

Abstract
The availability of adequate treatments for poxvirus infections would be valuable not only for human use but also for veterinary use. In the search for novel antiviral agents, a 1′-methyl-substituted 4′-thiothymidine nucleoside, designated KAY-2-41, emerged as an efficient inhibitor of poxviruses. In vitro, KAY-2-41 was active in the micromolar range against orthopoxviruses (OPVs) and against the parapoxvirus orf. The compound preserved its antiviral potency against OPVs resistant to the reference molecule cidofovir. KAY-2-41 had no noticeable toxicity on confluent monolayers, but a cytostatic effect was seen on growing cells. Genotyping of vaccinia virus (VACV), cowpox virus, and camelpox virus selected for resistance to KAY-2-41 revealed a nucleotide deletion(s) close to the ATP binding site or a nucleotide substitution close to the substrate binding site in the viral thymidine kinase (TK; J2R) gene. These mutations resulted in low levels of resistance to KAY-2-41 ranging from 2.7- to 6.0-fold and cross-resistance to 5-bromo-2′-deoxyuridine (5-BrdU) but not to cidofovir. The antiviral effect of KAY-2-41 relied, at least in part, on activation (phosphorylation) by the viral TK, as shown through enzymatic assays. The compound protected animals from disease and mortality after a lethal challenge with VACV, reduced viral loads in the serum, and abolished virus replication in tissues. In conclusion, KAY-2-41 is a promising nucleoside analogue for the treatment of poxvirus-induced diseases. Our findings warrant the evaluation of additional 1′-carbon-substituted 4′-thiothymidine derivatives as broad-spectrum antiviral agents, since this molecule also showed antiviral potency against herpes simplex virus 1 in earlier studies.
INTRODUCTION
Poxviruses are large double-stranded DNA viruses. Over the few last years, zoonotic orthopoxvirus (OPV) outbreaks have been widely reported, and they involved cowpox virus (CPXV), vaccinia virus (VACV), buffalopox virus, and monkeypox virus in, respectively, European countries (1), Brazil (2), India (3), and the Democratic Republic of the Congo (4). Also, orf disease, caused by the parapoxvirus orf, is a zoonosis that can be transmitted to humans by contact with infected ruminants (5, 6). Poxviruses also include the virus molluscum contagiosum, an obligate human pathogen which generally causes benign infections (7, 8). With the exception of monkeypox, which leads to generalized clinical symptoms with a life-threatening outcome, cowpox, vaccinia, orf, and molluscum contagiosum diseases are usually localized and self-limited in immunocompetent individuals. Clinical presentation can, however, be more severe when the immune status of the patient is impaired (9–12).
The access to antiviral therapies may be critical to manage such infections. There is so far no FDA- or EMA-approved drug for the treatment of poxvirus-induced diseases. Three compounds are, however, promising and have received investigational new drug status (IND) for emergency use. Among them, cidofovir {(S)-1-[3-hydroxy-2-(phosphonomethoxy)propyl]cytosine [(S)-HPMPC]; Vistide} and its prodrug, CMX001 [hexadecyloxypropyl-(S)-HPMPC (HDP-cidofovir)], are acyclic nucleoside phosphonates (ANPs). They are broad-spectrum antiviral molecules inhibiting poxviruses at the level of viral DNA replication by interacting with the viral DNA polymerase (E9L) (13, 14). Various studies have demonstrated their potency in inhibiting virus replication and resolving OPV-associated diseases (13–16). Cidofovir requires intravenous administration and can be nephrotoxic (13), whereas CMX001, which may be taken orally, displays some gastrointestinal toxicity (17, 18). Other ANPs containing a 2,4-diaminopyrimidine base moiety {(R)-9-[3-hydroxy-2-(phosphonomethoxy)propoxy]-2,4-diaminopyrimidine [(R)-HPMPO-DAPy]} or a 5-azacytosine base moiety {1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]-5-azacytosine [(S)-HPMP-5-azaC]} appear to be promising for further antiviral development on the basis of their anti-OPV activities in infected cells and in animal models of poxvirus infections (19, 20). 4-Trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl)-benzamide (ST-246) is an OPV-specific inhibitor with a distinct mode of action that targets the viral protein F13L and, as a consequence, inhibits the egress of the virus from cells (21, 22). Orally active, this compound has undergone phase 2 clinical development to assess safety, tolerability, and pharmacokinetics (23).
Nucleoside analogues are also extensively studied as therapeutic agents against the proliferation of cancer cells, against virus replication, and, recently, also against bacterial infections (24). Among them, the 2′-deoxy-4′-thiopyrimidine nucleosides have been reported to be inhibitors of some herpesviruses, and they are listed in Table 1 (25, 26). In particular, 4′-thiothymidine was active against herpes simplex virus 1 (HSV-1) and human cytomegalovirus (HCMV) (25), but its inhibitory activity against VACV was masked by its cytostatic effect (27). In 2003, a series of 5-substituted 2′-deoxy-4′-thiopyrimidine nucleosides was also shown to retain good antiviral activity against some herpesviruses, with the 2′-deoxy-5-ethyl-4′-thiouridine derivative being the most potent against alphaherpesviruses (28). The antiviral efficacy of 5-substituted 4′-thiopyrimidine against alphaherpesviruses and OPVs was reported in two studies (27, 29). The compound 5-iodo-4′-thio-2′-deoxyuridine (4′-thioIDU) inhibited both VACV and CPXV replication at concentrations in the nanomolar range (27).
TABLE 1.
Overview of 2′-deoxy-4′-thiopyrimidine compounds synthesized since 1991 and evaluated as potential antiviral agents
| Compound family | Compound | Antiviral activity (EC50 [μM])a | IC50 (μM)b | CC50 (μM)b,c | Reference(s) |
|---|---|---|---|---|---|
| 2′-Deoxy-4′-thiopyrimidine | 4′-Thiothymidine | Activity against HSV-1 (0.8) and HCMV (21) | ≥32 (MRC5 cells) | 2.0 (MRC5 cells) | 25 |
| 2′-Deoxy-4′-thiocytidine | No activity against HSV-1 or HCMV | 1.2 (L1210 cells) | |||
| 2′-Deoxy-4′-thiouridine | No activity against HSV-1 or HCMV | 2.7 (L1210 cells) | |||
| 2′-Deoxy-4′-thionucleoside | 4′-Thiothymidine | Activity against alphaherpesviruses (HSV-1, 0.37; HSV-2, 2.3; VZV, 10) and HCMV (0.98) | 7.1 (Vero cells) | 26 | |
| 3′-Azido-4′-thiothymidine | No activity against alphaherpesviruses (>100) | >100 (Vero cells) | |||
| (E)-5-(2-Bromovinyl)-4′-thio-2′-deoxyuridine (4′-S-BVDU) | Activity against alphaherpesviruses (HSV-1, 0.6; HSV-2, 10; VZV, 0.08) | >500 (Vero cells) | |||
| 5-Substituted-2′-deoxy-4′-thiopyrimidine | 2′-Deoxy-5-ethyl-4′-thiouridine | Activity against alphaherpesviruses (HSV-1, 0.33; HSV-2, 3.5; VZV, 0.99); no activity against HCMV (138) | >500 (Vero cells) | 28 | |
| Other 5-substituted derivatives, including 5-propyl, 5-isopropyl, 5-cyclopropyl, and 5-(2-chloroethyl) derivatives and the molecule 4′-thio-5-vinyluridine | Active against HSV-1 (range, 0.15 to 6.8) and VZV (range, 0.3 to 4.1) | Range, >100 to >500 (Vero cells) | |||
| 1′-Carbon-substituted 2′-deoxy-4′-thiothymidine | 4′-Thiothymidine | Activity against HSV-1 (0.031) | 34 (HEL cells) | 34 | |
| 1-(2-Deoxy-1-methyl-4-thio-β-d-ribofuranosyl)thymine (KAY-2-41) | Activity against HSV-1 (14.7); no activity against HIV-1 (>34.1) | 319 (HEL cells) | |||
| Other derivatives of 1′-carbon-substituted 4′-thiothymidine | No activity against HSV-1 (>74) or HIV-1 (>95.1) | >367 (HEL cells) | |||
| 5-Substituted-2′-deoxy-4′-thiopyrimidine | 1-(2-Deoxy-4-thio-beta-d-ribofuranosyl)-5-iodouracil (4′-thioIDU) | Activity against VACV-Cop (0.5), CPXV-BR (0.1), alphaherpesviruses (HSV-1, 0.08; HSV-2, 0.45; VZV, 2), and HCMV (5.9); no activity against HHV-6 (>100), EBV (>0.16), or HHV-8 (>4) | >100 (HFF cells) | 3.0 (HFF cells) | 27, 29 |
| 4′-Thiothymidine | Activity against VACV-Cop (0.03) and CPXV-BR (0.02) | >100 (HFF cells) | <0.03 (HFF cells) | ||
| Other derivatives of 5-substituted-2′-deoxy-4′-thiopyrimidine | Activity against HSV-2 (range, 0.3 to 15), VACV-Cop (range, 0.03 to 0.9), and CPXV-BR (range, 0.02 to 1.6) | >88 (HFF cells) | Ranging from <0.03 to 26 (HFF cells) |
HSV-1 and HSV-2, herpes simplex virus 1 and 2, respectively; VZV, varicella-zoster virus; HCMV, human cytomegalovirus; HHV-6, human herpesvirus 6; EBV, Epstein-Barr virus; HHV-8, human herpesvirus 8; HIV-1, human immunodeficiency virus type 1; VACV-Cop, vaccinia virus strain Copenhagen; CPXV-BR, cowpox virus strain Brighton.
MRC-5, human lung fibroblasts; L1210, skin lymphoblast mouse cells; Vero cells, African green monkey kidney cells; HEL, human embryonic lung fibroblasts; HFF, human foreskin fibroblasts.
Measured by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide assay as the inhibitory dose required to reduce the viability of the cells by 50%.
The inhibitory activity of such molecules may rely on an initial activation step by a nucleoside kinase, such as thymidine kinase (TK), which can be of cellular or viral origin. Herpesviruses encode a type I TK, active as a homodimer, whereas OPVs encode a type II TK (the J2R gene in VACV strain Copenhagen [VACV-Cop]) that is (i) active as a homotetramer and (ii) homologous to the cellular cytosolic TK (human cytosolic thymidine kinase [hTK1]). These enzymes differ significantly in their substrate specificities, ranging from broad for type I TK to narrow for type II TK, with only thymidine, 2′-deoxyuridine, and closely related analogues being preferentially phosphorylated (30, 31). OPVs also encode a thymidylate kinase (TMPK; A48R gene) preferentially involved in the phosphorylation of the monophosphate form of thymidine to its diphosphate form (32, 33).
Therapeutic agents with novel modes of action, potent and broad antiviral activity, and good pharmacokinetic properties are still needed to enlarge the pipeline of antipoxvirus agents and to mitigate the hazard of drug resistance. In 2004, a series of 1′-carbon-substituted 4′-thiothymidines was synthesized (34), and some molecules exhibiting inhibitory activity against HSV-1 replication (Table 1) were also evaluated in preliminary assays for their activity against poxviruses (our unpublished data). One particularly active compound, 1-(2-deoxy-1-methyl-4-thio-β-d-ribofuranosyl)thymine (KAY-2-41) (Fig. 1), has now been further studied, and we describe herein the antiviral efficacy of this 4′-thiothymidine derivative against poxvirus replication in cell culture and the benefit of KAY-2-41 treatment in a VACV lethal mouse model. The antiviral activity of KAY-2-41 against ANP-resistant OPVs and the genotypic characterization of three OPVs selected for resistance to KAY-2-41 enabled us to investigate its mode of action.
FIG 1.
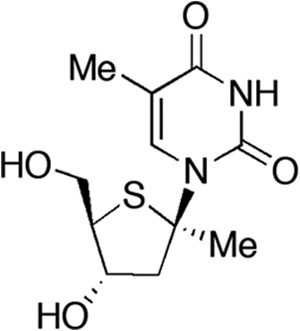
Structure of KAY-2-41 [1-(2-deoxy-1-methyl-4-thio-β-d-ribofuranosyl)thymine]. Me, methyl.
MATERIALS AND METHODS
Cells.
Human embryonic lung (HEL) fibroblast cells were grown in Earle's minimum essential medium (Earle's MEM; Life Technologies, Merelbeke, Belgium) containing 5% fetal calf serum (FCS) and supplemented as described previously (35). Cytosolic thymidine kinase-negative human osteosarcoma cells (H143B, ATCC CRL-8303) were grown in Dulbecco's modified Eagle medium (DMEM; Life Technologies) supplemented with 10% FCS. For virus infection, the percentage of FCS for all media was reduced to 2%.
Viruses.
The following virus strains were used: VACV strain Western Reserve (VACV-WR); VACV-Cop; VACV strain Lister (VACV-Lis); two VACV strains with a deletion of the J2R gene encoding thymidine kinase (VACV-Cop-ΔTK and VACV-WR-ΔTK); CPXV strain Brighton (CPXV-BR); various CPXV clinical isolates, including CPXV-AUS1999-867, CPXV-FIN2000-MAN, CPXV-GER1980-EP4, and CPXV-GER1991-3 (for the origins of the clinical isolates, see reference 36), kindly provided by H. Meyer (Bundeswehr Institute of Microbiology, Munich, Germany); camelpox virus (CMLV) strain Iran (CML1) and CMLV strain Dubai (CML14), kindly provided by H. Meyer (37, 38); the parapoxvirus orf strain NZ2 (ORFV-NZ2); drug-resistant VACV-WR, CML1, and CML14 harboring single or double amino acid substitutions within the E9L gene that result in resistance to cidofovir and/or (S)-1-(3-hydroxy-2-phosphonomethoxypropyl)-2,6-diaminopurine [(S)-HPMPDAP] (35); and VACV-WR with deletions of both ribonucleotide reductase subunits (the I4L large subunit and the F4L small subunit) (39).
Compounds.
The sources of the compounds were as follow: KAY-2-41 was synthesized and characterized at Showa University, Tokyo, Japan, as described in reference 34; cidofovir [(S)-HPMPC] was obtained from Gilead Sciences (Foster City, CA); CMX001 (HDP-cidofovir), (S)-HPMP-5-azaC, and (S)-HPMPDAP were synthesized by M. Krečmerová (Institute of Organic Chemistry and Biochemistry, Prague, Czech Republic); (E)-5-(2-bromovinyl)-2′-deoxyuridine (BVDU) was synthesized by P. Herdewijn (Rega Institute, Leuven, Belgium); 5-bromo-2′-deoxyuridine (5-BrdU) and 3′-azido-3′-deoxythymidine (AZT) were purchased from Sigma-Aldrich (Schnelldorf, Germany); and ST-246 was kindly provided by D. E. Hruby from SIGA Inc. (Corvallis, OR).
Antiviral and cytostatic assays.
Antiviral and cytostatic assays were performed as previously described (35). Briefly, confluent monolayers of HEL cells (in 96-well plates) were infected at a multiplicity of infection (MOI) of 0.01 PFU/cell for 2 h. Residual virus was removed and replaced with medium containing serial dilutions of the test compounds (in duplicate). After 2 to 3 days for VACV and CPXV or 4 to 5 days for CMLV, the viral cytopathic effect (CPE) was recorded under microscopic examination after ethanol fixation and Giemsa staining of 96-well plates. Scores were attributed on a scale of from 0 to 5, with a score of 0 corresponding to no CPE and a score of 5 corresponding to a 100% CPE. The 50% effective concentration (EC50) was defined as the concentration of a compound required to reduce the viral CPE by 50%. Cytostatic concentrations were determined as previously reported (40). HEL cells were seeded at a density of 3.5 ×103 cells/well (in 96-well plates), and serial dilutions of test compounds were added after 1 day. Three days later, cells were washed, trypsinized, and counted with a Beckman Coulter Counter (Analis, Suarlée, Belgium), and the concentration required to inhibit cell growth by 50% (the 50% cytostatic concentration [CC50]) was calculated. The minimum cytotoxic concentration (MCC) was also recorded on confluent cell monolayers and was defined as the minimum cytotoxic concentration required to alter the cell morphology.
Selection, isolation, and genotyping of viruses resistant to KAY-2-41.
The VACV-WR, CPXV-BR, and CML1 strains were repeatedly passaged in HEL cells for, respectively, 30, 21, and 44 rounds in the presence of increasing concentrations of KAY-2-41, starting at 0.2 μM. Viruses that replicated in the presence of a final concentration of 20 μM KAY-2-41 were cultured twice more in drug-free medium. Antiviral assays were performed to evaluate the resistance phenotypes of these viruses. Three clones per virus strain were isolated by plaque purification and selected for further genotyping and phenotyping. All genes cited have been named following the nomenclature for VACV-Cop genes. Sequencing of A48R (TMPK), E9L (viral DNA polymerase), D4R (uracil DNA glycosylase), B1R (Ser/Thr kinase), F10L (Ser/Thr kinase), and J2R (TK) of wild-type (WT) and drug-resistant clones was performed as follows: DNA was extracted from virus-infected HEL cells using a QIAamp DNA blood minikit (Qiagen Benelux B.V, Venlo, Netherlands) following the manufacturer's instructions. The genes A48R, D4R, B1R, F10L, and J2R were PCR amplified as one amplicon, and the full-length E9L gene was amplified as five (CMLV) or four (VACV and CPXV) overlapping amplicons using FastStart high-fidelity DNA polymerase (Roche Applied Science, Mannheim, Germany) (35). Sequencing reactions and data assembling were performed as previously reported (35).
Thymidine kinase assays.
The affinity of KAY-2-41 to TKs of various origins was obtained through the measurement of the activity of purified enzymes, including hTK1, human mitochondrial thymidine kinase (hTK2), and VACV-WR TK (J2R). The cytotoxic concentrations, or the 50% inhibitory concentrations (IC50s), were calculated as the concentrations of KAY-2-41 required to inhibit 50% of the phosphorylation of the natural radiolabeled substrate [methyl-3H]deoxythymidine ([methyl-3H]dThd). The assays were performed in a 50-μl reaction mixture containing 50 mM Tris-HCl, pH 8.0, 2.5 mM MgCl2, 10 mM dithiothreitol, 10 mM sodium fluoride, 1 mg/ml bovine serum albumin, 2.5 mM ATP, 1 μM [methyl-3H]dThd, and enzyme. The samples were incubated at 37°C for 30 min in the presence or absence of different concentrations (5-fold dilutions) of KAY-2-41. Aliquots of 45 μl of the reaction mixtures were spotted on Whatman DE-81 filter paper disks. The filters were washed three times for 5 min each time in 1 mM ammonium formate, once for 1 min in water, and once for 5 min in ethanol. The radioactivity was determined by scintillation counting.
In vivo experiments.
Animal work was approved by the KU Leuven Ethics Committee for Animal Care and Use (permit number P044-2010). Infections were performed while the mice were under anesthesia using ketamine (100 mg/kg)-xylazine (10 mg/kg) in saline, and euthanasia was done by administration of pentobarbital sodium. female NMRI mice [Rj:NMRI(Han); Elevage-Janvier, Le-Genest-St-Isle, France] 5 weeks old were used. All animal experimentations were completed in biosafety level 2 facilities. Groups were defined as uninfected (mock infected) or as VACV-WR infected. After anesthesia, mice were inoculated intranasally (i.n.) with 10 μl of phosphate-buffered saline (PBS) (uninfected) or with 10 μl of PBS containing 4,000 PFU of VACV-WR (5 μl per nostril). Treatment was given intraperitoneally (i.p.) once daily for 5 consecutive days, beginning 6 h after infection, at a concentration of 5 or 50 mg/kg of body weight of KAY-2-41 dissolved in PBS. Cohorts were then monitored for body weight, morbidity, and mortality. The reported mortality rates included both actual mortality (approximately 40% of the infected animals) and mortality due to euthanasia of animals that lost more than 30% of their body weight. To determine the extent of viral replication, five mice in the VACV-WR-infected groups that received no treatment or that received KAY-2-41 at 50 mg/kg were euthanized at day 5 postinfection (p.i.), and serum as well as lungs, liver, spleen, kidneys, and mesenteric lymph nodes were collected and processed as previously described (41). Briefly, after tissue disruption and homogenization, samples were used for virus titer and viral load determinations. The virus titers in lung, liver, spleen, and kidney tissue homogenates were determined on HEL cells. Real-time quantitative PCR (qPCR) was used to quantify viral DNA, as previously reported (41). Organs from one mouse of each group were used for histological examination (41).
Statistical analyses.
All statistical analyses were done with GraphPad Prism (version 6) software (GraphPad Software Inc., La Jolla, CA). Unpaired t tests were used to compare mean EC50s, obtained from at least three independent experiments, between WT and drug-resistant viruses. The Mann-Whitney test, two-tailed, was used to compare the ranks of the viral DNA loads or virus titers of KAY-2-41-treated mice to those of the PBS-treated group. Statistical significance was defined for the above-described tests as follows: P < 0.001, extremely significant; P < 0.01, very significant; P < 0.05, significant; and P > 0.05, not significant (NS).
RESULTS
Antiviral activity of KAY-2-41 against poxviruses.
Several 1′-carbon-substituted 4′-thiothymidines were synthesized, and due to its anti-HSV-1 activity (34), KAY-2-41 was further evaluated for its activity against poxviruses. In HEL cells, KAY-2-41 emerged as a potent inhibitor of OPVs and displayed EC50s of 0.48 ± 0.33 μM against VACV-Cop, 0.48 ± 0.15 μM against CPXV-BR, and 0.79 ± 0.40 μM against CML1 (Table 2). Similar KAY-2-41 EC50s were also observed with two other VACVs (VACV-Lis and VACV-WR). The compound appeared to be 10- to 15-fold more active than two nucleotide phosphonate analogues, i.e., cidofovir and (S)-HPMP-5-azaC, which had mean EC50s of 9.8 μM and 9.6 μM, respectively, against the different OPVs. The lipid prodrug derivative of cidofovir, CMX001, was 395-fold (VACV-Lis) to 1,380-fold (VACV-Cop) more active than its parent counterpart, cidofovir, and 18-fold (VACV-Lis) to 96-fold (VACV-Cop) more active than KAY-2-41. Also, the KAY-2-41 EC50 against CPXV-BR was in the range of that of ST-246, yet ST-246 was 32- to 80-fold more potent against the other OPV strains.
TABLE 2.
Cytostatic concentrations, antiviral activities, and selectivity indices of compounds against poxviruses
| Compound | CC50 (μM)a | VACV-Cop |
VACV-Lis |
VACV-WR |
CPXV-BR |
CML1 |
ORF-NZ2 |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (μM)b | SIc | EC50 (μM) | SI | EC50 (μM) | SI | EC50 (μM) | SI | EC50 (μM) | SI | EC50 (μM) | SI | ||
| KAY-2-41 | 14 ± 10 | 0.48 ± 0.33 | 29 | 0.41 ± 0.33 | 34 | 0.80 ± 0.37 | 18 | 0.48 ± 0.15 | 29 | 0.79 ± 0.40 | 18 | 3.5 ± 0.7 | 4 |
| Cidofovir | 397 ± 255 | 6.9 ± 3.0 | 57 | 9.1 ± 6.6 | 44 | 8.2 ± 2.6 | 48 | 13.9 ± 8.3 | 29 | 11.2 ± 4.5 | 35 | 0.8 ± 0.1 | 496 |
| (S)-HPMP-5-azaC | 125 ± 79 | 5.5 ± 2.7 | 23 | 10.0 ± 6.3 | 12 | 7.5 ± 1.4 | 17 | 11.5 ± 4.6 | 11 | 13.3 ± 6.4 | 9 | 0.5 ± 0.1 | 250 |
| CMX001 | ≥2.4 ± 1.8 | 0.005 ± 0.002 | ≥467 | 0.023 ± 0.021 | ≥106 | 0.013 ± 0.011 | ≥180 | 0.021 ± 0.026 | ≥114 | 0.024 ± 0.022 | ≥100 | —d | — |
CC50, 50% cytostatic concentration, or the drug concentration required to reduce HEL cell growth by 50%. Data represent the means ± standard deviations of at least three independent experiments.
EC50, 50% effective concentration, or the concentration of compound required to reduce the viral cytopathic effect by 50%. Data are shown as the means ± standard deviations of at least four independent experiments.
SI, selectivity index, or the ratio CC50/EC50.
—, not done.
We also examined whether KAY-2-41 was active against nonlaboratory OPV strains and assessed various CPXV clinical isolates collected from human and animal cowpox cases (36). The molecule potently inhibited CPXV-GER1991-3 (mean EC50, 0.46 ± 0.21 μM), CPXV-GER1980-EP4 (mean EC50, 0.28 ± 0.06 μM), CPXV-FIN2000-MAN (mean EC50, 0.42 ± 0.007 μM), and CPXV-GER1999-867 (mean EC50, 0.58 ± 0.22 μM).
KAY-2-41 was not found to alter the cell morphology of stationary cells (MCC value > 100 μM). The molecule was, however, cytostatic for growing HEL cells, with a CC50 value of 14 ± 10 μM, which accounted for the relatively low selectivity indices (SIs; ratio of CC50/EC50) ranging from 18 to 29 (Table 2). Similarly, the CC50 value of CMX001 was ≥2.4 ± 1.8 μM, but because of its potency, SIs were ≥100. The molecules cidofovir, (S)-HPMP-5-azaC, and ST-246 were less toxic for growing cells than KAY-2-41. Both ANPs had SIs comparable to the SI of KAY-2-41, while ST-246 was the most selective molecule with an SI of ≥1,696.
We could further demonstrate that KAY-2-41 had a 4-fold weaker activity against the parapoxvirus ORF-NZ2 than OPVs, with an EC50 of 3.5 ± 0.7 μM (Table 2). Both ANPs evaluated were potent inhibitors of ORF-NZ2, while ST-246 was not active, which is consistent with the specific anti-OPV activity of ST-246.
Antiviral activity of KAY-2-41 against cidofovir and (S)-HPMPDAP-resistant OPVs.
Being a thymidine analogue, the triphosphate form of KAY-2-41 was expected to interact ultimately with the viral DNA polymerase E9L in VACV-Cop to block virus replication. We therefore wondered whether KAY-2-41 could inhibit OPVs bearing mutations within E9L that are known to confer resistance to certain ANPs. In previous studies, we identified such amino acid substitutions in the exonuclease domain of E9L (A314V), in the polymerase domain (T831I), or in both domains (A314V plus A684V) (35). Recombinant CML1, CML14, and VACV harboring these amino acid substitutions were used, and the two ANPs cidofovir and (S)-HPMPDAP were included as control drugs to confirm the resistance phenotype of each strain (Table 3). The presence of the A314V or the T831I substitution did not alter the sensitivity of VACV, CML1, and CML14 to KAY-2-41, while these amino acid substitutions conferred resistance to cidofovir and/or (S)-HPMPDAP. Interestingly, mutating both exonuclease and polymerase domains of E9L (A314V plus A684V) appeared to render CML14 and VACV even more sensitive to the effect of KAY-2-41, albeit the shifts in EC50s, compared to those for the WT viruses, were only 4.6-fold (0.10 μM versus 0.46 μM [P = 0.0164] for CML14) and 3.85-fold (0.2 μM versus 0.77 μM [P = 0.0173] for VACV) (Table 3). On the other hand, the A314V and A684V substitutions were responsible for high levels of resistance to cidofovir and (S)-HPMPDAP. In conclusion, KAY-2-41 was endowed with potent antiviral efficacy against cidofovir-resistant viruses, and the molecule appeared to have a slightly increased activity when the A314V and A684V mutations were present.
TABLE 3.
Antiviral activity of KAY-2-41 against cidofovir and (S)-HPMPDAP-resistant CMLV and VACV
| Compound | EC50 (μM)a |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| CML14 | CML14 A314Vb | CML14 A314V + A684Vb | CML1 | CML1 T831Ib | VACV-WR | VACV-WR A314Vb | VACV-WR A314V + A684Vb | VACV-WR T831Ib | |
| (S)-HPMPC | 8.6 ± 3.2 | 24.4 ± 12.4* | 49.4 ± 21.4* | 7.1 ± 3.0 | 9.6 ± 3.6 | 16.7 ± 9.3 | 64.7 ± 30.2** | 148.4 ± 23.0*** | 108.7 ± 42.1*** |
| (S)-HPMPDAP | 0.5 ± 0.1 | 14.6 ± 4.2*** | 17.4 ± 10.6* | 0.2 ± 0.1 | 4.1 ± 2.8** | 2.0 ± 1.1 | 17.3 ± 9.7*** | 53.0 ± 13.9*** | 31.3 ± 9.4*** |
| KAY-2-41 | 0.46 ± 0.22 | 0.22 ± 0.11 | 0.10 ± 0.01* | 0.29 ± 0.11 | 0.26 ± 0.13 | 0.77 ± 0.46 | 0.44 ± 0.24 | 0.20 ± 0.12* | 1.09 ± 0.35 |
EC50, 50% effective concentration, or the concentration of compound required to reduce the viral cytopathic effect by 50%. Data are shown as the means ± standard deviations of at least four independent experiments. Asterisks indicate where the EC50 differs significantly from that of the corresponding WT virus, as determined by an unpaired t test: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Drug-resistant recombinant viruses previously characterized (35) and bearing amino acid substitutions, i.e., A314V, A314V plus A684V, or T831I, in the viral DNA polymerase (E9L).
Selection, genotyping, and phenotyping of OPVs resistant to KAY-2-41 (KAY-2-41r).
To elucidate the mode of action of KAY-2-41, drug-resistant viruses were isolated following 21 (CPXV-BR), 30 (VACV-WR), and 44 (CML1) passages in the presence of increasing amounts of the drug. Three clones of each virus stock were plaque purified and used for further analysis.
Genotypic characterization revealed the presence of a nucleotide deletion(s) and one nucleotide substitution in the TK (J2R) gene of each of the KAY-2-41r clones, whereas the gene sequences of the TMPK (A48R), two serine/threonine kinases (B1R and F10L), the uracil DNA glycosylase (D4R), and the viral DNA polymerase (E9L) were comparable to those of the corresponding WT clones. Figure 2 depicts the mutations found in the viral TK. Resistant VACV-WR clones had a deletion of a thymine at position 43 (Tdel43) leading to a frameshift mutation from amino acid 15, changing the serine into a glutamine, until the appearance of a stop codon at position 21. CPXV-BR clones harbored a double deletion of a guanine and a thymine at positions 73 and 74 (GTdel73/74) that resulted in a stop codon at position 25. These frameshift mutations by deletions were all found in the ATP binding region, localized in the N-terminal extremity of the TK, and led to the production of truncated viral TKs. In contrast, KAY-2-41r CML1 clones showed a substitution mutation at position 328 (G328T) that modified the aspartic acid 110 into a tyrosine (D110Y).
FIG 2.

Mapping of mutations found in KAY-2-41r viruses and overview of OPV thymidine kinase. (A) Nucleotide sequences of regions of the TK gene of the VACV-WR, CPXV-BR, and CML1 wild-type viruses that were found to be mutated in KAY-2-41r viruses. The position of mutations found in KAY-2-41r viruses are indicated in bold and with a star. The name of each resistant strain, i.e., VACV-WR-[T]del43, CPXV-BR-[GT]del73/74, and CML1-G328T, is given. (B) A schematic view of the TK protein highlighting the major regions involved in ATP, substrate, and metal binding is presented. A partial sequence of the VACV-WR TK is shown for the regions where amino acid substitutions were identified proximal to the β1 sheet-P loop-α1 helix or the β5 sheet. Protein sequences of the human cytosolic TK (hTK1) and of Thermotoga maritima TK (TmTK) are also shown for comparison purposes. The amino acid substitutions found in KAY-2-41r viruses are presented for each of the viruses and are represented by a star in the schematic view or by underlining in the amino acid sequences.
We further proceeded to the phenotypic characterization of the KAY-2-41r clones. As shown in Fig. 3A to C, relatively low levels of resistance to KAY-2-41 were found for the mutant viruses, as they were only 2.7-, 6.0-, and 5.8-fold greater for VACV-WR, CPXV-BR, and CML1 KAY-2-41r, respectively, than for their WT counterparts. These drug-resistant viruses displayed a trend in hypersensitivity toward the nucleoside analogue BVDU, while ANPs and ST-246 showed inhibitory activities in the range of those of WT viruses.
FIG 3.

Drug resistance profiles of plaque-purified VACV-WR (A), CPXV-BR (B), and CML1 (C) KAY-2-41r clones and of viruses with thymidine kinase deletions (D). (A to C) Three clones of each WT and drug-resistant virus were used for plaque reduction assays, and at least three to four independent experiments were performed for each compound tested. The data are presented as a dot plot of the EC50s of the KAY-2-41r clones (filled symbols) versus the EC50s of the WT parent clones (empty symbols). On top of each graph are shown the fold changes in EC50s, which were calculated as the ratio of the mean EC50s of the KAY-2-41r clones divided by the mean EC50s of the WT clones. Results are presented as means ± standard deviations. The statistical significance of the differences in drug sensitivity of the resistant viruses compared to that of the WT is indicated as follows: *, P < 0.05; ***, P < 0.001; ****, P < 0.0001. (D) Plaque reduction assays were done with each of the three KAY-2-41r strains, two VACV strains with TK deletions, and their corresponding WT viruses in three independent experiments. Results are plotted on a linear scale as the log10 of the ratios of the EC50s, and standard deviations are given. Resistance was observed with a ratio of >2 (log 0.3), and hypersensitivity was observed with a ratio of <0.5 (log −0.3).
To further investigate whether mutations within the TK gene may be one of the determinants responsible for the resistance phenotype, we used two VACV strains with deletions in TK (VACV-Cop-ΔTK and VACV-WR-ΔTK) and evaluated the profiles of their sensitivities to four drugs (Fig. 3D). Viruses with TK deletions showed resistance to KAY-2-41 and to 5-BrdU, another TK-dependent drug, and hypersensitivity to BVDU, a TMPK-dependent drug, while cidofovir kept its full activity. Similar observations were made with each of the KAY-2-41r OPVs, suggesting the requirement of viral TK in the antiviral effect of KAY-2-41. Cross-resistance to 5-BrdU reinforced this conclusion, as it partially relied on viral TK to be active. Additionally, the TK-negative phenotype of each of the KAY-2-41r OPVs was confirmed by the ability of these viruses to multiply in H143B TK-deficient cells in the presence of a KAY-2-41 or 5-BrdU concentration which was inhibitory for the WT viruses (data not shown).
We also evaluated the potential involvement of the ribonucleotide reductase in KAY-2-41 efficacy by using a VACV-WR strain with deletions in both ribonucleotide reductase subunits, I4L and F4L. KAY-2-41 conserved its inhibitory activity against this virus with an EC50 of 0.97 ± 0.18 μM, which is in the range of that for the WT virus. A similar observation was made with 5-BrdU, which had an EC50 of 0.2 μM against the virus with the I4L/F4L deletion versus an EC50 of 0.32 μM against the WT virus.
KAY-2-41 interacts with VACV TK.
Next, we wanted to know whether KAY-2-41 could alter the phosphorylating activity of VACV TK, hTK1, and hTK2 for their natural substrate. Enzymatic TK assays based on the competitive binding between the natural substrate dThd and KAY-2-41 demonstrated that KAY-2-41 was inhibitory to the phosphorylation of radiolabeled dThd by hTK2, VACV TK, and hTK1, in decreasing order of potency (Table 4). The inhibition was, however, much weaker than that observed for 5-BrdU and AZT, which appeared to be rather potent competitive inhibitors of dThd phosphorylation, with IC50s ranging, respectively, from 2.9 to 3.6 μM and from 2.4 to 16 μM. In contrast, BVDU strongly affected dThd phosphorylation by hTK2, but not by VACV TK or hTK1, at the highest concentration tested (50 μM).
TABLE 4.
Inhibitory activity of KAY-2-41, 5-BrdU, AZT and BVDU on dThd phosphorylation by VACV-TK, hTK1, and hTK2
| Compound | IC50 (μM)a |
||
|---|---|---|---|
| VACV TK | hTK1 | hTK2 | |
| KAY-2-41 | 490 ± 1 | ≥500 | 352 ± 55 |
| 5-BrdU | 2.9 ± 1.1 | 3.0 ± 0.0 | 3.6 ± 0.6 |
| AZT | 16 ± 0 | 5.8 ± 3.9 | 2.4 ± 0.6 |
| BVDU | >500 | >500 | 0.36 ± 0.05 |
IC50, 50% inhibitory concentrations, or the concentration of compound required to inhibit the binding of the natural substrate dThd by 50%. Data are shown as the means ± standard deviations of two independent experiments. VACV TK, vaccinia virus thymidine kinase; hTK1, human cytosolic thymidine kinase; hTK2, human mitochondrial thymidine kinase.
In vivo evaluation of KAY-2-41.
A mouse model of lethal VACV-WR infection was used to determine the efficacy of KAY-2-41 treatment in vivo. As shown in Fig. 4, all animals intranasally exposed to VACV-WR and treated with vehicle (PBS) died by day 7 p.i. In contrast, both VACV-WR-challenged cohorts that received KAY-2-41 systemically (i.p. injection) at a dose of 5 or 50 mg/kg once daily for 5 consecutive days, starting on the day of infection, survived the infection. The group of infected mice that received 5 mg/kg of the drug had a pronounced loss of body weight between days 5 and 10 p.i., but after this time point, animals progressively recovered. Among the animals receiving the 50-mg/kg treatment regimen, a slight loss of body weight was seen. The effect of a similar treatment with KAY-2-41 (50 mg/kg) was also assessed in mock-infected animals, and no apparent morbidity was observed, as evidenced by a body weight evolution comparable to that of untreated animals (Fig. 4).
FIG 4.

KAY-2-41 protects mice from VACV-WR-induced mortality. NMRI mice (5 mice per group) were challenged intranasally either with vehicle (mock infected) or with VACV-WR. Animals were then treated with PBS or with the drug at a dose of 5 or 50 mg/kg once per day for 5 consecutive days starting at 6 h after infection. Cohorts were monitored for 20 days for body weight (left) and survival (right). Body weight evolution is shown as the percentage of the change in the average weight for each group of mice.
On the basis of these results, we further evaluated the impact of the 50-mg/kg KAY-2-41 treatment on viral DNA loads and VACV-WR replication in various organs. Mice were infected and treated in the same way as in the first experiment. Four (VACV-WR-infected) or five (VACV-WR-infected and KAY-2-41-treated) animals per group were sacrificed at day 5 p.i. The efficacy of the compound was confirmed, as KAY-2-41 (50 mg/kg daily for 5 consecutive days) protected 100% of the animals from VACV-WR mortality, with no apparent morbidity (Fig. 5). As depicted in Fig. 5B, viral DNA was detected in all organs of infected untreated mice, with means of 5.1 (kidneys) to 6.5 (lungs) log DNA copies/g, and the sera were also positive (4.2 log DNA copies/50 μl serum) at this time point. Lung, liver, and spleen tissues showed the highest levels of viral DNA, which were equivalent to mean viral titers of, respectively, 3.8, 3.3, and 3.5 log 50% tissue culture infective doses (TCID50s)/g. Treating the mice with KAY-2-41 significantly reduced viral DNA loads in various organs, including lungs (means, 4.0 versus 6.5 log DNA copies/g), liver (means, 1.2 versus 6.6 log DNA copies/g), spleen (means, 4.5 versus 6.3 log DNA copies/g), and serum (means, 2.5 versus 4.2 log DNA copies/g). A similar trend was also noticed in the kidneys and mesenteric lymph nodes. Viral titrations further demonstrated the absence of replicating virus in lungs, liver, spleen, and kidneys. Histological examination of the lung tissue revealed the absence of inflammation in KAY-2-41-treated animals, which, strikingly, contrasts with the interstitial inflammation seen in infected untreated animals (Fig. 5C).
FIG 5.

KAY-2-41 treatment inhibits virus dissemination and replication in various organs. (A) Body weight evolution and survival curves of mock-infected and VACV-WR-infected mice that received vehicle (PBS) or KAY-2-41 treatment at 50 mg/kg once a day for 5 consecutive days beginning 6 h after virus exposure. Body weight evolution is shown as the percentage of the change in average weight for each group of mice (5 mice per group). dpi, day postinfection. (B) Viral DNA loads in various organs and in serum (left) and virus titers in lungs, liver, spleen, and kidneys (right) are shown. Animals were sacrificed at day 5 p.i. Viral loads were determined by qPCR and are expressed as VACV DNA copy numbers per g of tissue or per 50 μl of serum. Virus titers are shown in TCID50s per g tissue. The scatter plots show the viral DNA loads or virus titers from individual mice and the means (horizontal bars) for each group. Four (VACV-WR-infected) or five (VACV-WR-infected and KAY-2-41-treated [50 mg/kg]) individual mice were used lung. MLNs, mesenteric lymph nodes. Dashed lines, limit of detection. The viral DNA loads or virus titers of KAY-2-41-treated mice differed significantly from those of the PBS-treated group by the Mann-Whitney test (*, P < 0.05). (C) Lung tissue examination at 5 days p.i. While VACV-WR-treated mice exhibited interstitial inflammation (arrow), no inflammatory cells were seen after KAY-2-41 treatment.
DISCUSSION
To our knowledge, the in vitro and in vivo antipoxvirus activity of a 1′-carbon-substituted 4′-thiothymidine derivative has not yet been reported. Other laboratories have demonstrated the inhibitory potential of 5-substituted 4′-thiopyrimidine nucleoside derivatives against poxviruses (27, 29), despite the fact that the narrow substrate specificity of poxvirus TK remains a challenge for selecting potent nucleoside analogues. Our study demonstrated that OPVs (VACVs, CPXVs, and CML1) are efficiently inhibited by KAY-2-41 in vitro at concentrations in the submicromolar range. This activity was also displayed against a variety of cowpox clinical isolates, a finding which may have implications for clinical use in the current context of the increased incidence of cowpox virus infections (1, 42). KAY-2-41 appeared to be more potent than cidofovir and (S)-HPMP-5-azaC, albeit the prodrug of cidofovir, CMX001, and ST-246, a morphogenesis inhibitor, remained the most active molecules toward OPVs.
KAY-2-41 was effective in inhibiting the replication of mutant viruses resistant to cidofovir and/or (S)-HPMPDAP. If KAY-2-41 in its triphosphate metabolite form ultimately targets the viral DNA polymerase, our data suggest that the presence of the A314V or T831I mutation in the DNA polymerase gene (E9L) did not impact its ability to accept this substrate, whereas these changes clearly influenced the acceptance of acyclic nucleoside pyrimidine and purine analogues [i.e., cidofovir and (S)-HPMPDAP]. In fact, while these mutations were associated with different ANP resistance profiles when present in a CMLV or in a VACV backbone (in line with our published observations [35]), KAY-2-41 remained equally active against WT and mutant viruses bearing the A314V or T831I mutation. Interestingly, the CML14 and VACV A314V plus A684V double mutants appeared to be more sensitive to KAY-2-41 inhibition, suggesting that through these conformational changes, E9L might become more prone to accept the thymidine analogue triphosphate metabolite.
Being a nucleoside analogue, the involvement of the viral TK was expected to be important for the metabolic activation of the molecule, and the genotypic characterization of KAY-2-41r strains confirmed this hypothesis. Additionally, the poor but significant antiviral activity of KAY-2-41 against the parapoxvirus orf in comparison to that against OPVs was of interest, as it also pointed toward the need for TK for KAY-2-41 to inhibit OPVs. Indeed, this virus does not possess a specific TK gene (43), and in line with that, the EC50 against ORFV-NZ2 was in the range of the EC50s seen with KAY-2-41r viruses. Sequencing of KAY-2-41r clones allowed us to identify mutations in the TK gene that are most likely responsible for the drug-resistant phenotype since they were identified only in drug-resistant viruses. Five other viral genes investigated, including A48R (TMPK), B1R (Ser/Thr kinase), F10L (Ser/Thr kinase), E9L (DNA polymerase), and D4R (uracil DNA glycosylase), had sequences that were identical to those of the corresponding WT viruses. Also, a VACV-WR mutant with a deletion in ribonucleotide reductase subunits did not display resistance to KAY-2-41 or to 5-BrdU. In contrast, two viruses with TK deletions displayed resistance to both KAY-2-41 and 5-BrdU (which are TK dependent) and not to BVDU (which is TMPK dependent), mimicking the drug resistance phenotype of KAY-2-41r strains. We finally showed, in our TK competition assays, that KAY-2-41 competed with the dThd substrate of VACV TK. These findings demonstrate that the mutations found in the viral TK most probably occurred due to KAY-2-41 selection pressure and that viral TK may contribute to KAY-2-41 sensitivity, although it may not be the sole determinant. As a consequence, additional studies are still required (i) to determine whether the identified mutations or mutations in other genes contribute to drug resistance, (ii) to identify the metabolites of the compound, and (iii) to investigate whether cellular partners can be implicated in KAY-2-41 antiviral activity.
Both the KAY-2-41r viruses and viruses with TK deletions showed hypersensitivity to BVDU. While we and others have shown that VACV TK and hTK1 do not measurably phosphorylate BVDU in enzymatic assays (30), the mitochondrial homodimer hTK2 was efficient in this activity (Table 4). Also, the secondary phosphorylation of BVDU has been shown to rely on the viral TMPK (33), and we observed that KAY-2-41 still potently inhibited TMPK OPV mutants resistant to BVDU (our unpublished data). Although these two molecules seem to have a distinct mode of activation, how can we explain this hypersensitivity? The TK deletion could affect pyrimidine metabolism and, consequently, the pool of available nucleotides (i.e., dTTP) needed for viral replication. For these reasons, more BVDU triphosphate might then be accepted as a substrate for incorporation by the DNA polymerase (E9L), potentiating BVDU antiviral activity. Alternatively, it cannot be excluded that the mutations in the viral TK may make BVDU a better substrate for further phosphorylation.
We can further speculate about the impact of the identified mutations on TK enzymatic activity, albeit we did not prove, through marker rescue experiments, that such changes were responsible for KAY-2-41 resistance. In KAY-2-41-resistant VACV-WR, a single nucleotide deletion caused a codon frameshift starting at amino acid 15 (S15Q) and finishing at position 21 with a stop codon, and in the CPXV-BR strain, the double nucleotide deletion changed the valine codon at position 25 into a stop codon. These deletion mutations occurred in the amino-terminal region of the gene and presumably led to the production of a truncated TK (Fig. 2 and 6). This region is critical for TK enzymatic activity, as it includes the site where ATP binds to act as a γ-phosphoryl donor required to produce dThd monophosphate. In VACV, this region consists of a P loop (amino acids 11 to 16) preceded by a β sheet (β1, amino acids 4 to 10) and followed by an α helix (α1, amino acids 17 to 29) (44, 45). A similar organization is found in structurally related TKs, including the human cytosolic TK (hTK1) and the TK of Thermotoga maritima (TmTK) (45, 46).
FIG 6.

Potential impact of D110Y mutation on substrate binding and thymidine kinase monomer assembly. (A) Tridimensional structure of one monomer of VACV TK in complex with dTTP (Protein Data Bank accession number 2J87). Residue F113, localized in the loop between the β5 sheet and α4 helix and required for substrate binding, is highlighted (see also Fig. 2). On the same loop is also shown the tyrosine residue (red sticks) found at position 110 and identified as the D110Y mutation. The presence of the mutated residue may disturb the loop and, as a consequence, substrate recognition. (B to D) Two monomers in complex with dTTP are shown in green and yellow. The phenylalanine at position 113 is depicted in red on each monomer. The aspartic acid at position 110 of one monomer establishes a direct interaction with the isoleucine 125 of the other monomer (B). Modification of the aspartic acid 110 into a tyrosine provokes a steric hindrance that may destabilize the assembly of monomers (C and D). The figures were generated using PyMol software (version 0.99; DeLano Scientific LLC).
The amino acid residue mutated in drug-resistant CML1 (D110Y) mapped to a highly conserved residue in the type II TK family (D110) that is localized close to phenylalanine 113 (F113), which has been shown to interact with the substrate via its main-chain amide nitrogen (Fig. 2) (44). The substrate binding region has been well characterized in the model of VACV TK bound to dTTP (44), as well as in related hTK1 and TmTK bound to a bisubstrate analogue, TP4A {P1-(5′-adenosyl)P4-[5′-(2′-deoxy-thymidyl)]tetraphosphate} (45). While no direct interactions between D110 and the nucleoside substrate have been described, we observed that changing a negatively charged amino acid (i.e., aspartic acid [D]) into a residue with a hydrophobic aromatic side chain (i.e., tyrosine [Y]) may disturb the loop localized between the β5 sheet (amino acids 106 to 110) and the α4 helix (amino acids 121 to 128) and, as a consequence, the binding of the nucleoside substrate (Fig. 6). This mutation may have a second impact on VACV TK enzymatic activity, since this enzyme is catalytically active as a homotetramer. Based on the model of El Omari and colleagues (44), we observed that the D110 residue is located at the interface of two monomers (Fig. 6). The mutation D110Y may thus create a steric hindrance that can disrupt monomer assembly, explaining a loss of enzymatic activity.
KAY-2-41 was found to be cytostatic for growing cells, although no apparent morphological changes were seen on confluent (monolayer) cells. Also, the KAY-2-41r virus strains did not show high levels of resistance to the drug (from 2.7- to 6.0-fold increases in EC50s), and they remained partially sensitive to the inhibitory effect of KAY-2-41. These observations suggest that other cellular enzymes may be involved in the mode of action of the molecule as well. Similar observations have been made with 4′-thioIDU (27). Animal experiments, however, showed that a repetitive dose of 50 mg/kg once daily for 5 consecutive days did not visually affect the naive animals. However, toxicity studies in animals are required to investigate this in more detail. Additionally, the fact that the KAY-2-41r virus strains did not display high levels of drug resistance suggests that KAY-2-41-based antiviral treatment might still be efficacious if resistant mutants would emerge. Also, such TK-altered viruses might show a decreased pathogenicity, as noticed before in mice challenged with VACV with a TK deletion (47), albeit this property was not assessed here.
Finally, we demonstrated the potency of KAY-2-41 in protecting mice from disease and subsequent death after a lethal challenge with VACV-WR. This was observed with a daily treatment regimen of 50 mg/kg for 5 consecutive days, while reducing the drug concentration to 5 mg/kg protected against mortality but not weight loss. After i.p. administration of KAY-2-41 (50 mg/kg), virus replication was completely abolished in the primary target organs, i.e., lungs, as well as in the liver, spleen, and kidneys. Our results also showed that the levels of viral DNA in serum or in several organs were markedly reduced or even abolished. In contrast, oral administration of 4′-thioIDU (15 mg/kg twice daily for 5 consecutive days beginning at 24 h after infection) reduced lung virus titers by only 10-fold, as reported by Kern and colleagues (27), although differences in the doses and routes of administration of the compounds and/or the volume of virus inoculated might be at the origin of the discrepancy in the results. Histology of the lungs of VACV-WR-infected mice revealed signs only of inflammation and not of pneumonia, which could be due to (i) the time point of sacrifice of the animals, i.e., at day 5 p.i., or (ii) the use of a relatively small volume for virus inoculation, as discussed by Smee and colleagues (48).
The rationale of using viral TKs in the selective activation of nucleoside analogues mainly comes from research done with herpesviruses, which possess a type 1 TK that can phosphorylate a variety of substrates. Although OPVs encode a TK, its narrower substrate specificity and its resemblance to hTK1 have hampered the development of TK-specific drugs for OPVs. However, VACV TK might have a substrate specificity broader than was initially thought (44, 49). Indeed, the structure of VACV TK and the demonstration that a bulky nucleoside analogue can be OPV-TK specific, as shown by Fan et al. (49), have provided new evidence that conformational differences between OPV TK and hTK1 may be exploited to design selective poxvirus inhibitors (44, 49). Altogether, our study proves that nucleoside analogues such as KAY-2-41 and possibly also other 1′-substituted 4′-thiothymidine derivatives should not be neglected as potential antiviral agents, as they may show promise for future development of broad-spectrum therapy against both pox- and herpesviruses.
ACKNOWLEDGMENTS
We thank Steven Carmans, Anita Camps, Pierre Fiten, Barbara Mertens, Lizette van Berckelaer, Ria Van Berwaer, and Lieve Ophalvens for the excellent technical assistance that they provided.
These works were financed by two Belgian grants, FWO G-0608-08 and GOA 10/014, and the Japan Society for the Promotion of Science, KAKENHI no. 24590144, to K.H.
Footnotes
Published ahead of print 14 October 2013
REFERENCES
- 1.Vogel S, Sardy M, Glos K, Korting HC, Ruzicka T, Wollenberg A. 2012. The Munich outbreak of cutaneous cowpox infection: transmission by infected pet rats. Acta Derm. Venereol. 92:126–131. 10.2340/00015555-1227 [DOI] [PubMed] [Google Scholar]
- 2.Silva DC, Moreira-Silva EA, Gomes JA, Fonseca FG, Correa-Oliveira R. 2010. Clinical signs, diagnosis, and case reports of Vaccinia virus infections. Braz. J. Infect. Dis. 14:129–134. 10.1590/S1413-86702010000200003 [DOI] [PubMed] [Google Scholar]
- 3.Bhanuprakash V, Venkatesan G, Balamurugan V, Hosamani M, Yogisharadhya R, Gandhale P, Reddy KV, Damle AS, Kher HN, Chandel BS, Chauhan HC, Singh RK. 2010. Zoonotic infections of buffalopox in India. Zoonoses Public Health 57:e149–e155. 10.1111/j.1863-2378.2009.01314.x [DOI] [PubMed] [Google Scholar]
- 4.Reynolds MG, Damon IK. 2012. Outbreaks of human monkeypox after cessation of smallpox vaccination. Trends Microbiol. 20:80–87. 10.1016/j.tim.2011.12.001 [DOI] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention 2012. Human orf virus infection from household exposures—United States, 2009-2011. MMWR Morb. Mortal. Wkly. Rep. 61:245–248 [PubMed] [Google Scholar]
- 6.Al-Qattan MM. 2011. Orf infection of the hand. J. Hand Surg. Am. 36:1855–1858. 10.1016/j.jhsa.2011.08.019 [DOI] [PubMed] [Google Scholar]
- 7.de Carvalho CH, de Andrade AL, de Oliveira DH, Lima E, da Silveira EJ, de Medeiros AM. 2012. Intraoral molluscum contagiosum in a young immunocompetent patient. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 114:e57–e60. 10.1016/j.oooo.2011.10.009 [DOI] [PubMed] [Google Scholar]
- 8.Fiandeiro PT, Attard N. 2013. Koebnerized molluscum contagiosum. QJM 106:1043. 10.1093/qjmed/hcs183 [DOI] [PubMed] [Google Scholar]
- 9.Elsendoorn A, Agius G, Le Moal G, Aajaji F, Favier AL, Wierzbicka-Hainault E, Beraud G, Flusin O, Crance JM, Roblot F. 2011. Severe ear chondritis due to cowpox virus transmitted by a pet rat. J. Infect. 63:391–393. 10.1016/j.jinf.2011.06.004 [DOI] [PubMed] [Google Scholar]
- 10.Haase O, Moser A, Rose C, Kurth A, Zillikens D, Schmidt E. 2011. Generalized cowpox infection in a patient with Darier disease. Br. J. Dermatol. 164:1116–1118. 10.1111/j.1365-2133.2011.10226.x [DOI] [PubMed] [Google Scholar]
- 11.Lin HY, Linn G, Liu CB, Chen CJ, Yu KJ. 2010. An immunocompromised woman with severe molluscum contagiosum that responded well to topical imiquimod: a case report and literature review. J. Low Genit. Tract Dis. 14:134–135. 10.1097/LGT.0b013e3181bf1a50 [DOI] [PubMed] [Google Scholar]
- 12.Pelkonen PM, Tarvainen K, Hynninen A, Kallio ER, Henttonen K, Palva A, Vaheri A, Vapalahti O. 2003. Cowpox with severe generalized eruption, Finland. Emerg. Infect. Dis. 9:1458–1461. 10.3201/eid0911.020814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andrei G, Snoeck R. 2010. Cidofovir activity against poxvirus infections. Viruses 2:2803–2830. 10.3390/v2122803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hostetler KY. 2010. Synthesis and early development of hexadecyloxypropylcidofovir: an oral antipoxvirus nucleoside phosphonate. Viruses 2:2213–2225. 10.3390/v2102213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Centers for Disease Control and Prevention 2009. Progressive vaccinia in a military smallpox vaccinee—United States, 2009. MMWR Morb. Mortal. Wkly. Rep. 58:532–536 [PubMed] [Google Scholar]
- 16.Kaiser J. 2007. Smallpox vaccine. A tame virus runs amok. Science 316:1418–1419. 10.1126/science.316.5830.1418 [DOI] [PubMed] [Google Scholar]
- 17.Lanier R, Trost L, Tippin T, Lampert B, Robertson A, Foster S, Rose M, Painter W, O'Mahony R, Almond M, Painter G. 2010. Development of CMX001 for the treatment of poxvirus infections. Viruses 2:2740–2762. 10.3390/v2122740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Papanicolaou G, Kurtzberg J, Grimley M, Storch GA, Goyal R, Anderson M, Mommeja-Marin H, Painter W. 2011. CMX001 is not nephrotoxic or myelosuppressive in 183 patients with life threatening dsDNA infections including refractory cytomegalovirus, adenovirus, and BK virus, abstr V-499b. Abstr 51st Intersci. Conf. Antimicrob. Agents Chemother. American Society for Microbiology, Washington, DC [Google Scholar]
- 19.Gammon DB, Snoeck R, Fiten P, Krecmerova M, Holy A, De Clercq E, Opdenakker G, Evans DH, Andrei G. 2008. Mechanism of antiviral drug resistance of vaccinia virus: identification of residues in the viral DNA polymerase conferring differential resistance to antipoxvirus drugs. J. Virol. 82:12520–12534. 10.1128/JVI.01528-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stittelaar KJ, Neyts J, Naesens L, van Amerongen G, van Lavieren RF, Holy A, De Clercq E, Niesters HG, Fries E, Maas C, Mulder PG, van der Zeijst BA, Osterhaus AD. 2006. Antiviral treatment is more effective than smallpox vaccination upon lethal monkeypox virus infection. Nature 439:745–748. 10.1038/nature04295 [DOI] [PubMed] [Google Scholar]
- 21.Duraffour S, Snoeck R, de Vos R, van Den Oord JJ, Crance JM, Garin D, Hruby DE, Jordan R, De Clercq E, Andrei G. 2007. Activity of the anti-orthopoxvirus compound ST-246 against vaccinia, cowpox and camelpox viruses in cell monolayers and organotypic raft cultures. Antivir. Ther. 12:1205–1216 [PubMed] [Google Scholar]
- 22.Yang G, Pevear DC, Davies MH, Collett MS, Bailey T, Rippen S, Barone L, Burns C, Rhodes G, Tohan S, Huggins JW, Baker RO, Buller RL, Touchette E, Waller K, Schriewer J, Neyts J, DeClercq E, Jones K, Hruby D, Jordan R. 2005. An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus challenge. J. Virol. 79:13139–13149. 10.1128/JVI.79.20.13139-13149.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chinsangaram J, Honeychurch KM, Tyavanagimatt SR, Leeds JM, Bolken TC, Jones KF, Jordan R, Marbury T, Ruckle J, Mee-Lee D, Ross E, Lichtenstein I, Pickens M, Corrado M, Clarke JM, Frimm AM, Hruby DE. 2012. Safety and pharmacokinetics of the anti-orthopoxvirus compound ST-246 following a single daily oral dose for 14 days in human volunteers. Antimicrob. Agents Chemother. 56:4900–4905. 10.1128/AAC.00904-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Srivastav NC, Rai D, Tse C, Agrawal B, Kunimoto DY, Kumar R. 2010. Inhibition of mycobacterial replication by pyrimidines possessing various C-5 functionalities and related 2′-deoxynucleoside analogues using in vitro and in vivo models. J. Med. Chem. 53:6180–6187. 10.1021/jm100568q [DOI] [PubMed] [Google Scholar]
- 25.Secrist JA, III, Tiwari KN, Riordan JM, Montgomery JA. 1991. Synthesis and biological activity of 2′-deoxy-4′-thio pyrimidine nucleosides. J. Med. Chem. 34:2361–2366. 10.1021/jm00112a007 [DOI] [PubMed] [Google Scholar]
- 26.Dyson MR, Coe PL, Walker RT. 1991. The synthesis and antiviral activity of some 4′-thio-2′-deoxy nucleoside analogues. J. Med. Chem. 34:2782–2786. 10.1021/jm00113a016 [DOI] [PubMed] [Google Scholar]
- 27.Kern ER, Prichard MN, Quenelle DC, Keith KA, Tiwari KN, Maddry JA, Secrist JA., III 2009. Activities of certain 5-substituted 4′-thiopyrimidine nucleosides against orthopoxvirus infections. Antimicrob. Agents Chemother. 53:572–579. 10.1128/AAC.01257-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahim SG, Trivedi N, Bogunovic-Batchelor MV, Hardy GW, Mills G, Selway JW, Snowden W, Littler E, Coe PL, Basnak I, Whale RF, Walker RT. 1996. Synthesis and anti-herpes virus activity of 2′-deoxy-4′-thiopyrimidine nucleosides. J. Med. Chem. 39:789–795. 10.1021/jm950029r [DOI] [PubMed] [Google Scholar]
- 29.Prichard MN, Quenelle DC, Hartline CB, Harden EA, Jefferson G, Frederick SL, Daily SL, Whitley RJ, Tiwari KN, Maddry JA, Secrist JA, III, Kern ER. 2009. Inhibition of herpesvirus replication by 5-substituted 4′-thiopyrimidine nucleosides. Antimicrob. Agents Chemother. 53:5251–5258. 10.1128/AAC.00417-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prichard MN, Williams AD, Keith KA, Harden EA, Kern ER. 2006. Distinct thymidine kinases encoded by cowpox virus and herpes simplex virus contribute significantly to the differential antiviral activity of nucleoside analogs. Antiviral Res. 71:1–6. 10.1016/j.antiviral.2006.01.013 [DOI] [PubMed] [Google Scholar]
- 31.Solaroli N, Johansson M, Balzarini J, Karlsson A. 2006. Substrate specificity of three viral thymidine kinases (TK): vaccinia virus TK, feline herpesvirus TK, and canine herpesvirus TK. Nucleosides Nucleotides Nucleic Acids 25:1189–1192. 10.1080/15257770600894451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caillat C, Topalis D, Agrofoglio LA, Pochet S, Balzarini J, Deville-Bonne D, Meyer P. 2008. Crystal structure of poxvirus thymidylate kinase: an unexpected dimerization has implications for antiviral therapy. Proc. Natl. Acad. Sci. U. S. A. 105:16900–16905. 10.1073/pnas.0804525105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Topalis D, Collinet B, Gasse C, Dugue L, Balzarini J, Pochet S, Deville-Bonne D. 2005. Substrate specificity of vaccinia virus thymidylate kinase. FEBS J. 272:6254–6265. 10.1111/j.1742-4658.2005.05006.x [DOI] [PubMed] [Google Scholar]
- 34.Haraguchi K, Takahashi H, Tanaka H, Hayakawa H, Ashida N, Nitanda T, Baba M. 2004. Synthesis and antiviral activities of 1′-carbon-substituted 4′-thiothymidines. Bioorg. Med. Chem. 12:5309–5316. 10.1016/j.bmc.2004.07.057 [DOI] [PubMed] [Google Scholar]
- 35.Duraffour S, Andrei G, Topalis D, Krecmerova M, Crance JM, Garin D, Snoeck R. 2012. Mutations conferring resistance to viral DNA polymerase inhibitors in camelpox virus give different drug-susceptibility profiles in vaccinia virus. J. Virol. 86:7310–7325. 10.1128/JVI.00355-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carroll DS, Emerson GL, Li Y, Sammons S, Olson V, Frace M, Nakazawa Y, Czerny CP, Tryland M, Kolodziejek J, Nowotny N, Olsen-Rasmussen M, Khristova M, Govil D, Karem K, Damon IK, Meyer H. 2011. Chasing Jenner's vaccine: revisiting cowpox virus classification. PLoS One 6:e23086. 10.1371/journal.pone.0023086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfeffer M, Meyer H, Wernery U, Kaaden OR. 1996. Comparison of camelpox viruses isolated in Dubai. Vet. Microbiol. 49:135–146. 10.1016/0378-1135(95)00181-6 [DOI] [PubMed] [Google Scholar]
- 38.Ramyar H, Hessami M. 1972. Isolation, cultivation and characterization of camel pox virus. Zentralbl. Veterinarmed. B 19:182–189 [DOI] [PubMed] [Google Scholar]
- 39.Gammon DB, Gowrishankar B, Duraffour S, Andrei G, Upton C, Evans DH. 2010. Vaccinia virus-encoded ribonucleotide reductase subunits are differentially required for replication and pathogenesis. PLoS Pathog. 6:e1000984. 10.1371/journal.ppat.1000984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duraffour S, Snoeck R, Krecmerova M, Van den Oord J, de Vos R, Holy A, Crance JM, Garin D, De Clercq E, Andrei G. 2007. Activities of several classes of acyclic nucleoside phosphonates against camelpox virus replication in different cell culture models. Antimicrob. Agents Chemother. 51:4410–4419. 10.1128/AAC.00838-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duraffour S, Matthys P, van den Oord JJ, De Schutter T, Mitera T, Snoeck R, Andrei G. 2011. Study of camelpox virus pathogenesis in athymic nude mice. PLoS One 6:e21561. 10.1371/journal.pone.0021561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwarzer H, Kurth A, Hermel M, Plange N. 2013. Severe ulcerative keratitis in ocular cowpox infection. Graefes Arch. Clin. Exp. Ophthalmol. 251:1451–1452. 10.1007/s00417-012-2138-x [DOI] [PubMed] [Google Scholar]
- 43.Delhon G, Tulman ER, Afonso CL, Lu Z, Concha-Bermejillo A, Lehmkuhl HD, Piccone ME, Kutish GF, Rock DL. 2004. Genomes of the parapoxviruses ORF virus and bovine papular stomatitis virus. J. Virol. 78:168–177. 10.1128/JVI.78.1.168-177.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.El Omari K, Solaroli N, Karlsson A, Balzarini J, Stammers DK. 2006. Structure of vaccinia virus thymidine kinase in complex with dTTP: insights for drug design. BMC Struct. Biol. 6:22. 10.1186/1472-6807-6-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Segura-Pena D, Lutz S, Monnerjahn C, Konrad M, Lavie A. 2007. Binding of ATP to TK1-like enzymes is associated with a conformational change in the quaternary structure. J. Mol. Biol. 369:129–141. 10.1016/j.jmb.2007.02.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Segura-Pena D, Lichter J, Trani M, Konrad M, Lavie A, Lutz S. 2007. Quaternary structure change as a mechanism for the regulation of thymidine kinase 1-like enzymes. Structure 15:1555–1566. 10.1016/j.str.2007.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Puhlmann M, Brown CK, Gnant M, Huang J, Libutti SK, Alexander HR, Bartlett DL. 2000. Vaccinia as a vector for tumor-directed gene therapy: biodistribution of a thymidine kinase-deleted mutant. Cancer Gene Ther. 7:66–73. 10.1038/sj.cgt.7700075 [DOI] [PubMed] [Google Scholar]
- 48.Smee DF, Gowen BB, Wandersee MK, Wong MH, Skirpstunas RT, Baldwin TJ, Hoopes JD, Sidwell RW. 2008. Differential pathogenesis of cowpox virus intranasal infections in mice induced by low and high inoculum volumes and effects of cidofovir treatment. Int. J. Antimicrob. Agents 31:352–359. 10.1016/j.ijantimicag.2007.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fan X, Zhang X, Zhou L, Keith KA, Prichard MN, Kern ER, Torrence PF. 2006. Toward orthopoxvirus countermeasures: a novel heteromorphic nucleoside of unusual structure. J. Med. Chem. 49:4052–4054. 10.1021/jm060404n [DOI] [PMC free article] [PubMed] [Google Scholar]