4 h of T cell activation. Cells were surface stained, fixed in 4% paraformaldehyde for 10 min, and permeablized by 0.1% saponin. The staining was performed with anti-IFNγ antibody (PE or APC conjugated, Pharmingen) in 0.1% saponin. Cells were washed and subjected to FACS analysis.
ELISA
Cytokine levels in tissue culture supernatants were assayed by ELISA using the antibody pair for IFNγ (Pharmingen) according to the manufacturer's recommendations.
Production of anti-MTK1/MEKK4 antibodies
Rabbit polyclonal anti-MTK1/MEKK4 antibodies (anti-MTK1-N and -C) were raised against GST-MTK1 fusion protein corresponding to aa 58–415 mapping at the amino terminus of human MTK1 and a synthetic peptide derived from the carboxy-terminus of MTK1 (aa 1580–1595), respectively. These regions are highly conserved between human MTK1 and mouse MEKK4 sequences. Anti-MTK1-C antibody was purified by protein A and peptide affinity chromatography.
Western blot
Aliquots of 20 μg of protein extracts from T cells were separated on a SDS–PAGE gel, transferred to a polyvinylidene difluoride membrane (Millipore), and probed with anti-phospho-p38, anti-p38, anti-phospho-JNK, anti-JNK (all from Cell Signaling Technology), anti-phosphoserine-STAT4 (Santa Cruz Biotechnology), anti-phosphotyrosine-STAT4 (Zymed), anti-STAT4 (Santa Cruz Biotechnology), anti-actin (Santa Cruz Biotechnology), and anti-MTK1-N and C- antibodies.
Northern blot
RNA was isolated using the RNAeasy kit (Qiagen). RNA (2 μg) was resolved on a formaldehyde gel and blotted. The probes for GADD45β and GADD45γ were RT–PCR products.
Real-time PCR analysis
RNA (2 μg) was reverse-transcribed by Superscript II enzyme (Invitrogen). The PCR amplification was performed in an ICycler machine (Biorad Laboratories). The primers used in the real-time PCR of IFNγ mRNA were GGATGCATTCATGAGTATTGC (forward) and CCTTTTCCGCTTCCTGAGG (reverse) and the probe sequence is TTTGAGGTCAACAACCCACAGGTCCA (Biosearch Technologies). The level of IFNγ mRNA was normalized by the amount of HPRT, which was detected with the primers CTGGTGAAAAGGACCTCTCG (forward), TGAAGTACTCATTATAGTCAAGGGCA (reverse) and probe TGTTGGATACAGGCCAGACTTTGTTGGAT.
Acknowledgments
We thank L Evangelisti and C Hughes for the technical assistance with the creation of MEKK4−/− mice, Drs E Eynon, P Fields, E Jones, M Sarkisian, and E Tran for critical reading of the paper, and R Chenell and F Manzo for secretarial assistance. This work was supported by the National Institutes of Health grant (1 P01 AI36529) and the Howard Hughes Medical Institute, and also supported in part by grants from the Ministry of Education, Culture, Sports, Science, and Technology of the Japan government (to MT and Haruo Saito, University of Tokyo). RJD and RAF are Investigators of the Howard Hughes Medical Institute. HC is supported by a postdoctoral fellowship from the Arthritis Foundation.
References
- Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, Murphy TL, Murphy KM (2002) T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol 3: 549–557 [DOI] [PubMed] [Google Scholar]
- Avni O, Lee D, Macian F, Szabo SJ, Glimcher LH, Rao A (2002) T(H) cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nat Immunol 3: 643–651 [DOI] [PubMed] [Google Scholar]
- Avni O, Rao A (2000) T cell differentiation: a mechanistic view. Curr Opin Immunol 12: 654–659 [DOI] [PubMed] [Google Scholar]
- Bulavin DV, Kovalsky O, Hollander MC, Fornace AJ Jr (2003) Loss of oncogenic H-ras-induced cell cycle arrest and p38 mitogen-activated protein kinase activation by disruption of Gadd45a. Mol Cell Biol 23: 3859–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps M, Nichols A, Arkinstall S (2000) Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J 14: 6–16 [PubMed] [Google Scholar]
- Carrier F, Georgel PT, Pourquier P, Blake M, Kontny HU, Antinore MJ, Gariboldi M, Myers TG, Weinstein JN, Pommier Y, Fornace AJ Jr (1999) Gadd45, a p53-responsive stress protein, modifies DNA accessibility on damaged chromatin. Mol Cell Biol 19: 1673–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103: 239–252 [DOI] [PubMed] [Google Scholar]
- De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, Cong R, Franzoso G (2001) Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature 414: 308–313 [DOI] [PubMed] [Google Scholar]
- Fields PE, Kim ST, Flavell RA (2002) Cutting edge: changes in histone acetylation at the IL-4 and IFN-gamma loci accompany Th1/Th2 differentiation. J Immunol 169: 647–650 [DOI] [PubMed] [Google Scholar]
- Hildesheim J, Bulavin DV, Anver MR, Alvord WG, Hollander MC, Vardanian L, Fornace AJ Jr (2002) Gadd45a protects against UV irradiation-induced skin tumors, and promotes apoptosis and stress signaling via MAPK and p53. Cancer Res 62: 7305–7315 [PubMed] [Google Scholar]
- Kaplan MH, Sun YL, Hoey T, Grusby MJ (1996) Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature 382: 174–177 [DOI] [PubMed] [Google Scholar]
- Kaplan MH, Wurster AL, Grusby MJ (1998) A signal transducer and activator of transcription (Stat)4-independent pathway for the development of T helper type 1 cells. J Exp Med 188: 1191–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearsey JM, Coates PJ, Prescott AR, Warbrick E, Hall PA (1995) Gadd45 is a nuclear cell cycle regulated protein which interacts with p21Cip1. Oncogene 11: 1675–1683 [PubMed] [Google Scholar]
- Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA (2001) Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med 193: 741–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 81: 807–869 [DOI] [PubMed] [Google Scholar]
- Li B, Yu H, Zheng W, Voll R, Na S, Roberts AW, Williams DA, Davis RJ, Ghosh S, Flavell RA (2000) Role of the guanosine triphosphatase Rac2 in T helper 1 cell differentiation. Science 288: 2219–2222 [DOI] [PubMed] [Google Scholar]
- Lu B, Ferrandino AF, Flavell RA (2004) Gadd45beta is important for perpetuating cognate and inflammatory signals in T cells. Nat Immunol 5: 38–44, Epub 2003 Dec 2014.AQ [DOI] [PubMed] [Google Scholar]
- Lu B, Yu H, Chow C, Li B, Zheng W, Davis RJ, Flavell RA (2001) GADD45gamma mediates the activation of the p38 and JNK MAP kinase pathways and cytokine production in effector TH1 cells. Immunity 14: 583–590 [DOI] [PubMed] [Google Scholar]
- Lu HT, Yang DD, Wysk M, Gatti E, Mellman I, Davis RJ, Flavell RA (1999) Defective IL-12 production in mitogen-activated protein (MAP) kinase kinase 3 (Mkk3)-deficient mice. EMBO J 18: 1845–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons AB (2000) Analysing cell division in vivo and in vitro using flow cytometric measurement of CFSE dye dilution. J Immunol Methods 243: 147–154 [DOI] [PubMed] [Google Scholar]
- Morinobu A, Gadina M, Strober W, Visconti R, Fornace A, Montagna C, Feldman GM, Nishikomori R, O'Shea JJ (2002) STAT4 serine phosphorylation is critical for IL-12-induced IFN-gamma production but not for cell proliferation. Proc Natl Acad Sci USA 99: 12281–12286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon M, Enslen H, Raingeaud J, Recht M, Zapton T, Su MS, Penix LA, Davis RJ, Flavell RA (1998) Interferon-gamma expression by Th1 effector T cells mediated by the p38 MAP kinase signaling pathway. EMBO J 17: 2817–2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccani S, Pantano S, Natoli G (2002) p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol 3: 69–75 [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M (1999) Stress-induced JNK activation is independent of Gadd45 induction. J Biol Chem 274: 29595–29598 [DOI] [PubMed] [Google Scholar]
- Smith ML, Chen IT, Zhan Q, Bae I, Chen CY, Gilmer TM, Kastan MB, O'Connor PM, Fornace AJ Jr (1994) Interaction of the p53-regulated protein Gadd45 with proliferating cell nuclear antigen. Science 266: 1376–1380 [DOI] [PubMed] [Google Scholar]
- Takekawa M, Posas F, Saito H (1997) A human homolog of the yeast Ssk2/Ssk22 MAP kinase kinase kinases, MTK1, mediates stress-induced activation of the p38 and JNK pathways. EMBO J 16: 4973–4982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takekawa M, Saito H (1998) A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell 95: 521–530 [DOI] [PubMed] [Google Scholar]
- Takekawa M, Tatebayashi K, Itoh F, Adachi M, Imai K, Saito H (2002) Smad-dependent GADD45beta expression mediates delayed activation of p38 MAP kinase by TGF-beta. EMBO J 21: 6473–6482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodosiou A, Ashworth A (2002) MAP kinase phosphatases. Genome Biol 3, REVIEWS3009. Epub 2002 Jun 3026AQ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, Sangster MY, Vignali DA, Doherty PC, Grosveld GC, Ihle JN (1996) Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature 382: 171–174 [DOI] [PubMed] [Google Scholar]
- Trinchieri G (1995) Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu Rev Immunol 13: 251–276 [DOI] [PubMed] [Google Scholar]
- Usui T, Nishikomori R, Kitani A, Strober W (2003) GATA-3 suppresses Th1 development by downregulation of Stat4 and not through effects on IL-12Rbeta2 chain or T-bet. Immunity 18: 415–428 [DOI] [PubMed] [Google Scholar]
- Visconti R, Gadina M, Chiariello M, Chen EH, Stancato LF, Gutkind JS, O'Shea JJ (2000) Importance of the MKK6/p38 pathway for interleukin-12-induced STAT4 serine phosphorylation and transcriptional activity. Blood 96: 1844–1852 [PubMed] [Google Scholar]
- Wang X, Gorospe M, Holbrook NJ (1999) gadd45 is not required for activation of c-Jun N-terminal kinase or p38 during acute stress. J Biol Chem 274: 29599–29602 [DOI] [PubMed] [Google Scholar]
- Wu CC, Hsu SC, Shih HM, Lai MZ (2003) Nuclear factor of activated T cells c is a target of p38 mitogen-activated protein kinase in T cells. Mol Cell Biol 23: 6442–6454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Murphy TL, Ouyang W, Murphy KM (1999) Induction of interferon-gamma production in Th1 CD4+ T cells: evidence for two distinct pathways for promoter activation. Eur J Immunol 29: 548–555 [DOI] [PubMed] [Google Scholar]
- Yang J, Zhu H, Murphy TL, Ouyang W, Murphy KM (2001) IL-18-stimulated GADD45 beta required in cytokine-induced, but not TCR-induced, IFN-gamma production. Nat Immunol 2: 157–164 [DOI] [PubMed] [Google Scholar]
- Yi YW, Kim D, Jung N, Hong SS, Lee HS, Bae I (2000) Gadd45 family proteins are coactivators of nuclear hormone receptors. Biochem Biophys Res Commun 272: 193–198 [DOI] [PubMed] [Google Scholar]
- Yoo J, Ghiassi M, Jirmanova L, Balliet AG, Hoffman B, Fornace AJ Jr, Liebermann DA, Bottinger EP, Roberts AB (2003) Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J Biol Chem 278: 43001–43007 [DOI] [PubMed] [Google Scholar]
- Zhan Q, Antinore MJ, Wang XW, Carrier F, Smith ML, Harris CC, Fornace AJ Jr (1999) Association with Cdc2 and inhibition of Cdc2/Cyclin B1 kinase activity by the p53-regulated protein Gadd45. Oncogene 18: 2892–2900 [DOI] [PubMed] [Google Scholar]
- Zhang S, Kaplan MH (2000) The p38 mitogen-activated protein kinase is required for IL-12-induced IFN-gamma expression. J Immunol 165: 1374–1380 [DOI] [PubMed] [Google Scholar]