

Abstract
Since conventional 14-day primaquine (PMQ) radical cure of vivax malaria is associated with poor compliance, and as total dose, not therapy duration, determines efficacy, a preliminary pharmacokinetic study of two doses (0.5 and 1.0 mg/kg of body weight) was conducted in 28 healthy glucose-6-phosphate dehydrogenase-normal Papua New Guinean children, aged 5 to 12 years, to facilitate development of abbreviated high-dose regimens. Dosing was with food and was directly observed, and venous blood samples were drawn during a 168-h postdose period. Detailed safety monitoring was performed for hepatorenal function and hemoglobin and methemoglobin concentrations. Plasma concentrations of PMQ and its metabolite carboxyprimaquine (CPMQ) were determined by liquid chromatography-mass spectrometry and analyzed using population pharmacokinetic methods. The derived models were used in simulations. Both single-dose regimens were well tolerated with no changes in safety parameters. The mean PMQ central volume of distribution and clearance relative to bioavailability (200 liters/70 kg and 24.6 liters/h/70 kg) were within published ranges for adults. The median predicted maximal concentrations (Cmax) for both PMQ and CPMQ after the last dose of a 1.0 mg/kg 7-day PMQ regimen were approximately double those at the end of 14 days of 0.5 mg/kg daily, while a regimen of 1.0 mg/kg twice daily resulted in a 2.38 and 3.33 times higher Cmax for PMQ and CPMQ, respectively. All predicted median Cmax concentrations were within ranges for adult high-dose studies that also showed acceptable safety and tolerability. The present pharmacokinetic data, the first for PMQ in children, show that further studies of abbreviated high-dose regimens are feasible in this age group.
INTRODUCTION
Primaquine (PMQ) is an 8-aminoquinoline drug used for primary (causal) and terminal malaria prophylaxis, radical cure of Plasmodium vivax and P. ovale, and as a gametocytocidal agent in P. falciparum infections (1–4). It remains the only FDA-approved drug for elimination of liver stages (hypnozoites and schizonts) of P. vivax and P. ovale (2, 4, 5). In many non-African tropical countries, such as Papua New Guinea (PNG), there is hyperendemic transmission of P. vivax (5–7). Children carry the burden of repeated P. vivax infections in this geoepidemiologic setting (5, 8, 9). Therefore, a safe and effective radical cure would benefit personal well-being, growth, and development and lessen the economic impact of the infection on the community (10).
Primaquine is conventionally administered as a 14-day course for terminal prophylaxis and radical cure (2), but this regimen can be problematic due to poor compliance (1, 5). An abbreviated regimen would have advantages (11, 12) as long as it was safe and well tolerated. Pharmacokinetic studies of PMQ to date have been conducted only in adults (6). As the pharmacokinetic and pharmacodynamic profiles of antimalarial drugs can differ between adults and children (13), there is a need for a study of PMQ disposition in the pediatric age group (14, 15). The conventionally recommended PMQ regimen for radical cure of 0.5 mg/kg daily for 14 days is well tolerated in glucose-6-phosphate dehydrogenase (G6PD)-normal PNG children (12). To characterize the pharmacokinetic properties of PMQ and facilitate the development of higher-dose, shorter-course PMQ treatment in this patient population, we conducted an intensive-sampling pharmacokinetic study with 0.5 or 1.0 mg/kg of body weight given as a single dose to healthy PNG children aged 5 to 12 years.
MATERIALS AND METHODS
Study site, approvals, and subjects.
The present study was based at the Alexishafen Health Centre, Madang Province, on the north coast of PNG, where there is hyperendemic transmission of P. falciparum and P. vivax (8). The study was approved by the PNG Institute of Medical Research Institutional Review Board and the Medical Research Advisory Committee of the PNG Health Department. Subjects were recruited between August and September 2010 from local villages, where an explanation of study aims and procedures was given to community members. After written informed consent had been obtained from parents/guardians, eligible children were screened for G6PD status (WST-8 [lyophilized] method; Dojindo Molecular Technologies Inc., Japan), their demographic and anthropometric data were recorded, a hemoglobin concentration was measured (HemoCue, Ängelholm, Sweden), and a blood slide was taken. Children who had (i) normal G6PD status, (ii) a blood slide that was negative for malaria, (iii) no clinical features of illness, (iv) no history of PMQ allergy, (v) no evidence of severe malnutrition (weight-for-age nutritional Z score [WAZ], <60th percentile), and (vi) a hemoglobin concentration of ≥80 g/liter were recruited and admitted to the Alexishafen Health Center for the first 2 days of study procedures.
Baseline assessment, treatment allocation, and clinical procedures.
At enrollment, a detailed history and symptom questionnaire were completed with the assistance of the parents/guardians and a full physical examination was performed, including body weight, height, axillary temperature, supine and erect blood pressure and pulse rate, respiratory rate, mean upper arm circumference, and spleen size. A urine sample was tested for the presence of protein, blood, and/or glucose. Baseline methemoglobin levels were determined by pulse oximetry (Masimo Rad-57 pulse oximeter with SpMet; Masimo, Australia), and an electrocardiogram was taken. An intravenous cannula was inserted, and a venous blood sample was drawn for a full blood count (Coulter counter; Beckman Coulter, Australia). The remainder of the sample was centrifuged and the plasma stored at <−20°C for subsequent drug and biochemical assays. The red cell pellet was retained for parasite genotyping.
Each child was randomized to a single oral dose of PMQ at either (i) 0.5 mg/kg body weight (group A) or (ii) 1.0 mg/kg (group B) PMQ base given as diphosphate tablets (Shin Poon Pharmaceuticals, Seoul, South Korea). Participants were not required to fast. The drug was administered with water under direct observation, and to minimize adverse effects of PMQ when taken on an empty stomach, all participants were given a packet of crackers to consume directly afterwards. If a child vomited within 1 h of dosing, the same dose was to be readministered and the time documented. After dosing, all participants had additional 2-ml venous blood samples drawn at 1, 2, 3, 4, 6, 8, 12, 18, 24, 36, 48, 72, 120, and 168 h for drug assay. Aliquots of plasma taken at baseline and at 72 and 168 h were retained for biochemical analysis. All children were reassessed clinically on days 1, 2, 3, and 7 for symptoms and vital signs, as well as hemoglobin concentration (HemoCue), methemoglobin saturation (Masimo), and blood slide.
Laboratory methods.
Screening for G6PD deficiency was performed according to the manufacturer's instructions. In brief, a 250-μl finger prick blood sample was collected into a cooled tube containing EDTA as the anticoagulant. A 5-μl aliquot of whole blood was then mixed with buffer-dye solution, wrapped in aluminum foil, and incubated at 37°C for 25 min. A 10-μl aliquot of hydrochloric acid then was added, and the color change was assessed visually in reference to a standard color chart. Each sample was then loaded into a 96-well plate and read on a microplate reader at an absorbance of 460 nm to confirm visual interpretation. Participants who had G6PD deficiency were withdrawn from the study, and the result was recorded in the child's health book with a recommendation that PMQ therapy not be administered in the future. Other biochemical tests were performed at PathWest Laboratory Medicine, Fremantle Hospital, Western Australia, Australia, using methods that have been described previously (16).
After initial microscopy in the field, all blood films were reexamined independently by two skilled microscopists in a central laboratory with discrepancies adjudicated by a third microscopist. Parasite densities were calculated from the number of parasites per 200 or 500 white blood cells depending on parasitemia and an assumed total peripheral white cell count of 8,000/μl. Reinfection and recrudescence were distinguished by comparing PCR-restriction fragment length polymorphism-generated genotype patterns of merozoite surface protein 2 to PCR genotype patterns of merozoite surface protein 1 and glutamate-rich protein in pairs of samples obtained at enrollment and on the day of reappearance of parasitemia (17).
Plasma concentrations of PMQ and its principal metabolite, carboxyprimaquine (CPMQ), were determined simultaneously using a validated liquid chromatography-mass spectrometry (LC-MS) method as described previously (18). Intra- and interday precision for both PMQ and CPMQ were <10% across the concentration range of 5 to 1,000 μg/liter, with >85% recovery and sensitivity of 1 to 2 μg/liter. All venous blood samples for drug assay were centrifuged for 5 min at 3,000 × g, and the plasma was separated from the red cell pellet and stored in a foil-covered tube at <−20°C until assay within 6 months of sample collection. Primaquine content was determined in 20 tablets selected randomly from the single batch used in the present study. The stated content was 26.3 mg PMQ diphosphate (or 15 mg PMQ base), which was comparable to the assayed mean ± standard deviation (SD) content of 25.71 ± 0.85 mg.
Pharmacokinetic modeling.
Loge plasma concentration-time data sets for PMQ and CPMQ were analyzed by nonlinear mixed-effect modeling using NONMEM (v 7.2.0; ICON Development Solutions, Ellicott City, MD) with an Intel Visual FORTRAN 10.0 compiler. The first-order conditional estimation (FOCE) with interaction estimation method was used. The minimum value of the objective function (OFV), conditional weighted residual (CWRES) plots, and condition number (<1,000) were used to choose suitable models during the model-building process. Allometric scaling was employed a priori, with volume terms multiplied by (WT/70)1.0 and clearance terms by (WT/70)0.75, where WT is body weight in kg (19). Two structures for residual variability (RV), equivalent to proportional and combined RV structures on the normal scale, were used for the log-transformed data. Secondary pharmacokinetic parameters, including area under the curve (AUC0-∞) and elimination half-lives (t1/2) for the participants, were obtained from post hoc Bayesian prediction in NONMEM using the final model parameters. Models were parameterized using ka (absorption rate constant), VC/F (central volume of distribution [Vc] relative to bioavailability [F]), CL/F (clearance), and VP/F and Q/F (peripheral volumes of distribution[s] and their intercompartmental clearance[s], respectively).
PMQ was initially modeled alone using one- and two-compartment models (ADVAN 2 and 4, respectively) with first-order absorption and with and without lag time. Once a suitable model for PMQ was obtained, CPMQ and PMQ data sets were modeled simultaneously. All PMQ was assumed to be converted to CPMQ to allow identifiability in the model. One-, two- and three-compartment models were tested for CPMQ using user-defined linear mammillary models (ADVAN 5). Once the structure of the models was established, interindividual variability (IIV) and correlations between IIV terms were estimated when supported by the data. Finally, relationships between model parameters and the covariates dose group, dose (mg/kg), age, and sex were identified using correlation plots and subsequently evaluated within NONMEM.
Model evaluation and simulations.
A bootstrap using Perl speaks NONMEM (PSN) with 1,000 samples was performed, and the parameters derived from this analysis were summarized as median and 2.5th and 97.5th percentiles (95% empirical confidence intervals [CI]) to facilitate evaluation of final model parameter estimates. In addition, prediction-corrected visual predictive checks (pcVPCs) were performed with 1,000 data sets simulated from the final models, and these were stratified according to treatment group for PQ. The observed 10th, 50th, and 90th percentiles were plotted with their respective simulated 95% CIs. Numeric predictive checks (NPCs) were performed to complement the pcVPCs in assessing the predictive performance of the model.
Once a final model had been established, simulations were performed to assess three different multiple-dosing treatment regimens on the peak concentration (Cmax) of PMQ and CPMQ. These were (i) 0.5 mg/kg daily for 14 days, (ii) 1.0 mg/kg daily for 7 days, and (iii) 1.0 mg/kg twice daily for 7 doses (3.5 days). Each treatment regimen had the same total dose (7.0 mg/kg). Cmax was determined from simulated subjects, and their drug concentrations were obtained at 6-min intervals. For the simulations, 1,000 male and female subjects for each age between 5 and 10 years were used. Weights for each age group were based on sex and simulated from WHO weight-for-age data (20). Simulated data for male and female children were combined, and the resulting median for each dosing regimen was plotted against age. Simulated concentrations across all age groups were pooled, and the median concentration with 95% prediction intervals versus time was plotted for both PMQ and CPMQ for the three dosing regimens.
Data analysis.
Data are, unless otherwise stated, summarized as means and SD or medians and interquartile ranges (IQR). General linear modeling for repeated measures was used to determine whether variables differed significantly over time or by treatment group and whether there was a treatment group-time interaction.
RESULTS
Patient characteristics.
Thirty children were recruited, but two in group B were found to be parasitemic on review of the baseline blood smear and were excluded. Details of the remaining 28 children are summarized in Table 1. The two groups were well matched for demographic, anthropometric, and clinical characteristics. No child had a baseline value for hemoglobin, methemoglobin, or biochemical assays that was outside reference ranges for these analytes (16).
TABLE 1.
Admission details for the children in each primaquine dose group
| Parameter | Value for dose groupa: |
|
|---|---|---|
| A (n = 15) | B (n = 13) | |
| Age (mo) | 97 (72–120) | 80 (70–90) |
| Sex (no. [%] male) | 9 (60) | 6 (46) |
| Body wt (kg) | 19.3 ± 3.5 | 17.5 ± 2.2 |
| Height (cm) | 115 ± 10 | 111 ± 7.1 |
| Upper arm circumference (cm) | 15.9 ± 1.4 | 15.7 ± 1.6 |
| Axillary temp (°C) | 36.6 ± 0.2 | 36.5 ± 0.4 |
| Supine systolic/diastolic blood pressure (mmHg) | 82 (80–90)/54 (50–60) | 90 (80–92)/60 (53–61) |
| Standing systolic/diastolic blood pressure (mmHg) | 90 (80–100)/60 (50–64) | 90 (80–95)/60 (58–61) |
| Systolic/diastolic postural blood pressure change (mmHg) | −2 (−10–2)/0 (−6–0) | −2 (−4–0)/−1 (−5–0) |
| Respiratory rate (breaths/min) | 22 ± 3 | 25 ± 4* |
| Supine pulse rate (beats/min) | 83 ± 11 | 84 ± 13 |
| Hemoglobin (g/liter) | 117 ± 12 | 115 ± 12 |
| Methemoglobin (% of total hemoglobin) | 1.0 ± 0.2 | 1.1 ± 0.5 |
| Rate-corrected electrocardiographic QT interval (ms0.5) | 440 (414–447) | 431 (417–440) |
| White cell count (×109/liter) | 9.0 ± 2.7 | 9.5 ± 5.2 |
| Platelet count (×109/liter) | 275 ± 67 | 216 ± 74* |
| Serum alanine aminotransaminase (U/liter) | 9 (6–13) | 9 (8–16) |
| Serum total bilirubin (μmol/liter) | 1.9 (1.8–2.9) | 2.1 (1.8–2.9) |
| Serum creatinine (μmol/liter) | 47 (41–51) | 41 (40–46) |
Data are means ± SD or medians (IQR). *, P < 0.05 versus a single dose of 0.5 mg/kg PMQ.
Safety and tolerability.
Both doses were well tolerated. No child vomited after drug administration. There were no changes in symptoms and their severity during follow-up in either group, including nausea and abdominal pain, and no severe adverse events were reported. No children developed abnormal hepatorenal function on day 3 or 7 after treatment. Hemoglobin concentrations declined initially and then increased (trend P = 0.033) (Fig. 1), with no between-group differences (mean difference, −2 [−11 to 8]; P = 0.69). After pooling dose groups, the mean hemoglobin concentration at day 2 was significantly lower than that at day 7 (P < 0.023, Bonferroni corrected). There were no significant changes in methemoglobin levels in participants over time or between groups (trend P = 0.81; between-group mean difference [95% confidence interval], −0.1 [−0.2 to 0.1]; P = 0.29).
FIG 1.
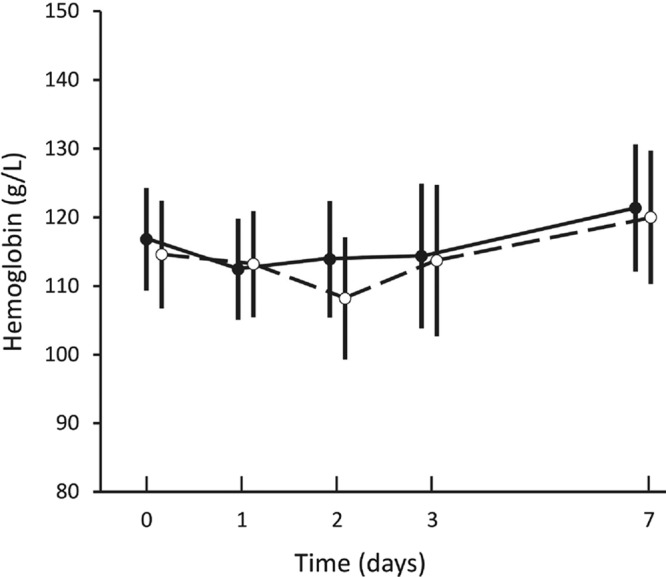
Means and 95% confidence intervals for hemoglobin concentrations measured before and for 7 days after dosing of 0.5 mg/kg (closed circles) and 1.0 mg/kg (open circles).
Pharmacokinetic modeling.
There were 246 PMQ and 360 CPMQ individual plasma concentrations available for analysis. Of these, 6 (2.4%) and 3 (0.8%) were below the limit of quantification for PMQ and CPMQ, respectively. For PMQ, a 1-compartment model with first-order absorption and no lag time adequately described the plasma concentration-time coordinates. An additional compartment did not improve the CWRES plot or result in a significant decrease in the objective function value (ΔOFV, −5.767; P > 0.05; df, 2). For CPMQ, a model with two CPMQ-specific compartments, central and peripheral (P1), was superior to a single additional compartment with an improved CWRES plot accompanied by a decrease in the OFV of 254.163 (P < 0.01; df, 2). The addition of a third compartment for CPMQ did not result in further improvement (ΔOFV, −1.435; P > 0.05; df, 2). Therefore, the structural model parameters were ka, VC/FPMQ, CL/FPMQ, VC/F*CPMQ, VP1/F*CPMQ, CL/F*CPMQ, and Q2/F*CPMQ, where F* represents the combination of the bioavailability of PMQ and the fraction of metabolic conversion of PMQ to CPMQ. As ka was poorly estimated in the combined model with a relative standard error (RSE) of >80%, it was fixed as the value obtained from modeling PMQ alone. IIV was estimable on ka, VC/FPMQ, and CL/FPMQ. VC/F*CPMQ and CL/F*CPMQ and the correlation between VC/FPMQ and CL/FPMQ, as well as that between VC/F*CPMQ and CL/F*CPMQ, were estimated. None of the tested covariates significantly improved the model.
The final model parameter estimates and the bootstrap results for PMQ and CPMQ are summarized in Table 2. Most (94%) bootstrap runs were successful. Bias was <5% for all structural and random model parameters. The condition number for the final model was 631. Figures 2 and 3 show goodness-of-fit plots for PMQ and CPMQ, respectively, and pcVPCs are shown in Fig. 4. From the NPCs, 10 and 9% of the data points were below and 7.5 and 8% of the data points were above the 80% prediction interval for PMQ and CPMQ, respectively.
TABLE 2.
Final population pharmacokinetic estimates and bootstrap results for PMQ and CPMQ
| Parameter | Mean | RSE (%) | Bootstrap median (95% CI) |
|---|---|---|---|
| Objective function value | −790.147 | −810.879 (−978.555-−668.075) | |
| Structural model | |||
| kaa (/h) | 2.18 | 30 | 2.2 (1.2–6.3) |
| CL/FPMQ (liters/h/70 kg) | 24.6 | 7 | 24.5 (21.7–28.1) |
| VC/FPMQ (liters/70 kg) | 200 | 6 | 199 (175–226) |
| CL/F*CPMQ (liters/h/70 kg) | 1.15 | 8 | 1.14 (0.98–1.34) |
| VC/F*CPMQ (liters/70 kg) | 7.19 | 27 | 7.08 (4.04–10.74) |
| Q/F*CPMQ (liters/h/70 kg) | 3.59 | 15 | 3.59 (2.77–4.47) |
| VP/F*CPMQ (liters/70 kg) | 14.2 | 8 | 14.2 (12.6–16.3) |
| Variable model (% shrinkage) | |||
| IIV in ka (%) | 138 (25) | 22 | 140 (89–245) |
| IIV in CL/FPMQ (%) | 33 (2) | 12 | 32 (24–39) |
| IIV in VC/FPMQ (%) | 31 (6) | 16 | 31 (20–41) |
| IIV in CL/F*CPMQ (%) | 95 (3) | 14 | 94 (70–124) |
| IIV in VC/F*CPMQ (%) | 40 (1) | 16 | 39 (27–49) |
| r(CL/FPMQ, VC/FPMQ) | 0.820 | 32 | 0.829 (0.633–0.934) |
| r(CL/FCPMQ, VC/FCPMQ) | 0.829 | 26 | 0.837 (0.409–1.62) |
| RV for PMQ (%) | 25.0 | 6 | 24 (21–27) |
| RV for CPMQ (%) | 20 | 9 | 20 (16–24) |
| RV for CPMQ (μg/liter) | 2.39 | 39 | 2.32 (1.09–4.77) |
ka was fixed in the combined model. RSE and bootstrap values were from the PMQ-only model. r is the Pearson product-moment correlation coefficient for the two variables in parentheses. The % shrinkage for IIV is a measure of parameterization of the data, with low percentages indicating an acceptable number of model parameters.
FIG 2.

Goodness-of-fit plots for primaquine.
FIG 3.

Goodness-of-fit plots for carboxyprimaquine.
FIG 4.
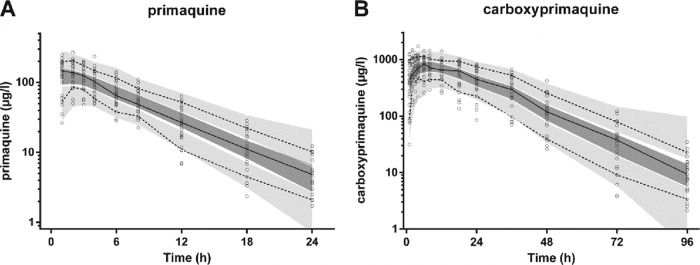
Prediction-corrected visual predictive check with observed 50th (solid line), 10th, and 90th (dotted lines) percentiles within their simulated 95% CI (gray shaded areas) for primaquine (A) and carboxyprimaquine (B) (μg/liter on a log10 scale) overlying the data points (○).
The simulated Cmax for PMQ and CPMQ for the three different dosing regimens are depicted by age in Fig. 5. Variability between the age groups was low (<7%) for both PMQ and CPMQ. Simulated plasma PMQ and CPMQ concentrations, together with 95% prediction intervals for the three different dosing regimens, are shown in Fig. 6 and are summarized in Table 3. The median Cmax for both PMQ and CMPQ after the last dose of the 1.0 mg/kg 7-day regimen were approximately double those at the end of 14 days of 0.5 mg/kg daily, while the 1.0 mg/kg twice-daily regimen resulted in a 2.38 and 3.33 times higher Cmax for PMQ and CPMQ, respectively.
FIG 5.
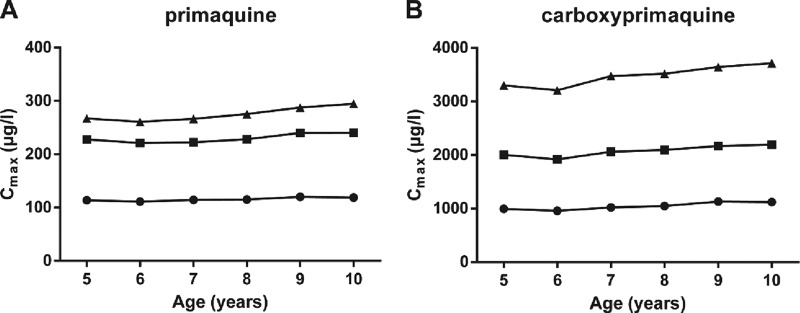
Simulated Cmax for PMQ (A) and CPMQ (B) for children aged 5 to 10 years for three different dosing regimens: (i) 0.5 mg/kg daily for 14 days (•), (ii) 1 mg/kg daily for 7 days (■), and (iii) 1 mg/kg twice daily for 7 doses (3.5 days; ▲).
FIG 6.

Simulated median (black lines) and 95% prediction intervals (gray lines) for plasma PMQ (A) and CPMQ (B) concentrations (μg/liter) for three different dosing regimens.
TABLE 3.
Median intervals for Cmax of PMQ and CPMQ for three different treatment regimens simulated for children aged 5 to 10 years
| Treatment regimen | Median (95% prediction) intervals for Cmax of: |
|
|---|---|---|
| Primaquine | Carboxyprimaquine | |
| 0.5 mg/kg daily for 14 days | 115 (56.5–226) | 1,046 (478–2,032) |
| 1 mg/kg daily for 7 days | 230 (112–442) | 2,073 (968–4,143) |
| 1 mg/kg twice daily for 3.5 days | 275 (142–508) | 3,477 (1,638–7,001) |
DISCUSSION
The present study was designed to generate novel pharmacokinetic data that could be used to develop an abbreviated PMQ dosing regimen for radical cure of pediatric vivax malaria. Both 0.5 mg/kg (conventional) and 1.0 mg/kg (double) doses were safe and well tolerated. Frequent blood sampling and a validated LC-MS assay allowed simultaneous population pharmacokinetic analysis of PMQ and CPMQ plasma concentration-time profiles. Based on those single-dose data, published efficacy and tolerability studies of high-dose PMQ in adults (21–23), and practical considerations, two short-course PMQ regimens were simulated, specifically 1.0 mg/kg daily for 7 days and 1.0 mg/kg twice daily for 3.5 days. The predicted plasma PMQ and CPMQ concentrations achieved during these two regimens were no greater than those seen in previous pharmacokinetic studies of adults, suggesting that both could be further assessed in safety and efficacy field studies.
A major barrier to the control of malaria in countries where vivax malaria is endemic, such as PNG, is the ability of the parasite to relapse from dormant hypnozoites (7), including in the aftermath of successful treatment of falciparum malaria (17). Repeated vivax infections contribute to substantially increased morbidity and mortality (5, 8, 9) and have an adverse socioeconomic impact (10). Radical cure of vivax malaria in this geoepidemiologic situation has, however, been hampered by several factors. First, PMQ induces hemolysis in G6PD-deficient patients, mandating pretreatment testing for G6PD status or the use of low-dose PMQ regimens that may not be effective, especially against the Chesson strain in PNG (24). The development of cost-effective point-of-care tests (25, 26) should help to overcome this problem. Second, prolonged treatment courses are associated with poor compliance (27, 28). Since the total dose of, rather than duration of exposure to, PMQ determines the efficacy of radical cure (2, 29, 30), abbreviated treatment courses have been developed. Third, PMQ is associated with gastrointestinal side effects that are related to dose but which are attenuated by coadministration with food (31) and also causes methemoglobinemia which is typically mild and transient (6).
Several adult studies have examined the safety and efficacy of abbreviated high-dose PMQ courses. In Caucasians taking 0.5 mg/kg twice daily with food for 7 days, side effects were generally nonsevere, although 5% of the subjects had methemoglobinemia sufficient to cause peripheral cyanosis without respiratory compromise (21). However, in Thai patients with vivax malaria, this same regimen was as well tolerated as the conventional 14-day, once-daily 0.5 mg/kg regimen (23). In another Thai study, patients with vivax malaria who received 1.0 mg/kg daily for 7 days had the same adverse effect profile as those receiving 0.5 mg/kg daily for 7 days, but P. vivax relapses were significantly fewer (22).
Extrapolation of the findings from available adult studies (21–23) suggests that a 7-day regimen of 1.0 mg/kg daily in children should also be well tolerated and effective. Because it has an elimination half-life of 4 to 6 h (32), there is no significant PMQ accumulation with daily dosing. Therefore, the Cmax and area under the plasma concentration-time curve at the beginning and end of a 14-day course of daily PMQ doses are similar (1, 33). This lack of time/dose-dependent kinetics improves the validity of extrapolation from single to multiple dosings in our simulations. The model-derived mean CL/F of 24.6 liters/h/70 kg in our children is within the range for adults of 20 to 40 liters/h, as is the mean VC/F of 200 liters/70 kg versus 200 to 300 liters in adults (34). These data suggest that the median PMQ Cmax predicted after the last dose of this regimen in our children (230 μg/liter) would be similar to that in adults. There are no adult Cmax data for single or multiple 1.0 mg/kg doses, but in one of the first papers to detail the pharmacokinetic properties of single-dose PMQ in adults (35), the Cmax increased from 53 μg/liter at 0.25 mg/kg to 104 μg/liter at 0.5 mg/kg and 176 μg/liter at 0.75 mg/kg, a trend consistent with our predicted Cmax.
Although no adult pharmacokinetic studies have utilized a PMQ dose as high as 2.0 mg/kg given as a once-daily or divided twice-daily dose, the 95% prediction intervals for PMQ Cmax after the last dose of the 3.5-day, 1.0 mg/kg twice-daily regimen (142 to 508 μg/liter) lie within the range of concentrations found in healthy Thai adults who were given a single 45-mg (0.75 mg/kg) dose (113 to 532 μg/liter) (36). This emphasizes the wide interindividual variability in PMQ disposition. In any case, single PMQ doses of up to 240 mg (6.0 mg/kg) in adults are not associated with significant gastrointestinal side effects as long as the drug is taken with food (32, 37). In addition, in the study of Caucasians who took 0.5 mg/kg twice daily for 7 days, the presence of peripheral cyanosis was not related to plasma PMQ or CPMQ concentrations (21).
Carboxyprimaquine, the principal metabolite of PMQ (38), has substantially less potent antimalarial and hemolytic activity than its parent compound (39, 40). Its relatively slow elimination means that it accumulates during multiple daily or twice-daily dosing. However, this should not have clinical consequences, even after a high-dose abbreviated regimen, since other PMQ metabolites are considered responsible for toxicity, and they appear to be minor and highly labile (41). In any case, the predicted median CPMQ Cmax after the last dose of the 1.0 mg/kg twice-daily regimen given over a 3.5-day period in the present study (3,477 μg/liter) were within the absolute range of equivalent median Cmax from healthy Vietnamese adults given 30 mg PMQ base daily for 14 days (42) and similar to those seen in Caucasians at the end of a week of 0.5 mg/kg twice daily (means ± SD, 3,824 ± 624 μg/liter) (21).
In the present study, the higher 1.0 mg/kg single dose was as well tolerated as the conventional 0.5 mg/kg dose. In particular, there was no between-group difference in hemoglobin concentrations over time. In a larger-scale Tanzanian study, single-dose PMQ was associated with a mild mean fall in hemoglobin of 5 g/liter in children who were G6PD replete (43), raising the possibility that longer courses promote the development of anemia even when G6PD deficiency has been excluded. In the Thai study of 0.5 mg/kg twice daily for 7 days in adults (22), there was a similar proportionate fall in hematocrit by day 3, with a plateau thereafter and recovery after cessation of drug, a pattern that was also seen in the conventional 14-day 0.5 mg/kg daily regimen. If this between-group similarity applies to PNG children, the 5 g/liter mean fall in hemoglobin observed with a conventional 14-day 0.5 mg/kg daily regimen (12) would be no greater with higher-dose abbreviated regimens, but this needs to be assessed in further studies.
Conventional 14-day primaquine therapy elevates methemoglobin levels to around 4% of total hemoglobin in healthy subjects, but levels of up to 20% are usually asymptomatic (32). There may be racial differences in the propensity to methemoglobinemia, since short-course high-dose PMQ caused peripheral cyanosis in 5% of Caucasians (21) but no Thai subjects were affected in two separate similar studies (22, 23). Although there appears to be no relationship between plasma PMQ or CPMQ concentrations and methemoglobinemia (21), which may reflect the activity of transient toxic metabolites (41), future studies of short-course, high-dose PMQ regimens should include serial methemoglobin monitoring to ensure that such regimens are safe.
Due to a lack of published data on the safety and tolerability of PMQ in very young children, the WHO recommends that PMQ not be given to children under the age of 4 years (12, 44). Since vivax malaria is a common infection in this age-group in countries where it is endemic, such as PNG, including severe cases and deaths (45), consideration should be given to including these children in future trials.
ACKNOWLEDGMENTS
We thank the children and their parents/guardians for their participation. We are also most grateful to Sister Valsi Kurian and the staff of Alexishafen Health Centre for their kind cooperation during the study. We are grateful to Wendy Davis for statistical advice.
The study was funded by the Australian Agency for International Development (AusAID) and the National Health and Medical Research Council (NHMRC) of Australia (project grant 634343). B.R.M. is supported by an NHMRC C. J. Martin Overseas Biomedical Fellowship and T.M.E.D. by an NHMRC Practitioner Fellowship.
We have no conflicts of interest to declare.
Footnotes
Published ahead of print 4 November 2013
REFERENCES
- 1.Bhatia SC, Saraph YS, Revankar SN, Doshi KJ, Bharucha ED, Desai ND, Vaidya AB, Subrahmanyam D, Gupta KC, Satoskar RS. 1986. Pharmacokinetics of primaquine in patients with P. vivax malaria. Eur. J. Clin. Pharmacol. 31:205–210. 10.1007/BF00606660 [DOI] [PubMed] [Google Scholar]
- 2.Baird JK, Hoffman S. 2004. Primaquine therapy for malaria. Clin. Infect. Dis. 39:1336–1345. 10.1086/424663 [DOI] [PubMed] [Google Scholar]
- 3.Kim Y-R, Kuh H-J, Kim M-Y, Kim Y-S, Chung W-C, Kim S-I, Kang M-W. 2004. Pharmacokinetics of primaquine and carboxyprimaquine in Korean patients with vivax malaria. Arch. Pharm. Res. 27:576–580. 10.1007/BF02980134 [DOI] [PubMed] [Google Scholar]
- 4.Fernando D, Rodrigo C, Rajapakse S. 2011. Primaquine in vivax malaria: an update and review on management issues. Malar. J. 10:351. 10.1186/1475-2875-10-351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mueller I, Galinski MR, Baird JK, Carlton JM, Kochar DK, Alonso PL, del Portillo HA. 2009. Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. Lancet Infect. Dis. 9:555–566. 10.1016/S1473-3099(09)70177-X [DOI] [PubMed] [Google Scholar]
- 6.Hill DR, Baird JK, Parise ME, Lewis LS, Ryan ET, Magill AJ. 2006. Primaquine: report from CDC expert meeting on malaria chemoprophylaxis I. Am. J. Trop. Med. Hyg. 75:402–415 [PubMed] [Google Scholar]
- 7.Kazura JW, Siba PM, Betuela I, Mueller I. 2012. Research challenges and gaps in malaria knowledge in Papua New Guinea. Acta Trop. 121:274–280. 10.1016/j.actatropica.2011.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Müller I, Bockarie M, Alpers M, Smith T. 2003. The epidemiology of malaria in Papua New Guinea. Trends Parasitol. 19:253–259. 10.1016/S1471-4922(03)00091-6 [DOI] [PubMed] [Google Scholar]
- 9.Poespoprodjo JR, Fobia W, Kenangalem E, Lampah DA, Hasanuddin A, Warikar N, Sugiarto P, Tjitra E, Anstey NM, Price RN. 2009. Vivax malaria: a major cause of morbidity in early infancy. Clin. Infect. Dis. 48:1704–1712. 10.1086/599041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mendis K, Sina BJ, Marchesini P, Carter R. 2001. The neglected burden of Plasmodium vivax malaria. Am. J. Trop. Med. Hyg. 64:97–106 [DOI] [PubMed] [Google Scholar]
- 11.Barnadas C, Koepfli C, Karunajeewa HA, Siba PM, Davis TME, Mueller I. 2011. Characterization of treatment failure in efficacy trials of drugs against Plasmodium vivax by genotyping neutral and drug resistant-associated markers. Antimicrob. Agents Chemother. 55:4479–4481. 10.1128/AAC.01552-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Betuela I, Bassat Q, Kiniboro B, Robinson LJ, Rosanas-Urgell A, Stanisic D, Siba PM. 2012. Tolerability and safety of primaquine in Papua New Guinean children 1 to 10 years of age. Antimicrob. Agents Chemother. 56:2146–2149. 10.1128/AAC.05566-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartelink IH, Rademaker CM, Schobben AF, van den Anker JN. 2006. Guidelines on paediatric dosing on the basis of developmental physiology and pharmacokinetic considerations. Clin. Pharmacokinet. 45:1077–1097. 10.2165/00003088-200645110-00003 [DOI] [PubMed] [Google Scholar]
- 14.Tod M, Jullien V, Pons G. 2008. Facilitation of drug evaluation in children by population methods and modelling. Clin. Pharmacokinet. 47:231–243. 10.2165/00003088-200847040-00002 [DOI] [PubMed] [Google Scholar]
- 15.John GK, Douglas NM, Von Seidlein L, Nosten F, Baird JK, White NJ, Price RN. 2012. Primaquine radical cure of Plasmodium vivax: a critical review of literature. Malar. J. 11:280. 10.1186/1475-2875-11-280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manning L, Laman M, Townsend M-A, Chubb SP, Siba PM, Mueller I, Davis TME. 2011. Reference intervals for common laboratory tests in Melanesian children. Am. J. Trop. Med. Hyg. 85:50–54. 10.4269/ajtmh.2011.11-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karunajeewa HA, Mueller I, Senn M, Lin E, Law I, Gomorrai PS, Oa O, Griffin S, Kotab K, Suano P, Tarongka N, Ura A, Lautu D, Page-Sharp M, Wong R, Salman S, Siba P, Ilett KF, Davis TME. 2008. A trial of combination antimalarial therapies in children from Papua New Guinea. N. Engl. J. Med. 359:2545–2557. 10.1056/NEJMoa0804915 [DOI] [PubMed] [Google Scholar]
- 18.Page-Sharp M, Ilett KF, Betuela I, Davis TME, Batty KT. 2012. Simultaneous determination of primaquine and carboxyprimaquine in plasma using solid phase extraction and LC-MS assay. J. Chromatogr. B 902:142–146. 10.1016/j.jchromb.2012.06.019 [DOI] [PubMed] [Google Scholar]
- 19.Anderson BJ, Holford NH. 2009. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab. Pharmacokinet. 24:25–36. 10.2133/dmpk.24.25 [DOI] [PubMed] [Google Scholar]
- 20.World Health Organization 2009. WHO child growth standards and the identification of severe acute malnutrition in infants and children. World Health Organization, Geneva, Switzerland: [PubMed] [Google Scholar]
- 21.Ebringer A, Heathcote G, Baker J, Waller M, Shanks GD, Edstein MD. 2011. Evaluation of the safety and tolerability of a short higher-dose primaquine regimen for presumptive anti-relapse therapy in healthy subjects. Trans. R. Soc. Trop. Med. Hyg. 105:568–573. 10.1016/j.trstmh.2011.07.001 [DOI] [PubMed] [Google Scholar]
- 22.Pukrittayakamee S, Imwong M, Chotivanich K, Singhasivanon P, Day NP, White NJ. 2010. A comparison of two short-course primaquine regimens for the treatment and radical cure of Plasmodium vivax malaria in Thailand. Am. J. Trop. Med. Hyg. 82:542–547. 10.4269/ajtmh.2010.09-0428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krudsood S, Tangpukdee N, Wilairatana P, Phophak N, Baird JK, Brittenham GM, Looareesuwan S. 2008. High-dose primaquine regimens against relapse of Plasmodium vivax malaria. Am. J. Trop. Med. Hyg. 78:736–740 [PMC free article] [PubMed] [Google Scholar]
- 24.Cooper WC, Myatt AV, Hernandez T, Jeffery GM, Coatney GR. 1953. Studies in human malaria. XXXI. Comparison of primaquine, isopentaquine, SN-3883, and pamaquine as curative agents against Chesson strain vivax malaria. Am. J. Trop. Med. Hyg. 2:949–957 [PubMed] [Google Scholar]
- 25.Kim S, Nguon C, Guillard B, Duong S, Chy S, Sum S, Nhem S, Bouchier C, Tichit M, Christophel E, Taylor WR, Baird JK, Menard D. 2011. Performance of the CareStart G6PD deficiency screening test, a point-of-care diagnostic for primaquine therapy screening. PLoS One 6:e28357. 10.1371/journal.pone.0028357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tinley KE, Loughlin AM, Jepson A, Barnett ED. 2010. Evaluation of a rapid qualitative enzyme chromatographic test for glucose-6-phosphate dehydrogenase deficiency. Am. J. Trop. Med. Hyg. 82:210–214. 10.4269/ajtmh.2010.09-0416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leslie T, Rab MA, Ahmadzai H, Durrani N, Fayaz M, Kolaczinski J, Rowland M. 2004. Compliance with 14-day primaquine therapy for radical cure of vivax malaria–a randomized placebo-controlled trial comparing unsupervised with supervised treatment. Trans. R. Soc. Trop. Med. Hyg. 98:168–173. 10.1016/S0035-9203(03)00041-5 [DOI] [PubMed] [Google Scholar]
- 28.Pereira EA, Ishikawa EA, Fontes CJ. 2011. Adherence to Plasmodium vivax malaria treatment in the Brazilian Amazon region. Malar. J. 10:355. 10.1186/1475-2875-10-355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clyde DF, McCarthy VC. 1977. Radical cure of Chesson strain vivax malaria in man by 7, not 14, days of treatment with primaquine. Am. J. Trop. Med. Hyg. 26:562–563 [DOI] [PubMed] [Google Scholar]
- 30.Schmidt LH, Fradkin R, Vaughan D, Rasco J. 1977. Radical cure of infections with Plasmodium cynomolgi: a function of total 8-aminoquinoline dose. Am. J. Trop. Med. Hyg. 26:1116–1128 [DOI] [PubMed] [Google Scholar]
- 31.Clyde DF. 1981. Clinical problems associated with the use of primaquine as a tissue schizonticidal and gametocytocidal drug. Bull. World Health Organ. 59:391–395 [PMC free article] [PubMed] [Google Scholar]
- 32.Baird JK, Fryauff DJ, Hoffman SL. 2003. Primaquine for prevention of malaria in travelers. Clin. Infect. Dis. 37:1659–1667. 10.1086/379714 [DOI] [PubMed] [Google Scholar]
- 33.Ward SA, Mihaly GW, Edwards G, Looareesuwan S, Phillips RE, Chanthavanich P, Warrell DA, Orme ML, Breckenridge AM. 1985. Pharmacokinetics of primaquine in man. II. Comparison of acute vs chronic dosage in Thai subjects. Br. J. Clin. Pharmacol. 19:751–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edstein MD. 2010. Primaquine, p 2049–2058 In Grayson ML. (ed), Kucers' the use of antibiotics, 6th ed, vol 2 Hodder Arnold/ASM Press, Washington, DC [Google Scholar]
- 35.Mihaly GW, Ward SA, Edwards G, Nicholl DD, Orme ML, Breckenridge AM. 1985. Pharmacokinetics of primaquine in man. I. Studies of the absolute bioavailability and effects of dose size. Br. J. Clin. Pharmacol. 19:745–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edwards G, McGrath CS, Ward SA, Supanaranond W, Pukrittayakamee S, Davis TM, White NJ. 1993. Interactions among primaquine, malaria infection and other antimalarials in Thai subjects. Br. J. Clin. Pharmacol. 35:193–198. 10.1111/j.1365-2125.1993.tb05685.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clayman CB, Arnold J, Hockwald RS, Yount EH, Jr, Edgcomb JH, Alving AS. 1952. Toxicity of primaquine in Caucasians. JAMA 149:1563–1568. 10.1001/jama.1952.72930340022010b [DOI] [PubMed] [Google Scholar]
- 38.Mihaly GW, Ward SA, Edwards G, Orme ML, Breckenridge AM. 1984. Pharmacokinetics of primaquine in man: identification of the carboxylic acid derivative as a major plasma metabolite. Br. J. Clin. Pharmacol. 17:441–446. 10.1111/j.1365-2125.1984.tb02369.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bates MD, Meshnick SR, Sigler CI, Leland P, Hollingdale MR. 1990. In vitro effects of primaquine and primaquine metabolites on exoerythrocytic stages of Plasmodium berghei. Am. J. Trop. Med. Hyg. 42:532–537 [DOI] [PubMed] [Google Scholar]
- 40.Link CM, Theoharides AD, Anders JC, Chung H, Canfield CJ. 1985. Structure-activity relationships of putative primaquine metabolites causing methemoglobin formation in canine hemolysates. Toxicol. Appl. Pharmacol. 81:192–202. 10.1016/0041-008X(85)90155-3 [DOI] [PubMed] [Google Scholar]
- 41.Nanayakkara NP, Ager AL, Jr, Bartlett MS, Yardley V, Croft SL, Khan IA, McChesney JD, Walker LA. 2008. Antiparasitic activities and toxicities of individual enantiomers of the 8-aminoquinoline 8-[(4-amino-1-methylbutyl)amino]-6-methoxy-4-methyl-5-[3,4-dichlorophenoxy]quinoline succinate. Antimicrob. Agents Chemother. 52:2130–2137. 10.1128/AAC.00645-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Binh VQ, Chinh NT, Thanh NX, Cuong BT, Quang NN, Dai B, Travers T, Edstein MD. 2009. Sex affects the steady-state pharmacokinetics of primaquine but not doxycycline in healthy subjects. Am. J. Trop. Med. Hyg. 81:747–753. 10.4269/ajtmh.2009.09-0214 [DOI] [PubMed] [Google Scholar]
- 43.Shekalaghe SA, ter Braak R, Daou M, Kavishe R, van den Bijllaardt W, van den Bosch S, Koenderink JB, Luty AJ, Whitty CJ, Drakeley C, Sauerwein RW, Bousema T. 2010. In Tanzania, hemolysis after a single dose of primaquine coadministered with an artemisinin is not restricted to glucose-6-phosphate dehydrogenase-deficient (G6PD A−) individuals. Antimicrob. Agents Chemother. 54:1762–1768. 10.1128/AAC.01135-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.World Health Organization 2010. Guidelines for the treatment of malaria, 2nd ed. World Health Organization, Geneva, Switzerland [Google Scholar]
- 45.Manning L, Laman M, Law I, Bona C, Aipit S, Teine D, Warrell J, Rosanas-Urgell A, Lin E, Kiniboro B, Vince J, Hwaiwhanje I, Karunajeewa H, Michon P, Siba P, Mueller I, Davis TM. 2011. Features and prognosis of severe malaria caused by Plasmodium falciparum, Plasmodium vivax and mixed Plasmodium species in Papua New Guinean children. PLoS One 6:e29203. 10.1371/journal.pone.0029203 [DOI] [PMC free article] [PubMed] [Google Scholar]