

Abstract
Dengue virus (DENV) causes disease globally, resulting in an estimated 25 to 100 million new infections per year. No effective DENV vaccine is available, and the current treatment is only supportive. Thus, there is an urgent need to develop therapeutic agents to cure this epidemic disease. In the present study, we identified a potential small-molecule inhibitor, BP13944, via high-throughput screening (HTS) of 60,000 compounds using a stable cell line harboring an efficient luciferase replicon of DENV serotype 2 (DENV-2). BP13944 reduced the expression of the DENV replicon reporter in cells, showing a 50% effective concentration (EC50) of 1.03 ± 0.09 μM. Without detectable cytotoxicity, the compound inhibited replication or viral RNA synthesis in all four serotypes of DENV but not in Japanese encephalitis virus (JEV). Sequencing analyses of several individual clones derived from BP13944-resistant RNAs purified from cells harboring the DENV-2 replicon revealed a consensus amino acid substitution (E66G) in the region of the NS3 protease domain. Introduction of E66G into the DENV replicon, an infectious DENV cDNA clone, and recombinant NS2B/NS3 protease constructs conferred 15.2-, 17.2-, and 3.1-fold resistance to BP13944, respectively. Our results identify an effective small-molecule inhibitor, BP13944, which likely targets the DENV NS3 protease. BP13944 could be considered part of a more effective treatment regime for inhibiting DENV in the future.
INTRODUCTION
Dengue virus (DENV) (serotypes 1 to 4) belongs to the family Flaviviridae, a group of enveloped RNA viruses that includes the genera Hepacivirus, Flavivirus, and Pestivirus. The genus Flavivirus consists of arthropod-borne disease agents such as yellow fever virus (YFV), Japanese encephalitis virus (JEV), West Nile virus (WNV), tick-borne encephalitis virus (TBEV), and DENV (1). More than 70 members of the Flavivirus genus are important human pathogens that cause significant morbidity and mortality (2). DENV is a public health threat to an estimated 2.5 billion people living in areas where dengue is epidemic, leading to 50 to 100 million human infections each year (3, 4). DENV infection frequently leads to dengue fever, life-threatening dengue hemorrhagic fever (DHF), or dengue shock syndrome (DSS) (5–7). Approximately 500,000 cases of DHF and DSS have been reported among more than 100 countries, causing approximately 12,500 deaths per year (3). Despite the tremendous efforts invested in anti-DENV research, no clinically approved vaccine or antiviral therapeutic agents are available for humans, and disease treatment is limited to supportive care (4, 8, 9). Considering the spread of this epidemic and the severity of DENV, an effective anti-DENV drug is urgently needed.
DENV is an enveloped RNA virus consisting of a 10.7-kb single-stranded, positive-polarity RNA genome associated with multiple copies of capsid proteins. DENV RNA is translated as a single polyprotein upon entering the host cell and is cleaved by host proteases and the virus-encoded two-component protease (NS2B/NS3pro) into three structural proteins (C, M, and E) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) to initiate viral replication (10–12). Three structural proteins and host cell membranes form the viral particles. The nonstructural (NS) proteins take part in viral RNA replication, virion assembly (13–15), and evasion of the innate immune response (16–20). The NS3 protein is a multifunctional enzyme, acting as a protease (with NS2B as a cofactor) for polyprotein processing, an RNA triphosphatase for capping nascent viral RNA, and a helicase for unwinding the double-stranded replicative form of RNA (21–23). The viral protease (NS2B/NS3pro) consists of an N-terminal 184-residue NS3 protease domain (NS3pro), which acts as a trypsin-like protease, with a serine protease catalytic triad (His51, Asp75, and Ser135) (24–26). The RNA helicase and the NTPase domain are located in the C terminus of NS3 (27). The recently reported crystal structure of the full-length DENV NS3 molecule (28, 29) suggested that the protease and helicase domains of DENV NS3 are likely to be highly interdependent in the viral life cycle. The pivotal role of NS3 in viral replication and virion assembly makes it an attractive target for the development of anti-DENV inhibitors.
Viral proteases are considered good antiviral targets because of their known enzymatic activities, as demonstrated by the nine protease inhibitors of human immunodeficiency virus (HIV) currently in clinical use (30) and the two protease inhibitor drugs approved for the treatment of hepatitis C virus (HCV), a member of the Flaviviridae family (31–33). By analogy with the success of the HIV and HCV protease inhibitors, a number of approaches have been employed to search for DENV NS3 protease inhibitors (reviewed in references 8 and 34–37). One strategy used for this purpose involves a specific target-based approach, e.g., an enzyme-based biochemical assay or structure-based virtual screen (38–49). An alternative strategy is to use a replication-based approach, e.g., screening through virus infection assays or replicon cell lines (50–52). Although no drug is currently approved for the treatment of DENV infection, similar approaches have been successfully applied in the search for novel HCV inhibitors, suggesting the feasibility of these strategies.
Replicons are subgenomic, self-replicating RNA molecules that contain the nucleotide sequences required for RNA replication, transcription, and translation but are not infectious (53). The HCV replicon was successfully developed and used in a broad-based, proprietary antiviral screening platform, leading to the rapid development of anti-HCV drugs (reviewed in references 54–56). A number of potential novel HCV inhibitors derived from the high-throughput (HTS) HCV replicon are currently in clinical trials (57–59). Similarly, a subgenomic DENV RNA replicon system has been established and used for testing and screening inhibitors of DENV replication (8, 52, 60–68). Although no DENV inhibitors derived from HTS with the DENV replicon are in clinical trials at present, a number of HCV drug candidates discovered using the HCV replicon are in different stages of clinical trials, demonstrating that HTS is powerful screening tool for searching for antiviral inhibitors using replicons.
In this study, we performed HTS using the BHK-21 cell line containing a stable reporter-DENV replicon to identify a small-molecule inhibitor of DENV: BP13944, 1-hexadecanaminium, N-ethyl-N,N-bis(2-hydroxyethyl)-bromide (1:1). BP13944 specifically inhibited all four serotypes of DENV but did not inhibit JEV. Drug resistance assays suggested that BP13944 might target the DENV NS3 protease and affect NS3 protease activity. The data presented here may provide an alternative method for the development of an NS3 protease inhibitor.
MATERIALS AND METHODS
Cell lines and virus strains.
BHK-21 cells (ATCC CCL-10) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 4.5 g/liter glucose and 5% fetal bovine serum (FBS). The cells were cultured at 37°C in a 5% CO2 incubator. Aedes albopictus C6/36 cells (ATCC CRL-1660) were cultured in RPMI 1640 medium supplemented with 5% FBS. These cells were maintained at 28°C in a 5% CO2 incubator. Virus-infected cells were grown in the respective media supplemented with 2% FBS. Dengue virus type 2 (DENV-2) (Taiwanese strain PL046) (69) and Japanese encephalitis virus (JEV) (strain RP-9) (70) were provided by C. L. Liao (Institute of Microbiology and Immunology, National Defense Medical Center, Taiwan). DENV-2 and JEV stocks were prepared in C6/36 cells by infecting the cells at a multiplicity of infection (MOI) of 0.1 and 0.01 PFU/cell, respectively, with RPMI 1640 medium containing 2% FBS, followed by incubation at 28°C until a cytopathic effect was observed. The supernatant was harvested and stored with 20% FBS at −80°C. The viral titer was determined in a plaque-forming assay in the BHK-21 cells. The titers of DENV-2 and JEV stocks are generally approximately 1 × 108 and 1 × 109 PFU/ml, respectively.
E. coli and yeast strains.
Competent cells of Escherichia coli strain C41, a derivative of BL21(DE3) (71), were purchased frozen from OverExpress Inc. (Brooklyn, NY). A standard yeast medium and standard transformation methods were used in this study (72). Saccharomyces cerevisiae YPH857 was purchased from ATCC. The YPH857 genotype is MATα ade2-101 lys2-801 ura3-52 trp1-Δ63 HIS5 CAN1 his3-Δ200 leu2-Δ1 cyh2. Competent yeast cells were prepared using the lithium acetate procedure (72).
High-throughput screening.
HTS was conducted at the Division of Biotechnology and Pharmaceutical Research, National Health Research Institutes, Taiwan. The compound libraries used in this primary screen were purchased from Chemical Diversity Lab (San Diego, CA) and had a purity of >95%. Compounds from a compound library with diverse structures were provided as dimethyl sulfoxide (DMSO) stock solutions at a concentration of 10 mM. Stable dengue replicon cells were plated at a density of 4 × 103 cells per well in 96-well plates. The following day, the stable replicon cells were treated with various compounds at 10 μM or with DMSO as a control at 37°C for 24 h. The treated stable dengue replicon cells were washed with phosphate-buffered saline (PBS) and lysed with 20 μl/well of 1× lysis buffer (Promega; Renilla luciferase assay system E2820), and the resultant luminescence was measured using Paradigm Luminescence (Beckman). Nucleoside analog inhibitors of NS5, such as 2′-C-methyladenosine (50% effective concentration [EC50] = 4.58 ± 0.77 μM), BP2109 (EC50 = 1.21 ± 0.30 μM), and mycophenolic acid (EC50 = 0.90 ± 0.11 μM), were used as positive controls (73). The luminescence readings were plotted against the log transformation of the concentration of the compound, and a sigmoidal curve fit with a variable slope was generated to determine the EC50 using Prism version 5 software (GraphPad Software, San Diego, CA). Each data set was fitted separately. The obtained data were reproducible, with less than a 15% difference being observed in independent experiments. The standard deviation for each data point was below 15% of the EC50. The results are presented as the means ± standard errors of the means (SEM) of duplicate determinations from three independent experiments.
Cytotoxicity assay.
The sensitivity of the cell lines to BP13944 was examined using an MTS-based tetrazolium reduction assay, the CellTiter 96 AQueous nonradioactive cell proliferation assay (Promega G5430). The BHK-21 cells were plated at a density of 1 × 104 cells per well in 96-well plates containing 120 μl of culture medium followed by incubation for 6 h. Then, one of the serially diluted compounds or DMSO (positive control) was added, followed by incubation for an additional 72 h. Next, the MTS reagent was added to each well, followed by incubation for 1 h at 37°C in a humidified 5% CO2 atmosphere prior to reading at a wavelength of 490 nm using an enzyme-linked immunosorbent assay (ELISA) plate reader. All data are presented as the means ± SEM from three independent experiments.
Plaque-forming assay.
BHK-21 cells were plated at a density of 2 × 105 cells per well in 6-well plates containing 1 ml of culture medium and incubated overnight, after which 0.1 ml of a serially diluted virus solution was added to ∼70 to 80% confluent BHK-21 cells. After adsorption for 2 h, the virus solutions were replaced with either 0.75% methyl cellulose (Sigma; M-0512)-containing DMEM with 2% FBS for DENV-2-infected cells or 1.2% methyl cellulose-containing DMEM with 2% FBS for JEV-infected cells. On the sixth day postinfection, the methyl cellulose solution was removed from the wells, and the cells were fixed and stained with crystal violet solution (1% crystal violet, 0.64% NaCl, and 2% formaldehyde) (74).
Viral yield reduction assay in cultured cells.
BHK-21 cells were plated at a density of 1 × 105 cells per well in 12-well plates containing 0.5 ml of culture medium, followed by incubation for 4 h at 37°C. The tested compounds (500 μl) were added to the wells 16 h prior to the addition of DENV-2 or JEV. The plates were then incubated for an additional 72 h at 37°C in a humidified 5% CO2 atmosphere. To quantify the viral yield in the cells in the presence of BP13944, the supernatant of cells treated with this compound were harvested and subjected to viral titer determination via a plaque-forming assay conducted in BHK-21 cells. The mean values and SEM were calculated from three independent experiments. The detection limit was set to 10 PFU/ml.
Quantitative RT-PCR.
To quantify the viral positive-strand RNA present in the infected BHK-21 cells, RNA was isolated from the above-mentioned cell samples using the Qiagen RNeasy kit, as described in the manufacturer's protocol. The viral RNA was reverse transcribed to cDNA using the Invitrogen ThermoScript reverse transcription (RT) kit with the specific primer DV2.PS-R for detection of the positive-sense viral RNA, based on the manufacturer's protocol and as described previously (75), with slight modifications (all primer sequences are in Table S1 in the supplemental material). Real-time quantitative PCR was conducted to quantify viral RNA. Each reaction was conducted in a 10-μl volume comprising 5 μl of cDNA, 1× TaqMan master mix (Roche), 200 nM each primer (DV2.U2-F and DV2.L1-R), and a 50 nM concentration of the hydrolysis probe, DV2.P1 (76) (TIB MOLBIOL). The LightCycler TaqMan master kit (Roche Biochemicals) and LightCycler 1.5 instrument (Roche Biochemicals) were utilized in this study under the following conditions: preincubation at 95°C for 10 min, followed by 45 cycles of three-step incubations at 95°C for 15 s (denaturation), 60°C for 30 s (annealing and elongation), and 72°C for 1 s (to complete elongation, with a single fluorescence measurement). A linear relationship was established between the RNA copy numbers per milliliter and the corresponding threshold cycle (CT) value over seven logs of the RNA concentration (correlation coefficient, r = 0.99) (76).
RNA transcription and transfection.
The plasmid containing the replicon cDNA was linearized with XbaI. The DNA was then phenol-chloroform extracted, precipitated, and used as a template for in vitro transcription with the SP6 Message mMachine kit (Ambion). RNA transcript levels were quantified with a spectrophotometer, and the RNA was stored at −80°C. The RNA was subsequently transfected into BHK-21 cells using Lipofectamine 2000 (Invitrogen), according to the manufacturer's protocol.
To perform luciferase assays, at each indicated time point, the medium was removed, and the cells were washed with PBS, lysed by adding 100 μl of lysis buffer (Promega; Renilla luciferase assay system), and assayed according to the manufacturer's protocol (Promega; Renilla luciferase assay system). Duplicate wells were lysed at the indicated times and measured using a GloMAX 20/20 luminometer (Promega). To quantify the replication efficiency, the relative luciferase activity was normalized to the signal at 4 h posttransfection.
Transient replicon assay.
A transient replicon assay was performed to quantify the compound-mediated inhibition of viral translation and the reduction of the viral RNA synthesis (77). BHK-21 cells were seeded in 24-well plates (2 × 104 cells per well), followed by incubation overnight. Next, Lipofectamine 2000 (Invitrogen) was mixed with 0.5 μg of the wild type or substitution dengue replicon RNA for one-well transfection, and the cells were transfected according to the manufacturer's instructions. Either BP13944 (8 μM) or the control medium was then added to each well, which were assayed for luciferase activities at the indicated times. Duplicate wells were lysed to perform luminometry. For the luciferase assays, 10 μl of lysate was mixed with 50 μl of the Renilla luciferase assay reagent (Promega). For quantification of compound-mediated inhibition, the relative luciferase activity derived from the mock-treated cells was set as 100% (78).
Isolation and characterization of resistant replicons.
The selection of resistant replicon cells was performed by growing the dengue replicon cells in media containing an appropriate concentration of hit compounds. Media containing the compounds were added to monolayers of dengue replicon cells at ∼25% confluence in the presence of 0.5 to 1 mg/ml G418. Replicon cells maintained in the presence of DMSO were used as a control. After 6 to 8 weeks, total RNA from a pool of BP13944-resistant replicon cells was isolated from the control replicon cells, and the homogeneous cell lines were treated with the compounds using TRIzol (Invitrogen, Carlsbad, CA), according to the manufacturer's protocol. The RNA was then amplified via RT-PCR, and the NS1 to NS5B PCR products were gel purified and subcloned to the pCMV-DV2Rep vector (73) to replace the parental NS1 to NS5B sequence through homologous recombination in yeast. Finally, BP13944 drug-resistant replicon plasmids were purified from the yeast cells and reamplified in E. coli strain C41. After enzyme digestion, only replicon plasmids harboring the right inserts were subjected to DNA sequencing analyses.
Construction of the DENV-2 replicon and an infectious clone containing E66G substitution in the NS3 protease domain.
To generate the E66G substitution reporter replicon and infectious clone (full-length DENV cDNA clone), the E66G substitution was introduced into the NS3 protease domain of the replicon and infectious clone. Two primer pairs, D2/3817-F and E66G-R and E66G-F and D2/5800-R, were used to generate the fragments 3817-E66G and E66G-5800. These two PCR products were gel purified and joined via overlapping PCR to form the fragment containing the E66G substitution (3817-E66G-5800) for homologous recombination with the linearized DV2Rep (39) and DENV-2 infectious clone plasmids (79) (digested with MfeI and SmaI). The E66G substitution replicon and infectious clone plasmids purified from yeast cells were reamplified and maintained in E. coli strain C41. All the constructs were sequenced to confirm the presence of the desired substitution and to exclude external changes.
Construction of the recombinant NS2B/NS3 E66G substitution protease.
The soluble recombinant NS2B/NS3pro protease complex is comprised of the partial NS2B cofactor (residues 49 to 92) and the partial NS3 protein, NS3 protease domain (residues 1 to 184). To generate the E66G substitution protease plasmid, the E66G substitution was introduced into the protease domain of the pGEX4T.NS2B/NS3pro plasmid using the E66G-F and E66G-R primers. The parental and E66G substitution proteases were then purified as described previously (39).
NS2B/NS3pro protease activity assay.
Protease activity was assayed in 50 mM Tris buffer, pH 9.0, containing 10 mM NaCl and 20% glycerol. Parental or E66G substitution protease (1 μM) was preincubated with various test compounds (10 μM) at 37°C for 10 min and initiated by the addition of synthetic peptide substrate Ac-TTSTRR-pNA (38) to a final 800 μM for an additional 2 h of incubation time. Reaction was stopped by sodium citrate, pH 4.5 (625 μM), and read at A405 on a Multilabel HTS counter of Wallac Victor2 (PerkinElmer). Aprotinin (3 μM) was used as a positive control. Fifty-percent inhibitory concentrations (IC50s) were determined as described previously (38) using Prism version 5 software (GraphPad Software, San Diego, CA) and measured at least twice from independent triplicate experiments. The results represent the means ± SEM from duplicate determinations from three independent experiments.
Statistical analysis.
The data were compared in parallel with Prism version 5 software (GraphPad Software, San Diego, CA). The statistical significance of the differences between the means of the experimental groups was analyzed using Student's t test with Welch's correction to measure the two-tailed P values. P values less than 0.05 were considered statistically significant.
RESULTS
Identification of BP13944 as an inhibitor of the DEN virus.
To search for novel inhibitors of the dengue virus, we performed a cell-based high-throughput screening (HTS) assay, as described previously (73), and screened approximately 60,000 small molecules at a single concentration of 10 μM. A BHK-21 cell line containing a DENV-2 Renilla luciferase replicon was used in the HTS. HTS was performed in a 96-well format with a signal-to-noise ratio of 466.7 and a Z′ value of 0.74. The compounds that reduced luciferase activity by ≥90% with a 50% cytotoxic concentration (CC50) of ≥15 μM were defined as “hits.” The applied HTS procedure resulted in a hit rate of 0.02%.
About 12 inhibitor candidates were obtained after a second screening by their inhibitory effect on DENV yield. One of the inhibitor candidates, BP13944 (a quaternary ammonium salt; Fig. 1), is similar to the structure of BP2109, an NS2B/3 protease inhibitor, in our previous report (39). BP13944 caused inhibition in the cell-based replicon assay (EC50 = 1.03 ± 0.09 μM) and anti-DENV effects in the virus yield reduction assays (Fig. 2). The DMSO- and BP13944-treated BHK-21 cells were infected with the DENV-2 virus (MOI of 0.1 or 1), and the viral titers in the culture medium were assayed at 72 h postinfection. The compound reduced viral yields in the cells infected at MOIs of both 0.1 and 1.0 in a dose-dependent manner. A more than 10,000-fold reduction in the viral titer was observed when the medium contained 12 μM BP13944. These results indicate that BP13944 is a potential inhibitor of DENV-2.
FIG 1.
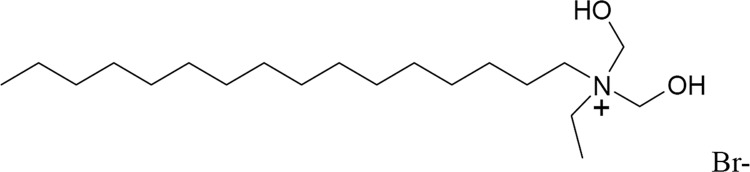
Structure of BP13944. A potential DENV inhibitor identified from the screening.
FIG 2.

BP13944 suppresses the viral yields of DENV-2. Mock- and compound-pretreated BHK-21 cells were infected with DENV-2 at an MOI of 0.1 (filled circles) or 1.0 (open circles) in the absence or presence of BP13944 and analyzed to determine the viral yield in the culture medium at 72 h postinfection. Error bars indicate the standard errors of the means (SEM) from three independent experiments.
Dose-dependent inhibition of all four DENV serotypes by BP13944.
To verify that the primary observed antiviral activity was not due to compound-mediated cytotoxicity, we performed a cytotoxicity assay based on the cellular metabolism of an MTS-based tetrazolium salt. No suppression of cell viability was observed when the cells were incubated with 40 μM BP13944. The CC50 of the compound was found to be 72.40 ± 0.95 μM. To eliminate any possible BP13944-mediated cytotoxicity, the antiviral activity of BP13944 was tested at concentrations below 15 μM (resulting in 100% cell viability). BP13944 must be active against all four serotypes of DENV for it to be considered a valuable anti-DENV agent. We performed the viral yield reduction assay in parallel against all four serotypes of DENV. As illustrated in Fig. 3, 8 μM BP13944 greatly decreased the viral titers of all four DENV serotypes in the culture medium. The viral yields of DENV-1, -2, -3, and -4 were decreased 457-, 14,333-, 801-, and 3,826-fold, respectively, compared to the respective DMSO-treated controls. These results indicate that BP13944 is a potentially therapeutic compound against all four serotypes of DENV.
FIG 3.

BP13944 inhibits all four serotypes of DENV. BHK-21 cells were incubated with 8 μM BP13944 and infected with the four serotypes of DENV at an MOI of 0.1. The viral yield in culture medium was determined via plaque formation assays at 72 h postinfection. The mean values and SEM from three independent experiments are plotted. **, P < 0.0001.
BP13944 selectively reduces the viral yield of DENV.
To examine the antiviral spectrum of BP13944, the viral yield reduction assay was also performed against JEV. BHK-21 cells were infected with JEV or DENV (MOI of 0.1) in the presence or absence of 12 μM BP13944. When the effects of the compound on the two viruses were compared, it was found that the viral yield of JEV did not show the marked suppression observed for DENV (Fig. 4).
FIG 4.

BP13944 selectively inhibits the viral yield and NS2B/NS3 protease activity of DNEV. BHK-21 cells infected with JEV or DENV-2 at an MOI of 0.1 were maintained in the absence or presence of 12 μM BP13944. The viral yields in the culture medium at 72 h postinfection were determined via plaque formation assays. The mean values and SEM from three independent experiments are plotted. ***, P < 0.0001.
BP13944 reduces the level of viral RNAs in cells infected with DENV-2.
To determine which steps of the DENV life cycle are suppressed by BP13944, we performed quantitative RT-PCR to verify whether BP13944 affects viral RNA replication. After BHK-21 cells had been treated with 12 μM BP13944 for 16 h, the cells were infected with DENV-2 and then analyzed to determine the viral RNA levels in the cells at 72 h postinfection. As shown in Fig. 5, the cells infected at MOIs of 0.1 and 1.0 and treated with BP13944 displayed 7- and 17-fold reductions in positive-strand viral RNA levels, respectively. The results implied that BP13944 reduced the replication of DENV-2 viral RNA by affecting the synthesis of positive-strand DENV-2 RNA.
FIG 5.

Effect of BP13944 on the replication of DENV-2 viral RNA. BHK-21 cells infected with DENV-2 at an MOI of 0.1 and 1.0 were maintained in the absence (open bars) or presence (filled bars) of 12 μM BP13944 for 72 h. The accumulation of positive-strand DENV-2 RNA in the cells was determined via TaqMan fluorogenic quantitative RT-PCR. RNA copy numbers were determined using standards, which were measured with a spectrophotometer. The mean values and SEM from three independent experiments are plotted. **, P < 0.005.
BP13944 inhibits the replication activity of cells harboring the DENV-2 replicon.
To further validate the mechanistic function of BP13944, the DENV-2 replicon was transcribed in vitro and transfected into BHK-21 cells to distinguish the inhibition of the viral translation from the inhibition of RNA synthesis. Luciferase activity was monitored at 4, 8, 24, 48, and 72 h posttransfection (Fig. 6). The luciferase activity reached a peak level at approximately 48 h posttransfection and was maintained at that level until 72 h posttransfection in the absence of BP13944. Because the luciferase activity peaked both within the first 8 h posttransfection and after 24 h posttransfection (representing viral translation and RNA replication, respectively [61]), we measured luciferase activity at 4, 8, 24, 48, and 72 h posttransfection. BP13944 had a minimal effect on the Rluc signal detected at 4 and 8 h posttransfection. However, the signal was significantly reduced by 49%, 73%, and 91% at 24, 48, and 72 h posttransfection, respectively (Fig. 6). Our data demonstrated strong suppression of viral RNA synthesis by BP13944.
FIG 6.

Inhibition of DENV-2 viral RNA replication by BP13944. Analysis of BP13944 through transient replicon assays. BP13944 inhibits viral replication stages (24 to 72 h) rather than viral translation stages (4 to 8 h). The replicon was transfected into BHK-21 cells maintained in the absence or presence of 8 μM BP13944. Luciferase activity was monitored at the indicated times posttransfection. The numbers above the BP13944 treatment times represent the percentages of the luciferase signals relative to the mock-treated controls (100%), and the error bars represent the SEM from three independent experiments. *, P < 0.05; **, P < 0.005.
Selection and characterization of BP13944-resistant replicon cells.
To identify the antiviral mechanism and the molecular target of BP13944, we obtained the BP13944-resistant replicon cells by serially passaging DENV-2 replicon cells in the presence of 0.5 mg/ml G418 and increasing the concentrations of BP13944 (1 to 5 μM). Replicon cells maintained in the presence of DMSO were used as a control. Compared to the parental cells, the EC50s for the parental and a pool of BP13944-resistant replicon cells were calculated as 1.03 ± 0.09 μM and 13.93 ± 0.92 μM, respectively, in experiments performed in parallel. The BP13944-resistant replicon cells exhibited 13.5-fold higher resistance than the parental replicon cells. The total viral RNAs from a pool of BP13944-resistant replicon cells was isolated and amplified by RT-PCR, and the PCR products for the NS1 to NS5 region were cloned into yeast cells. Plasmids were then purified from 10 independent yeast colonies and amplified in E. coli cells. Only five plasmids from 10 E. coli colonies harboring the full-length NS1 to NS5 insert were identified and subjected to DNA sequencing analyses. The obtained sequencing data (NS1 to NS5) revealed that three of the five clones had accumulated a consensus amino acid substitution (E66G) in the central region of the NS3 protease domain (Table 1). The other mutations were found to be sporadic mutations among the NS1 to NS5 region. Interestingly, 12 out of a total of 25 mutations were located within the NS3 gene. The DNA sequencing results implied that the consensus E66G substitution may be related to the drug resistance to BP13944.
TABLE 1.
Sequencing analyses of the dengue NS1 to NS5 genes from clones derived from BP13944-resistant DENV replicons
| Sequence | Encoded amino acid at the indicated position |
||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NS1 |
NS2A |
NS2B | NS3 |
NS4B | NS5 |
||||||||||||||||||
| 9 | 150 | 185 | 257 | 68 | 122 | 201 | 17 | 13 | 30 | 66 | 137 | 203 | 224 | 279 | 410 | 473 | 600 | 239 | 67 | 289 | 399 | 402 | |
| Parental | K | W | S | R | E | L | L | M | I | E | S | Y | T | N | I | I | I | E | E | F | K | ||
| Clone | F | ||||||||||||||||||||||
| 1 | R | G | H | G | K | ||||||||||||||||||
| 2 | L | L | G | P | V | S | E | ||||||||||||||||
| 3 | R | G | S | T | G | T | F | ||||||||||||||||
| 4 | V | P | S | ||||||||||||||||||||
| 5 | G | V | |||||||||||||||||||||
The E66G substitution in the NS3 protease domain confers resistance to BP13944 on the DENV replicon, recombinant DENV, and recombinant NS2B/NS3 protease complex.
To identify the determinant of BP13944 resistance, the E66G substitution was introduced into the NS3 protease domain of the parental DV2Rep replicon (39). The E66G substitution in the NS3 protease region reduced the BP13944 susceptibility of the E66G substitution replicon. The EC50s for the parental and substitution replicons were calculated as 0.76 ± 0.25 μM and 11.54 ± 2.91 μM, respectively, in the transient replicon assay. The E66G substitution replicon exhibited 15.2-fold-higher resistance than the parental replicon (Fig. 7A). The E66G substitution was also introduced into the NS3 protease domain of the parental infectious DENV-2 cDNA clone (79), and similar results were observed in this DENV-2 infectious clone system. The recombinant E66G substitution DENV showed a resistance level that was higher than that of the parental DENV. The EC50s for the parental and recombinant E66G substitution DENV strains were calculated as 0.23 ± 0.01 μM and 3.95 ± 0.72 μM, respectively (Fig. 7B). The EC50 of the recombinant E66G substitution DENV was approximately 17.2-fold higher than that of the parental DENV. We further prepared a recombinant NS2B/NS3 protease consisting of the partial NS2B cofactor (residues 49 to 92), the NS3 protease domain (residues 1 to 184) with or without the E66G substitution. The amino acid E66G substitution in the NS3 protease region reduced the BP13944 susceptibility of the E66G substitution protease. The IC50s for the parental and E66G substitution proteases were calculated as 22.63 ± 0.74 μM and 71.10 ± 0.79 μM, respectively (Fig. 7C). The E66G substitution protease exhibited a 3.1-fold-higher resistance than that of the parental protease. The glycine substitution at residue 66 in the NS3 protease region is therefore crucial for drug resistance. Thus, our results from three different assay systems indicated that the residue at position 66 in the NS3 protease region is the major determinant of BP13944 resistance and suggested that the molecular target of BP13944 may be the NS3 protein, possibly NS3 protease.
FIG 7.

The E66G substitution within the DENV NS3 protease domain in the DENV replicon, infectious cDNA clone, or NS2B/NS3 protease results in BP13944 resistance. (A) BP13944 susceptibility of the parental and E66G substitution DENV replicon. The curves were fitted based on the transient replication activity assay as percentages of the control value (DMSO-treated controls). BHK-21 cells were electroporated with equal amounts (100 ng/2 × 105 cells) of parental or E66G substitution replicon RNA and treated with BP13944 at the indicated concentrations. (B) Resistance analyses of the recombinant parental and E66G substitution DENV. BHK-21 cells were infected with parental and E66G substitution DENVs at an MOI of 0.1 and treated with BP13944 at the indicated concentrations. The EC50s were determined by calculating the viral yield at 72 h postinfection via plaque formation assays. (C) BP13944 susceptibility of the parental and E66G substitution NS2B/NS3 proteases. The curves were fitted based on the observed enzyme activity as a percentage of the control value (DMSO treated). The IC50s of the parental and E66G substitution NS2B/NS3 proteases, corresponding to a 50% reduction in the protease activity assay, were calculated as 22.63 ± 0.74 μM and 71.10 ± 0.79 μM, respectively. (D) Multiplication kinetics of parental and E66G substitution DENVs in BHK-21 cells. Growth curves were conducted at an MOI of 0.1, and the limit of detection was ≦10 PFU/ml. The viral yields in culture medium at each time point were determined by plaque formation assays. The mean values and SEM from three independent experiments are plotted.
DISCUSSION
In this study, we used a stable reporter-DENV replicon cell line to identify BP13944 as a DENV inhibitor that shows a selective antiviral spectrum and inhibitory activity in vitro. We found that BP13944 was an effective inhibitor of the viral yield and viral RNA replication of the four DENV serotypes and of the cytopathology induced by these serotypes. Sequencing analyses of isolated BP13944-resistant replicons revealed that an E66G substitution was frequently present within the NS3 protease region. Introduction of the E66G substitution into parental DENV replicon, infectious cDNA, and NS2B/NS3 protease constructs conferred resistance to BP13944. Because the E66 residue is located within the interface between the protease and helicase domains of the DENV NS3 protein (28, 29), BP13944 likely affects protease function by binding to the interface, thus reducing DENV replication. BP13944 may therefore provide a novel approach for inhibiting the DENV NS3 protease.
The replicon-based approach for screening small-molecule inhibitors of DENV has the potential to identify inhibitors of viral and host targets. The results of two different experimental methods indicate that BP13944 might target the DENV NS3 protease. First, BP13944 selectively inhibits all four serotypes of DENV but does not inhibit the closely related JEV (Fig. 3 and 4). This specific antiviral activity excludes the possibility that DENV inhibition by BP13944 occurs because of compound-mediated cytotoxicity. Second, resistance analyses of BP13944 revealed a consensus amino acid substitution (E66G) in the NS3 protease domain (NS3pro) (Table 1) that was able to confer resistance to BP13944 on the DENV replicon, infectious clone, and recombinant NS2B/NS3 protease constructs (Fig. 7). The fact that the inhibitory effect of BP13944 on DENV-2 and -4 was greater than that on DENV-1 and -3 (Fig. 3) indicates that BP13944 exerts a different degree of inhibition in the different serotypes of DENV and JEV. We suspected that the different susceptibilities of the various serotypes of DENV and JEV to BP13944 may result from differences in the amino acid sequences of the NS3 protease. We aligned and investigated the region of DENV NS3pro that may be responsible for the differential susceptibility of DENV and JEV to BP13944 (see Fig. S1 in the supplemental material). The degree of conservation of the NS3pro amino acid sequence of DENV-1 compared to those of DENV-2, DENV-3, DENV-4, and JEV was shown to correspond to 69% identity (81% similarity), 74% identity (84% similarity), 64% identity (75% similarity), and 53% identity (65% similarity), respectively. In general, the amino acid sequence of NS3pro (residues 1 to 184) is more conserved among DENV serotypes than between DENV and JEV. This finding may explain the differential susceptibilities of DENV and JEV to BP13944. Analysis of the phylogenetic tree constructed from the DENV NS3pro amino acid sequences revealed that two clusters exist, with DENV-2 and DENV-4 being located in one cluster and DENV-1 and DENV-3 in another (data not shown). The more effective inhibitory effect of BP13944 on the viral yields of DENV-2 or -4 than that on DENV-1 or -3 (Fig. 2) may result from the degree of conservation of the NS3pro amino acid sequences of NS3pro in DENV. Further studies are required to identify the binding region of BP13944 to the DENV-2 NS3 protein and to explain the differential susceptibilities of DENV-2 and JEV to BP13944.
The chemical structure of BP13944 consists of a long aliphatic side chain and one quaternary ammonium bromide, which is similar to the structure of the reported DENV NS2B/NS3 protease inhibitor BP2109 (39). BP2109 was identified via HTS using a DENV enzyme-based protease assay in a previous study by our group. There is one interesting observation that the pretreatment of BP13944 in cells is essential for its maximum inhibitory effect of BP13944 on DENV yield (see Fig. S2 in the supplemental material), which is similar to the inhibitory effect of BP2109 on DENV yield (data not shown). We suspected that the requirement of pretreatment in cells of BP13944 and BP2109 may be related to their characteristics of quaternary ammonium salt since charged small molecules affect the diffusion through lipid bilayer environment with difficulty (80). Resistance analyses of BP2109 suggested that an E80K substitution in the NS2B region could confer resistance to BP2109. We found that the E80K substitution does not confer resistance to BP13944 on the DENV replicon in a transient replicon assay (data not shown). In these transient replicon assays, replicons with the E66G substitution in the NS3 protease region did not show altered susceptibility to BP2109 (data not shown). These results implied that BP13944 and BP2109 inhibit the function of the DENV NS3 protease possibly through binding to different amino acid residues of the NS3 protease.
BP13944 appeared to exert a much more potent effect in the DENV-2 viral yield reduction assay (EC50 = 0.23 ± 0.01 μM) than in the enzyme-based protease assay (IC50 = 22.63 ± 0.74 μM) (Fig. 7). A similar phenomenon was observed regarding the inhibition of the DENV replication by BP2109. This inconsistency likely occurred because of the applied artificial enzyme-based protease assay, which is performed using the soluble recombinant NS2B/NS3pro protease complex, consisting of only the partial NS2B cofactor (residues 49 to 92) and the partial NS3 protein (protease domain, residues 1 to 184). The activity of the artificial NS2B/NS3pro protease complex may not fully represent the physiological function of the full-length NS2B/NS3 in vivo. The finding that glycerol or gelatin enhances the activity of the NS2B/NS3pro protease complex is consistent with this hypothesis (38). Because the three transmembrane helices of NS2B that anchor the active NS2B/NS3 protease to the endoplasmic reticulum (ER) are absent (8), the in vivo environment of the protease is completely different from the in vitro artificial assay system, which supports the above-given hypothesis. The recently reported crystal structure of full-length DENV NS3 (28, 29) suggests that the protease domain of NS3 increases its affinity for nucleotides, participates in RNA binding, and enhances the helicase activity of the protein. Studies addressing the HCV NS3 protease (81), in which the helicase domain of NS3 has been found to enhance the enzyme-based protease activity of NS3/NS4A, support the interdependency of the protease domain and helicase domain of the NS3 protein complex. It is likely that the two enzymatic domains of DENV NS3 are highly interdependent in the virus life cycle. These results may explain why BP13944 appeared to be more potent in the viral yield reduction assay than in the enzyme-based protease assay.
Our findings that BP13944 strongly blocks DENV RNA synthesis (>91%) without suppressing viral translation in transient DENV-2 replicon-based kinetics assays (Fig. 6) and that the E66G substitution in the NS3 region confers resistance to BP13944 (Fig. 7) suggested that BP1944 inhibits DENV by targeting the NS3 protease. Further kinetic analysis suggested that BP13944 may inhibit the NS2B/NS3 protease-substrate complex through an uncompetitive mechanism (data not shown), followed by the suppression of polypeptide processing and blockage of viral RNA replication. A similar inhibitory mechanism was suggested in a study addressing a novel noncompetitive inhibitor of the HCV NS3/NS4A protease discovered in an X-ray crystallographic fragment screen using the crystal structure of the full-length HCV NS3/NS4A holoenzyme (82). This HCV protease inhibitor blocks the function of the NS3 protein by binding to the interface between the protease and helicase domains of the HCV NS3 protein. Two facts support the idea that BP13944 may have a similar inhibitory mechanism as the HCV NS3/NS4A protease inhibitor (82). The first is that the key BP13944-resistant residue E66 was located at the interface between the protease and helicase domain of the DENV NS3 protein (83). The second is that BP13944 was not apparently affecting the DENV NS3 helicase activity (IC50 > 50 µM) (see Fig. S3 in the supplemental material). We suspect that the molecular mechanism by which BP13944 inhibits DENV is either through interfering with NS3 protease activity by binding to the interface between NS3pro and NS3 helicase domains or acting as a protease inhibitor that abolishes proteolytic activity by directly binding to the active site. Interestingly, the results (Fig. 5) showed that BP13944 reduced the level of DENV RNA accumulation and the production of virus particles by ∼1 log and ∼3 logs, respectively. It implied that BP13944 possibly also interferes with a stage of virus life cycle other than the stage of viral RNA replication. Further studies are needed to clarify the mode of action of BP13944.
The identification and characterization of BP13944 represents the first step in the development of this compound as a potential anti-DENV therapeutic agent. Based on recent experimental, crystallographic, computational docking, and mutagenesis analyses of HCV and/or DENV NS3 proteins, we propose that BP13944 displays an inhibitory mechanism, interfering with NS3 protease activity, possibly rather than binding to the active site of the NS3 protease. Further work is required to determine the detailed molecular mechanism by which BP13944 interacts with NS3 protein and inhibits DENV replication. Understanding the interaction between BP13944 and the NS3 protein will facilitate the development of additional novel DENV inhibitors.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Science Council of the Republic of China (grant no. NSC 101-2325-B-400-006).
Footnotes
Published ahead of print 21 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01281-13.
REFERENCES
- 1.Gubler DJ, Kuno G, Markoff L. 2007. Flaviviruses, p 1153–1253 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Ye J, Zhu B, Fu ZF, Chen H, Cao S. 2012. Immune evasion strategies of flaviviruses. Vaccine 31:461–471. 10.1016/j.vaccine.2012.11.015 [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization 2012. Dengue and severe dengue. World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs117/en/ [Google Scholar]
- 4.World Health Organization 2009. Dengue: guidelines for diagnosis, treatment, prevention and control. World Health Organization, Geneva, Switzerland: http://whqlibdoc.who.int/publications/2009/9789241547871_eng.pdf [PubMed] [Google Scholar]
- 5.Halstead SB. 1980. Dengue haemorrhagic fever—a public health problem and a field for research. Bull. World Health Organ. 58:1–21 [PMC free article] [PubMed] [Google Scholar]
- 6.Gubler DJ. 1989. Aedes aegypti and Aedes aegypti-borne disease control in the 1990s: top down or bottom up. Charles Franklin Craig lecture. Am. J. Trop. Med. Hyg. 40:571–578 [DOI] [PubMed] [Google Scholar]
- 7.Pinheiro FP. 1989. Dengue in the Americas. 1980–1987. Epidemiol. Bull. 10:1–8 [PubMed] [Google Scholar]
- 8.Noble CG, Chen YL, Dong H, Gu F, Lim SP, Schul W, Wang QY, Shi PY. 2010. Strategies for development of dengue virus inhibitors. Antiviral Res. 85:450–462. 10.1016/j.antiviral.2009.12.011 [DOI] [PubMed] [Google Scholar]
- 9.Simmons CP, Farrar JJ, Nguyen v V, Wills B. 2012. Dengue. N. Engl. J. Med. 366:1423–1432. 10.1056/NEJMra1110265 [DOI] [PubMed] [Google Scholar]
- 10.Rice CM, Lenches EM, Eddy SR, Shin SJ, Sheets RL, Strauss JH. 1985. Nucleotide sequence of yellow fever virus: implications for flavivirus gene expression and evolution. Science 229:726–733. 10.1126/science.4023707 [DOI] [PubMed] [Google Scholar]
- 11.Chambers TJ, Hahn CS, Galler R, Rice CM. 1990. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 44:649–688. 10.1146/annurev.mi.44.100190.003245 [DOI] [PubMed] [Google Scholar]
- 12.Falgout B, Pethel M, Zhang YM, Lai CJ. 1991. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J. Virol. 65:2467–2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kummerer BM, Rice CM. 2002. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J. Virol. 76:4773–4784. 10.1128/JVI.76.10.4773-4784.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu WJ, Chen HB, Khromykh AA. 2003. Molecular and functional analyses of Kunjin virus infectious cDNA clones demonstrate the essential roles for NS2A in virus assembly and for a nonconservative residue in NS3 in RNA replication. J. Virol. 77:7804–7813. 10.1128/JVI.77.14.7804-7813.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patkar CG, Kuhn RJ. 2008. Yellow fever virus NS3 plays an essential role in virus assembly independent of its known enzymatic functions. J. Virol. 82:3342–3352. 10.1128/JVI.02447-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Best SM, Morris KL, Shannon JG, Robertson SJ, Mitzel DN, Park GS, Boer E, Wolfinbarger JB, Bloom ME. 2005. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J. Virol. 79:12828–12839. 10.1128/JVI.79.20.12828-12839.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo JT, Hayashi J, Seeger C. 2005. West Nile virus inhibits the signal transduction pathway of alpha interferon. J. Virol. 79:1343–1350. 10.1128/JVI.79.3.1343-1350.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu WJ, Wang XJ, Mokhonov VV, Shi PY, Randall R, Khromykh AA. 2005. Inhibition of interferon signaling by the New York 99 strain and Kunjin subtype of West Nile virus involves blockage of STAT1 and STAT2 activation by nonstructural proteins. J. Virol. 79:1934–1942. 10.1128/JVI.79.3.1934-1942.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munoz-Jordan JL, Laurent-Rolle M, Ashour J, Martinez-Sobrido L, Ashok M, Lipkin WI, Garcia-Sastre A. 2005. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J. Virol. 79:8004–8013. 10.1128/JVI.79.13.8004-8013.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Munoz-Jordan JL, Sanchez-Burgos GG, Laurent-Rolle M, Garcia-Sastre A. 2003. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. U. S. A. 100:14333–14338. 10.1073/pnas.2335168100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wengler G, Wengler G. 1991. The carboxy-terminal part of the NS 3 protein of the West Nile flavivirus can be isolated as a soluble protein after proteolytic cleavage and represents an RNA-stimulated NTPase. Virology 184:707–715. 10.1016/0042-6822(91)90440-M [DOI] [PubMed] [Google Scholar]
- 22.Falgout B, Miller RH, Lai CJ. 1993. Deletion analysis of dengue virus type 4 nonstructural protein NS2B: identification of a domain required for NS2B-NS3 protease activity. J. Virol. 67:2034–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wengler G, Wengler G. 1993. The NS3 nonstructural protein of flaviviruses contains an RNA triphosphatase activity. Virology 197:265–273. 10.1006/viro.1993.1587 [DOI] [PubMed] [Google Scholar]
- 24.Bazan JF, Fletterick RJ. 1989. Detection of a trypsin-like serine protease domain in flaviviruses and pestiviruses. Virology 171:637–639. 10.1016/0042-6822(89)90639-9 [DOI] [PubMed] [Google Scholar]
- 25.Gorbalenya AE, Donchenko AP, Koonin EV, Blinov VM. 1989. N-terminal domains of putative helicases of flavi- and pestiviruses may be serine proteases. Nucleic Acids Res. 17:3889–3897. 10.1093/nar/17.10.3889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valle RP, Falgout B. 1998. Mutagenesis of the NS3 protease of dengue virus type 2. J. Virol. 72:624–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu T, Sampath A, Chao A, Wen D, Nanao M, Chene P, Vasudevan SG, Lescar J. 2005. Structure of the dengue virus helicase/nucleoside triphosphatase catalytic domain at a resolution of 2.4 A. J. Virol. 79:10278–10288. 10.1128/JVI.79.16.10278-10288.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo D, Xu T, Hunke C, Gruber G, Vasudevan SG, Lescar J. 2008. Crystal structure of the NS3 protease-helicase from dengue virus. J. Virol. 82:173–183. 10.1128/JVI.01788-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luo D, Wei N, Doan DN, Paradkar PN, Chong Y, Davidson AD, Kotaka M, Lescar J, Vasudevan SG. 2010. Flexibility between the protease and helicase domains of the dengue virus NS3 protein conferred by the linker region and its functional implications. J. Biol. Chem. 285:18817–18827. 10.1074/jbc.M109.090936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Menendez-Arias L. 2010. Molecular basis of human immunodeficiency virus drug resistance: an update. Antiviral Res. 85:210–231. 10.1016/j.antiviral.2009.07.006 [DOI] [PubMed] [Google Scholar]
- 31.Venkatraman S. 2012. Discovery of boceprevir, a direct-acting NS3/4A protease inhibitor for treatment of chronic hepatitis C infections. Trends Pharmacol. Sci. 33:289–294. 10.1016/j.tips.2012.03.012 [DOI] [PubMed] [Google Scholar]
- 32.Casey LC, Lee WM. 2013. Hepatitis C virus therapy update 2013. Curr. Opin. Gastroenterol. 29:243–249. 10.1097/MOG.0b013e32835ff972 [DOI] [PubMed] [Google Scholar]
- 33.Marks KM, Jacobson IM. 2012. The first wave: HCV NS3 protease inhibitors telaprevir and boceprevir. Antivir. Ther. 17:1119–1131. 10.3851/IMP2424 [DOI] [PubMed] [Google Scholar]
- 34.Noble CG, Shi PY. 2012. Structural biology of dengue virus enzymes: towards rational design of therapeutics. Antiviral Res. 96:115–126. 10.1016/j.antiviral.2012.09.007 [DOI] [PubMed] [Google Scholar]
- 35.Sampath A, Padmanabhan R. 2009. Molecular targets for flavivirus drug discovery. Antiviral Res. 81:6–15. 10.1016/j.antiviral.2008.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lescar J, Luo D, Xu T, Sampath A, Lim SP, Canard B, Vasudevan SG. 2008. Towards the design of antiviral inhibitors against flaviviruses: the case for the multifunctional NS3 protein from dengue virus as a target. Antiviral Res. 80:94–101. 10.1016/j.antiviral.2008.07.001 [DOI] [PubMed] [Google Scholar]
- 37.Bollati M, Alvarez K, Assenberg R, Baronti C, Canard B, Cook S, Coutard B, Decroly E, de Lamballerie X, Gould EA, Grard G, Grimes JM, Hilgenfeld R, Jansson AM, Malet H, Mancini EJ, Mastrangelo E, Mattevi A, Milani M, Moureau G, Neyts J, Owens RJ, Ren J, Selisko B, Speroni S, Steuber H, Stuart DI, Unge T, Bolognesi M. 2010. Structure and functionality in flavivirus NS-proteins: perspectives for drug design. Antiviral Res. 87:125–148. 10.1016/j.antiviral.2009.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leung D, Schroder K, White H, Fang NX, Stoermer MJ, Abbenante G, Martin JL, Young PR, Fairlie DP. 2001. Activity of recombinant dengue 2 virus NS3 protease in the presence of a truncated NS2B co-factor, small peptide substrates, and inhibitors. J. Biol. Chem. 276:45762–45771. 10.1074/jbc.M107360200 [DOI] [PubMed] [Google Scholar]
- 39.Yang CC, Hsieh YC, Lee SJ, Wu SH, Liao CL, Tsao CH, Chao YS, Chern JH, Wu CP, Yueh A. 2011. Novel dengue virus-specific NS2B/NS3 protease inhibitor, BP2109, discovered by a high-throughput screening assay. Antimicrob. Agents Chemother. 55:229–238. 10.1128/AAC.00855-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deng J, Li N, Liu H, Zuo Z, Liew OW, Xu W, Chen G, Tong X, Tang W, Zhu J, Zuo J, Jiang H, Yang CG, Li J, Zhu W. 2012. Discovery of novel small molecule inhibitors of dengue viral NS2B-NS3 protease using virtual screening and scaffold hopping. J. Med. Chem. 55:6278–6293. 10.1021/jm300146f [DOI] [PubMed] [Google Scholar]
- 41.Xu S, Li H, Shao X, Fan C, Ericksen B, Liu J, Chi C, Wang C. 2012. Critical effect of peptide cyclization on the potency of peptide inhibitors against dengue virus NS2B-NS3 protease. J. Med. Chem. 55:6881–6887. 10.1021/jm300655h [DOI] [PubMed] [Google Scholar]
- 42.Rothan HA, Han HC, Ramasamy TS, Othman S, Rahman NA, Yusof R. 2012. Inhibition of dengue NS2B-NS3 protease and viral replication in Vero cells by recombinant retrocyclin-1. BMC Infect. Dis. 12:314. 10.1186/1471-2334-12-314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chanprapaph S, Saparpakorn P, Sangma C, Niyomrattanakit P, Hannongbua S, Angsuthanasombat C, Katzenmeier G. 2005. Competitive inhibition of the dengue virus NS3 serine protease by synthetic peptides representing polyprotein cleavage sites. Biochem. Biophys. Res. Commun. 330:1237–1246. 10.1016/j.bbrc.2005.03.107 [DOI] [PubMed] [Google Scholar]
- 44.Ganesh VK, Muller N, Judge K, Luan CH, Padmanabhan R, Murthy KH. 2005. Identification and characterization of nonsubstrate based inhibitors of the essential dengue and West Nile virus proteases. Bioorg. Med. Chem. 13:257–264. 10.1016/j.bmc.2004.09.036 [DOI] [PubMed] [Google Scholar]
- 45.Kiat TS, Pippen R, Yusof R, Ibrahim H, Khalid N, Rahman NA. 2006. Inhibitory activity of cyclohexenyl chalcone derivatives and flavonoids of fingerroot, Boesenbergia rotunda (L.), towards dengue-2 virus NS3 protease. Bioorg. Med. Chem. Lett. 16:3337–3340. 10.1016/j.bmcl.2005.12.075 [DOI] [PubMed] [Google Scholar]
- 46.Yin Z, Patel SJ, Wang WL, Wang G, Chan WL, Rao KR, Alam J, Jeyaraj DA, Ngew X, Patel V, Beer D, Lim SP, Vasudevan SG, Keller TH. 2006. Peptide inhibitors of dengue virus NS3 protease. Part 1: warhead. Bioorg. Med. Chem. Lett. 16:36–39. 10.1016/j.bmcl.2005.09.062 [DOI] [PubMed] [Google Scholar]
- 47.Yin Z, Patel SJ, Wang WL, Chan WL, Ranga Rao KR, Wang G, Ngew X, Patel V, Beer D, Knox JE, Ma NL, Ehrhardt C, Lim SP, Vasudevan SG, Keller TH. 2006. Peptide inhibitors of dengue virus NS3 protease. Part 2: SAR study of tetrapeptide aldehyde inhibitors. Bioorg. Med. Chem. Lett. 16:40–43. 10.1016/j.bmcl.2005.09.049 [DOI] [PubMed] [Google Scholar]
- 48.Bodenreider C, Beer D, Keller TH, Sonntag S, Wen D, Yap L, Yau YH, Shochat SG, Huang D, Zhou T, Caflisch A, Su XC, Ozawa K, Otting G, Vasudevan SG, Lescar J, Lim SP. 2009. A fluorescence quenching assay to discriminate between specific and nonspecific inhibitors of dengue virus protease. Anal. Biochem. 395:195–204. 10.1016/j.ab.2009.08.013 [DOI] [PubMed] [Google Scholar]
- 49.Tomlinson SM, Malmstrom RD, Russo A, Mueller N, Pang YP, Watowich SJ. 2009. Structure-based discovery of dengue virus protease inhibitors. Antiviral Res. 82:110–114. 10.1016/j.antiviral.2009.02.190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Byrd CM, Grosenbach DW, Berhanu A, Dai D, Jones KF, Cardwell KB, Schneider C, Yang G, Tyavanagimatt S, Harver C, Wineinger KA, Page J, Stavale E, Stone MA, Fuller KP, Lovejoy C, Leeds JM, Hruby DE, Jordan R. 2013. Novel benzoxazole inhibitor of dengue virus replication that targets the NS3 helicase. Antimicrob. Agents Chemother. 57:1902–1912. 10.1128/AAC.02251-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu W, Gill T, Wang L, Du Y, Ye H, Qu X, Guo JT, Cuconati A, Zhao K, Block TM, Xu X, Chang J. 2012. Design, synthesis, and biological evaluation of N-alkylated deoxynojirimycin (DNJ) derivatives for the treatment of dengue virus infection. J. Med. Chem. 55:6061–6075. 10.1021/jm300171v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chao B, Tong XK, Tang W, Li DW, He PL, Garcia JM, Zeng LM, Gao AH, Yang L, Li J, Nan FJ, Jacobs M, Altmeyer R, Zuo JP, Hu YH. 2012. Discovery and optimization of 2,4-diaminoquinazoline derivatives as a new class of potent dengue virus inhibitors. J. Med. Chem. 55:3135–3143. 10.1021/jm2015952 [DOI] [PubMed] [Google Scholar]
- 53.Jones CT, Patkar CG, Kuhn RJ. 2005. Construction and applications of yellow fever virus replicons. Virology 331:247–259. 10.1016/j.virol.2004.10.034 [DOI] [PubMed] [Google Scholar]
- 54.Horscroft N, Lai VC, Cheney W, Yao N, Wu JZ, Hong Z, Zhong W. 2005. Replicon cell culture system as a valuable tool in antiviral drug discovery against hepatitis C virus. Antivir. Chem. Chemother. 16:1–12 [DOI] [PubMed] [Google Scholar]
- 55.Rice CM. 2011. New insights into HCV replication: potential antiviral targets. Top. Antivir. Med. 19:117–120 [PMC free article] [PubMed] [Google Scholar]
- 56.Tariq H, Manzoor S, Parvaiz F, Javed F, Fatima K, Qadri I. 2012. An overview: in vitro models of HCV replication in different cell cultures. Infect. Genet. Evol. 12:13–20. 10.1016/j.meegid.2011.10.009 [DOI] [PubMed] [Google Scholar]
- 57.Yang PL, Gao M, Lin K, Liu Q, Villareal VA. 2011. Anti-HCV drugs in the pipeline. Curr. Opin. Virol. 1:607–616. 10.1016/j.coviro.2011.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O'Boyle DR, II, Lemm JA, Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA, Hamann LG. 2010. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465:96–100. 10.1038/nature08960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Einav S, Sobol HD, Gehrig E, Glenn JS. 2010. The hepatitis C virus (HCV) NS4B RNA binding inhibitor clemizole is highly synergistic with HCV protease inhibitors. J. Infect. Dis. 202:65–74. 10.1086/653080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pang X, Zhang M, Dayton AI. 2001. Development of dengue virus type 2 replicons capable of prolonged expression in host cells. BMC Microbiol. 1:18. 10.1186/1471-2180-1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Puig-Basagoiti F, Tilgner M, Forshey BM, Philpott SM, Espina NG, Wentworth DE, Goebel SJ, Masters PS, Falgout B, Ren P, Ferguson DM, Shi PY. 2006. Triaryl pyrazoline compound inhibits flavivirus RNA replication. Antimicrob. Agents Chemother. 50:1320–1329. 10.1128/AAC.50.4.1320-1329.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ng CY, Gu F, Phong WY, Chen YL, Lim SP, Davidson A, Vasudevan SG. 2007. Construction and characterization of a stable subgenomic dengue virus type 2 replicon system for antiviral compound and siRNA testing. Antiviral Res. 76:222–231. 10.1016/j.antiviral.2007.06.007 [DOI] [PubMed] [Google Scholar]
- 63.Masse N, Davidson A, Ferron F, Alvarez K, Jacobs M, Romette JL, Canard B, Guillemot JC. 2010. Dengue virus replicons: production of an interserotypic chimera and cell lines from different species, and establishment of a cell-based fluorescent assay to screen inhibitors, validated by the evaluation of ribavirin's activity. Antiviral Res. 86:296–305. 10.1016/j.antiviral.2010.03.010 [DOI] [PubMed] [Google Scholar]
- 64.Qing M, Liu W, Yuan Z, Gu F, Shi PY. 2010. A high-throughput assay using dengue-1 virus-like particles for drug discovery. Antiviral Res. 86:163–171. 10.1016/j.antiviral.2010.02.313 [DOI] [PubMed] [Google Scholar]
- 65.Shum D, Smith JL, Hirsch AJ, Bhinder B, Radu C, Stein DA, Nelson JA, Fruh K, Djaballah H. 2010. High-content assay to identify inhibitors of dengue virus infection. Assay Drug Dev. Tech. 8:553–570. 10.1089/adt.2010.0321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xie X, Wang QY, Xu HY, Qing M, Kramer L, Yuan Z, Shi PY. 2011. Inhibition of dengue virus by targeting viral NS4B protein. J. Virol. 85:11183–11195. 10.1128/JVI.05468-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hsu YC, Chen NC, Chen PC, Wang CC, Cheng WC, Wu HN. 2012. Identification of a small-molecule inhibitor of dengue virus using a replicon system. Arch. Virol. 157:681–688. 10.1007/s00705-012-1224-z [DOI] [PubMed] [Google Scholar]
- 68.Stevens AJ, Gahan ME, Mahalingam S, Keller PA. 2009. The medicinal chemistry of dengue fever. J. Med. Chem. 52:7911–7926. 10.1021/jm900652e [DOI] [PubMed] [Google Scholar]
- 69.Lin YL, Liao CL, Chen LK, Yeh CT, Liu CI, Ma SH, Huang YY, Huang YL, Kao CL, King CC. 1998. Study of dengue virus infection in SCID mice engrafted with human K562 cells. J. Virol. 72:9729–9737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen LK, Lin YL, Liao CL, Lin CG, Huang YL, Yeh CT, Lai SC, Jan JT, Chin C. 1996. Generation and characterization of organ-tropism mutants of Japanese encephalitis virus in vivo and in vitro. Virology 223:79–88. 10.1006/viro.1996.0457 [DOI] [PubMed] [Google Scholar]
- 71.Miroux B, Walker JE. 1996. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260:289–298. 10.1006/jmbi.1996.0399 [DOI] [PubMed] [Google Scholar]
- 72.Burke D. 2000. Methods in yeast genetics: a Cold Spring Harbor Laboratory course manual, 2000 ed. Cold Spring Harbor Laboratory Press, Plainview, NY [Google Scholar]
- 73.Yang CC, Tsai MH, Hu HS, Pu SY, Wu RH, Wu SH, Lin HM, Song JS, Chao YS, Yueh A. 2013. Characterization of an efficient dengue virus replicon for development of assays of discovery of small molecules against dengue virus. Antiviral Res. 98:228–241. 10.1016/j.antiviral.2013.03.001 [DOI] [PubMed] [Google Scholar]
- 74.Lin YL, Liu CC, Lei HY, Yeh TM, Lin YS, Chen RM, Liu HS. 2000. Infection of five human liver cell lines by dengue-2 virus. J. Med. Virol. 60:425–431. 10.1002/(SICI)1096-9071(200004)60:4<425::AID-JMV10>3.0.CO;2-A [DOI] [PubMed] [Google Scholar]
- 75.Wu SF, Lee CJ, Liao CL, Dwek RA, Zitzmann N, Lin YL. 2002. Antiviral effects of an iminosugar derivative on flavivirus infections. J. Virol. 76:3596–3604. 10.1128/JVI.76.8.3596-3604.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Houng HH, Hritz D, Kanesa-thasan N. 2000. Quantitative detection of dengue 2 virus using fluorogenic RT-PCR based on 3′-noncoding sequence. J. Virol. Methods 86:1–11. 10.1016/S0166-0934(99)00166-4 [DOI] [PubMed] [Google Scholar]
- 77.Deas TS, Binduga-Gajewska I, Tilgner M, Ren P, Stein DA, Moulton HM, Iversen PL, Kauffman EB, Kramer LD, Shi PY. 2005. Inhibition of flavivirus infections by antisense oligomers specifically suppressing viral translation and RNA replication. J. Virol. 79:4599–4609. 10.1128/JVI.79.8.4599-4609.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zou G, Puig-Basagoiti F, Zhang B, Qing M, Chen L, Pankiewicz KW, Felczak K, Yuan Z, Shi PY. 2009. A single-amino acid substitution in West Nile virus 2K peptide between NS4A and NS4B confers resistance to lycorine, a flavivirus inhibitor. Virology 384:242–252. 10.1016/j.virol.2008.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pu SY, Wu RH, Yang CC, Jao TM, Tsai MH, Wang JC, Lin HM, Chao YS, Yueh A. 2011. Successful propagation of flavivirus infectious cDNAs by a novel method to reduce the cryptic bacterial promoter activity of virus genomes. J. Virol. 85:2927–2941. 10.1128/JVI.01986-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Palmer M, Chan A, Dieckmann T, Honek J. 2012. Biochemical pharmacology, 1st ed. John Wiley & Sons, Hoboken, NJ [Google Scholar]
- 81.Beran RK, Pyle AM. 2008. Hepatitis C viral NS3-4A protease activity is enhanced by the NS3 helicase. J. Biol. Chem. 283:29929–29937. 10.1074/jbc.M804065200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yao N, Reichert P, Taremi SS, Prosise WW, Weber PC. 1999. Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure 7:1353–1363. 10.1016/S0969-2126(00)80025-8 [DOI] [PubMed] [Google Scholar]
- 83.Wang CC, Huang ZS, Chiang PL, Chen CT, Wu HN. 2009. Analysis of the nucleoside triphosphatase, RNA triphosphatase, and unwinding activities of the helicase domain of dengue virus NS3 protein. FEBS Lett. 583:691–696. 10.1016/j.febslet.2009.01.008 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.