

Abstract
GSK2336805 is an inhibitor of hepatitis C virus (HCV) with picomolar activity on the standard genotype 1a, 1b, and 2a subgenomic replicons and exhibits a modest serum shift. GSK2336805 was not active on 22 RNA and DNA viruses that were profiled. We have identified changes in the N-terminal region of NS5A that cause a decrease in the activity of GSK2336805. These mutations in the genotype 1b replicon showed modest shifts in compound activity (<13-fold), while mutations identified in the genotype 1a replicon had a more dramatic impact on potency. GSK2336805 retained activity on chimeric replicons containing NS5A patient sequences from genotype 1 and patient and consensus sequences for genotypes 4 and 5 and part of genotype 6. Combination and cross-resistance studies demonstrated that GSK2336805 could be used as a component of a multidrug HCV regimen either with the current standard of care or in combination with compounds with different mechanisms of action that are still progressing through clinical development.
INTRODUCTION
Hepatitis C virus (HCV) is a 9.6-kb flavivirus that infects over 160 million people worldwide (1). HCV infection can lead to fibrosis, cirrhosis, and hepatocellular carcinoma, although these outcomes are generally observed only after years of chronic infection (2). Originally classified as non-A, non-B hepatitis, the pathogen was first recognized during the 1970s and successfully identified by a team of scientists working at Chiron in 1989 (3). Since its identification, substantial progress has been made to define the replication cycle of the virus as well as molecular mechanisms involved in its pathogenesis (reviewed in reference 4). Additionally, surrogate model systems of viral replication have enabled drug discovery efforts to identify specific inhibitors of HCV replication (5–10).
Until 2011, the standard of care (SOC) for HCV-infected patients was limited to a 24- or 48-week regimen of pegylated interferon (PEG-IFN) and ribavirin. This regimen could result in a sustained virologic response (SVR), equivalent to a virologic cure of HCV, but it was poorly tolerated and SVR rates varied greatly depending upon genotype. The length of treatment was dependent on HCV genotype, with genotype 2- and 3-infected patients having higher SVR rates on 24-week therapy than genotype 1 patients on the longer 48-week regimen. In 2011, the addition of HCV NS3 protease inhibitors (Incivek or Victrelis) to the SOC led to improved rates of SVR, up to 75% for genotype 1 in non-black patients and up to 53% in black patients, resulting in successful treatment of more patients (11, 12). Although the protease inhibitors increased successful treatment rates, they still resulted in failures in treated patients, highlighting the need for either more targeted therapies to add to the current regimen or a new treatment strategy. Ideally, the regimen would be composed of multiple specific inhibitors of the virus that reduced treatment duration and had fewer side effects.
HCV has a 9.6-kb RNA genome, encoding 10 proteins, that replicates via a polyprotein strategy on intracellular membranes by utilizing both host and viral proteins (reviewed in reference 4). Characterization of the virus replication cycle has identified many points for potential intervention that could be part of a new treatment regimen. The HCV NS2/3 and NS3 proteases work in conjunction with cellular proteases to liberate the individual viral proteins from the polyprotein, while the HCV NS5B RNA-dependent RNA polymerase is responsible for reproducing the viral genome. The other viral proteins lack enzymatic activity but are critical for completion of the full virus replication cycle. These include the two viral glycoproteins (E1 and E2), an ion channel (p7), the capsid protein (core), a protease accessory factor (NS4A), and two proteins of unknown function (NS4B and NS5A). In addition to virally encoded proteins, virus replication is dependent on host factors that could also be targets for antiviral drugs (13–15).
This report details the characterization of GSK2336805 (see Fig. S1 in the supplemental material), a potent small-molecule inhibitor of HCV replication with multigenotype activity that acts via a viral NS5A-mediated mechanism. GSK2336805 is currently in clinical development for the treatment of chronic HCV as part of a combination therapy.
MATERIALS AND METHODS
Cells.
Stable cell lines carrying a bicistronic genotype 1a (H77), genotype 1b (Con-1 ET with cell culture-adapted mutations), or genotype 2a (JFH-1) replicon were created in-house, licensed from ReBLikon GmbH (Mainz, Germany), or constructed in-house from HCVcc virus licensed from Apath, LLC (Brooklyn, NY), respectively (6, 16). All three replicons express luciferase and neomycin phosphotransferase. Cells were maintained as subconfluent monolayers and were split 1:4 to 1:6 twice a week in Dulbecco modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS), GlutaMAX-1, nonessential amino acids, and 500 μg/ml Geneticin. ET cured cells are a derivative of Con-1 ET cells generated by treating Con-1 ET cells with IFN-α for several passages until HCV RNA levels were undetectable. Huh7 Lunet and Huh7.5 cells were licensed from ReBLikon GmbH (Mainz, Germany) and Apath, LLC (Brooklyn, NY), respectively. Cell lines were maintained as subconfluent monolayers and were split 1:4 to 1:6 twice a week in DMEM supplemented with 10% FBS, nonessential amino acids, glutamine, and penicillin-streptomycin (complete medium).
Replicon activity assays.
Stable replicon cell lines were seeded at a density of 2 × 104 cells per well in a final volume of 200 μl of assay medium (DMEM supplemented with 5% FBS, penicillin-streptomycin, and nonessential amino acids) in 96-well assay plates containing compounds or dimethyl sulfoxide (DMSO). For assays performed in 384-well assay plates, 5 × 103 cells were added per well. Cells were incubated at 37°C with 5% CO2. Plates were removed from the incubator at 48 h after dosing and allowed to equilibrate to room temperature. HCV replication was monitored by determining firefly luciferase activity using Steady-Glo (Promega), and luminescence was measured in an EnVision 2103 multilabel reader (Perkin-Elmer). Cytotoxicity was measured on parallel plates using CellTiter-Glo (Promega). Replicon 50% effective concentration (EC50) and 50% cytotoxic concentration (CC50) values, the concentration of compound required to inhibit 50% of the assay response, were calculated by curve fitting data to the Hill equation, using a nonlinear least-squares curve-fitting program. Transient transfections used plasmid constructs containing wild-type (WT), variant, or chimeric replicons as templates for in vitro transcription reactions using the T7 RiboMAX Express large-scale production system (Promega). The in vitro transcripts were aliquoted and stored at −80°C before use. For genotype 1a constructs, 5 × 106 Huh7 Lunet cells were electroporated with 15 μg RNA in Cytomix supplemented with 2 mM ATP and 5 mM glutathione (17). Electroporation was carried out in 0.4-cm cuvettes using a Bio-Rad Gene Pulser II at 270 V, 950 μF, and infinite resistance. Transient transfections with genotype 1b constructs were performed similarly except that 5 × 106 ET cured cells were electroporated with 5 μg RNA in phosphate-buffered saline (PBS). Electroporated cells were resuspended in complete medium, and 2 × 104 cells were transferred to wells of a 96-well plate containing compounds or DMSO. Cells were incubated at 37°C with 5% CO2 for 3 days, and inhibition of HCV replication was measured as for the stable replicon cells using Bright-Glo (Promega).
Preparation of replicon variants.
A modified genotype 1a replicon, referred to as the 1a fit replicon (18), and the Con-1 ET replicon were mutagenized by site-directed mutagenesis. Single point mutations were generated with a QuikChange mutagenesis kit (Stratagene) according to the manufacturer's instructions or by GenScript USA Inc. (Piscataway, NJ), an external contract organization, using standard molecular biology techniques. Plasmid isolation was performed using mini- or maxiprep kits (Qiagen, Valencia, CA) according to the manufacturer's instructions.
Plating and dosing of HCVcc.
HCVcc was constructed from a plasmid licensed from Apath, LLC (Brooklyn, NY). HCVcc, also known as Jc1p7Fluc2a, is a genotype 2a/2a chimera consisting of J6CF- and JFH-1-derived sequences fused at a junction after the first transmembrane domain in NS2. The plasmid was modified such that the Gaussia luciferase gene was replaced with a DNA cassette carrying the firefly luciferase gene in tandem with the foot-and-mouth disease virus (FMDV) 2a peptide using standard cloning techniques (19). Linearized plasmid was purified using a QIAquick spin kit (Qiagen). In vitro-transcribed RNA was prepared using a T7 RiboMAX Express large-scale production system (Promega) and purified using an RNeasy kit (Qiagen) according to the manufacturer's instructions. Huh7.5 cells (1 × 107) were mixed with 10 μg of Jc1p7Fluc2A RNA and electroporated at 270 V and 950 μF with infinite resistance using a Bio-Rad Gene Pulser II. Following a 10-min incubation at room temperature, the cells were transferred to 30 ml fresh medium and seeded into a T150 tissue culture dish. The medium was changed 1 day after electroporation. The supernatants were collected every 12 h up to 5 days postelectroporation, after which they were pooled and the titer measured. Six thousand Huh7.5 cells and Jc1p7Fluc2a virus (multiplicity of infection [MOI] of 0.03) were mixed in DMEM supplemented with 10% FBS, nonessential amino acids, and penicillin-streptomycin (total volume, 40 μl) and added to 96-well assay-ready plates containing 1 μl of compound dilution. Cells were incubated at 37°C with 5% CO2. Assay plates were removed from the incubator after 4 days and allowed to equilibrate to room temperature. HCV replication was monitored by determining firefly luciferase activity using Steady-Glo (Promega), and luminescence was measured in an EnVision 2103 multilabel reader (Perkin-Elmer).
Assessment of GSK2336805 activity on a panel of RNA and DNA viruses.
The antiviral activity of GSK2336805 against a panel of 22 RNA and DNA viruses was determined by Southern Research Institute (Birmingham, AL). The cytoprotective and cytotoxic effects of GSK2336805 were determined at a single concentration of 10 μM. Detailed methods are provided in the supplemental material.
Selection of replicons resistant to GSK2336805.
Genotype 1a and 1b replicon cells were plated in flasks with compound diluted in DMSO or an equivalent amount of DMSO in DMEM supplemented with 10% FBS, penicillin-streptomycin, GlutaMax-1, nonessential amino acids, and 1 mg/ml Geneticin. The cells were fed every 3 to 4 days. Selected cells that reached confluence within the first week of selection were split 1:3 into the same medium plus compound. Generally, a few days of growth was observed after addition of compound and Geneticin, followed by an extended period of cell death. Following this period, the surviving cells proliferated and repopulated the flasks, generally taking 3 to 4 weeks from the beginning to the end of selection. After a sufficient number of cells were present, RNA was isolated using TRIzol (Invitrogen) according to the manufacturer's instructions. One microgram of RNA was mixed with HCV-RT primer (24 μM final concentration) and H2O to a final volume of 12.5 μl. The mixture was incubated at 65°C for 10 min. Four microliters of Transcriptor buffer (Roche), 2 μl deoxynucleoside triphosphates (dNTPs) (1 mM), 1 μl RNasin (Promega), and 0.5 μl reverse transcriptase (RT) (Transcriptor; Roche) were then added to the mixture, followed by a 1-hour incubation at 55°C. A PCR master mix (4 μM forward primer [1a, 5′-TCCGGTTCCTGGCTAAGGGAC-3′; 1b, 5′-TCCGGCTCGTGGCTAAGAGATG-3′], 4 μM reverse primer [1a, 5′-GCAGCACACGACATCTTCCGTG-3′; 1b, 5′-GCAGCAGACGACGTCCTCAC-3′], 5% DMSO, Pfu Turbo master mix [Stratagene, La Jolla, CA], water) was prepared, and then 47 μl was placed in each 0.2-ml PCR tube. Three microliters of the reverse transcription reaction mixture was added, mixed, and placed in the thermocycler. The PCR conditions were one round of 94°C for 5 min followed by 40 cycles of 94°C for 45 s, 55°C for 1 min, and 72°C for 4 min. At the end of cycling, the thermocycler incubated the reaction mixtures at 72°C for 7 min and then cooled the samples to 4°C. PCR products were separated on a 1.2% E-Gel (Invitrogen) to verify PCR product sizes.
Population sequencing of NS5A from GSK2336805-resistant cells.
PCR products (20 μl), generated as described in “Selection of replicons resistant to GSK2336805” above, were purified using Agencourt Ampure magnetic beads on the Biomek FX liquid handling system (Beckman Coulter). Purified samples were quantitated with a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies) and normalized to 40-ng/μl stocks. Gene-specific primers were designed in both the sense and antisense directions using GSK PrimerD software (in-house developed by GlaxoSmithKline [GSK]) to achieve double-strand coverage. DNA sequencing reactions were set up using 40 ng of PCR product, 1.0 μl BigDye terminator (v3.1), 2.0 μl sequencing buffer (5×), 4.0 pmol sequencing primer, and 0.5 μl DMSO (5%). Cycling conditions were 2 min of incubation at 98°C followed by 25 rounds of 98°C for 10 s, 50°C for 10 s, and 60°C for 2 min. Amplified sequencing products were purified using Agencourt CleanSEQ magnetic beads on the Beckman FX system followed by analysis on the Applied Biosystems 3730xl DNA sequencer. Electropherograms were edited and assembled to HCV reference sequences using GeneCodes Sequencher (v4.9) software. Mutations detected in both sense and antisense strands were reported.
Identification of NS5A sequences for chimeras containing sequences from patients with genotypes 1a and 1b.
Full-length sequences were retrieved from the European HCV database (http://euhcvdb.ibcp.fr/euHCVdb/) and then aligned using the program MUSCLE (20). The frequency of each residue in each column was calculated. Totals of 564 and 1,490 NS5A sequences for genotypes 1a and 1b, respectively, were used to generate polymorphism reports. Polymorphisms which had less than 60% conservation were deemed required to be present in the selected clinical isolates. A phylogenetic tree using maximum-likelihood methods was generated based on the alignments. Nine or 10 clinical sequences were identified as representative of the breadth of each subtype using a clustering program to cluster the sequences in each alignment. The clustering program clusters sequences based on a percent identity and also identifies a representative sequence for each cluster. The percent identity was adjusted until roughly eight to 10 clusters were generated, and a representative sequence from each cluster was chosen. Accession numbers are included in Table S2 in the supplemental material. Full-length patient sequences identified were synthesized and cloned into a plasmid containing the bicistronic genotype 1b Con-1 ET NS3-5B subgenomic replicon using standard DNA synthesis and cloning techniques. All constructs were sequence verified.
Method for aligning patient sequences and building consensus sequences for genotypes 2 to 6.
Full-length NS5A sequences from genotypes 1a, 1b, 2a, 2b, 3a, 4a, 5a, and 6 were downloaded separately by genotype from the European HCV database (http://euhcvdb.ibcp.fr/euHCVdb/) and then aligned using the program MUSCLE (20). The consensus for each gene was then generated using the Advanced Consensus Maker (http://www.hiv.lanl.gov/content/sequence/CONSENSUS/AdvCon.html). Only full-length gene sequences were included in the alignment, as including partial sequences introduced gap characters when computing the consensus. The parameters used were as follows: unanimous value = 1.00, majority value = 0.50, use most common character, and break ties with IUPAC characters. Unanimous values were indicated by uppercase consensus characters. Majority-rule values were indicated by lowercase consensus characters. The consensuses were then translated to ensure that no stop codons were introduced. Clinical isolates that were the closest to the consensus sequences were identified by Blast. The consensus gene sequence was used as the query and the sequence with the highest E value selected as that of the closest clinical isolate.
Since sequences from genotype 6 are extremely divergent, a phylogenetic tree was generated using the neighbor-joining method from reference sequences from the Viral Bioinformatics Resource Center (http://www.hcvdb.org/). The tree revealed that genotype 6 falls into three clades, from which three consensuses were made: ConSeq 1 (6a and b), ConSeq 2 (6c to g and 6o to t), and ConSeq 3 (6h to n). For ConSeq 2 and ConSeq 3, the gene sequences for the genotypes in each clade were aligned, and then a Markov model (which is a statistical description of a sequence family's consensus) was generated using the alignment. The sequence in the alignment that gave the highest score in a search using the model was then identified. The highest-scoring sequences in each gene were then joined together to form the consensus sequence. The nucleotide version of the protein-model consensus was used as the final consensus. Accession numbers for patient sequences are included in the Table S2 in the supplemental material. The alignment of NS5A consensus sequences for genotypes 2 to 6 is included in Fig. S9 in the supplemental material. Full-length NS5A sequences for the genotype 2a, 2b, 3a, 4a, 5a, 6ab, 6cg, and 6hn consensus and the patient closest to the consensus were synthesized and cloned into a plasmid containing the bicistronic genotype 2a JFH-1 subgenomic replicon using standard DNA synthesis and cloning techniques. All chimeric replicon plasmids were sequence verified.
Combination studies.
Interferon alpha (IFN-α), ribavirin, and cyclosporine were purchased from Sigma. All other inhibitors were obtained from the GSK compound collection as solids. The NS3 protease inhibitor was BILN-2061 (21), the NS4B inhibitor was GSK2358853A (compound 1a in reference 22), the NS5A inhibitor was GSK2336805, the NS5B nucleoside inhibitor was NM-107 (23) or MK-0608 (24), the NS5B NNI thumb pocket 1 inhibitor was a Merck NNI (compound A in reference 25), the NS5B NNI palm site 2 inhibitor was HCV-796 (26), and the NS5B NNI palm site 1 inhibitor was SB-711845 (compound 2 in reference 27). The HCV NS5B inhibitor is compound D in Fig. S1 in the supplemental material. Solid compounds, with the exception of IFN-α, were dissolved in DMSO. IFN-α was dissolved in PBS supplemented with bovine serum albumin (BSA), aliquoted, and stored at −80°C. The starting concentration for each compound was ≅ 4× EC50 determined in the genotype 1b Con-1 ET replicon assay. Two test compounds were serially diluted in DMSO to create separate 7-point dose-response plates. The dilutions of one compound were transposed to create a 7-point dose-response curve vertically. The diluted compounds from both compound plates were transferred to three 96-well assay plates using the Biomek FX liquid handling system (Beckman Coulter). Con-1 ET replicon cells were trypsinized and resuspended in medium containing 5% FBS, supplemented with GlutaMAX-1, penicillin-streptomycin, and nonessential amino acids (assay medium). Con-1 ET replicon cells (1.5 × 104) were added to the wells of three 96-well assay plates. Plates were incubated at 37°C with 5% CO2. Assay plates were removed from the incubator after 2 days and allowed to equilibrate to room temperature. HCV replication was monitored by determining firefly luciferase activity using Steady-Glo (Promega), and luminescence was measured in an EnVision 2103 multilabel reader (Perkin-Elmer).
Calculations for synergy/antagonism volume.
The MacSynergy II program was used to calculate the synergy and antagonism volumes (28). This program compares the theoretical drug interactions, which are based on the Bliss independence model, to the experimentally observed interactions (29). The Bliss independence model assumes that both compounds act independently on different targets, and this has been elaborated into a three-dimensional model for analyzing drug-drug interactions (28, 29). Buckwold et al. (30) have provided a detailed description of the analysis approach. Ninety-five percent confidence intervals were calculated for each point of the experimental dose-response surfaces. If the lower limit of the 95% confidence interval was greater than the response expected under the additive drug interaction model, synergy at this experimental response was considered to be significant. A similar argument was used for antagonism. Buckwold et al. note that the 95% synergy/antagonism volumes are the summation of the “peaks of statistically significant synergy or antagonism that deviate significantly from the expected additive drug interactions derived from 95% confidence interval data” (30). The volumes and corresponding volume descriptions at 95% confidence for the results of MacSynergy II analysis are as follows for synergism: volumes of <25 = insignificant synergism, volumes between 25 and 50 = minor but significant synergism, volumes between 50 and100 = moderate synergism (maybe important in vivo), volumes of >100 = strong synergism (probably important in vivo), and volumes of >1,000 = probably errors. The volumes and corresponding volume descriptions at 95% confidence for the results of MacSynergy II analysis are as follows for antagonism: volumes of <−25 = insignificant antagonism, volumes between −25 and −50 = minor but significant antagonism, volumes between −50 and −100 = moderate antagonism (maybe important in vivo), volumes of >−100 = strong antagonism (probably important in vivo), and volumes of >−1,000 = probably errors.
RESULTS
Characterization of activity.
The effect of GSK2336805 on HCV replication was initially assessed in the HCV subgenomic replicon system, the model system for HCV RNA replication. Replicon cells contain the nonstructural region of HCV as either a mono- or bicistronic RNA that continuously replicates as a result of the coordinated expression of the HCV proteins and their ability to set up replication complexes mimicking RNA replication during virus infection. GSK2336805 had EC50s of 58.5, 7.4, and 53.8 pM on genotype 1a (H77), genotype 1b (Con-1 ET), and genotype 2a (JFH-1) replicon cells, respectively (Table 1). Cytotoxicity was measured in parallel, and GSK2336805 had CC50 values of 43 and 47 μM on genotype 1a and 1b replicon cells, respectively, yielding a selectivity index of >10,000 (Table 1). The activity was modestly impacted by the addition of 40% human serum to GSK2336805, giving an EC50 of 25.7 pM, a 3.4-fold shift in potency (Table 1). The activity was further characterized by determining the potency on the HCVcc Jc1 virus. GSK2336805 had an average EC50 of 63.7 pM on genotype 2a Jc1 virus, showing that GSK2336805 can inhibit the HCV replication cycle and production of virus (Table 1).
TABLE 1.
Activity of GSK2336805 in HCV replicon and live virus assaysa
| HCV replicon or virus | Mean EC50, pM (95% CI) | n | CC50, pM |
|---|---|---|---|
| Genotype 1a (H77) | 58.5 (42.3–80.9) | 17 | 43,000,000 |
| Genotype 1b (Con-1) | 7.4 (6.5–8.4) | 44 | 47,000,00 |
| Genotype 1b (Con-1) + 40% HS | 25.7 (23.0–28.7) | 8 | ND |
| Genotype 2a (JFH-1) | 53.8 (37.7–77.0) | 20 | ND |
| HCVcc (Jc1) | 63b | 2 | ND |
CI, confidence interval; HS, human serum; ND, not determined.
Results of two independent experiments performed in triplicate.
To determine if the antiviral activity of GSK2336805 is specific for HCV, we profiled the activity of the compound against a custom panel of 22 RNA and DNA viruses. The most significant impact was observed in the adenovirus type 1 cytopathic effect (CPE) assay, where 10 μM GSK2336805 reduced the CPE by 35.2% (Table 2). This level of inhibition is less than the 50% inhibition required to generate an EC50, so no dose-response determination was performed. Additionally the concentration required for this level of activity is 5 to 6 log10 units higher than the picomolar activity observed for HCV, demonstrating the specificity of this inhibitor for HCV.
TABLE 2.
Activity of 10 μM GSK2336805 on a panel of RNA and DNA viruses
| Virus | % Reduction in CPE | % Cell viability |
|---|---|---|
| Adenovirus type 1 | 35.2 | 100 |
| Adenovirus type 3 | 2.4 | 100 |
| Adenovirus type 40 | 6.5 | 99.6 |
| BVDV | 10.3 | 100 |
| Coxsackie A7 virus | 3.8 | 98.8 |
| Coxsackie B4 virus | 1.5 | 98.5 |
| Dengue virus strain 1 | 0 | 99.1 |
| Dengue virus strain 2 | 3.1 | 98.1 |
| Dengue virus strain 3 | 3.8 | 95.8 |
| Dengue virus strain 4 | 4.5 | 94.6 |
| Enterovirus | 9.5 | 98.3 |
| Herpes simplex virus 1 | 7.2 | 95 |
| Herpes simplex virus 2 | 3.1 | 97 |
| Influenza A virus | 4.5 | 99.4 |
| Influenza B virus | 3 | 100 |
| Measles virus | 1.2 | 100 |
| Parainfluenza virus | 0 | 94.8 |
| Poliovirus | 2.2 | 99.3 |
| Respiratory syncytial virus | 26.8 | 99.6 |
| Rhinovirus | 29.7 | 100 |
| Rotavirus | 0 | 97.4 |
| Yellow fever virus | 2.4 | 98.6 |
Resistance profiling.
HCV encodes an error-prone RNA-dependent RNA polymerase, leading to the accumulation of mutations and the existence of a viral quasispecies, a population of similar but distinct viruses (31). Small-molecule inhibitors of HCV RNA replication can select for a population of RNA that is resistant to inhibition due to either preexisting mutations or the accumulation of mutations, resulting in a decrease in compound potency. We characterized the resistance profiles of GSK2336805 on genotype 1a and 1b replicon cells by performing resistance screens. Replicon cells were exposed to GSK2336805 at concentrations equivalent to 5, 10, or 20 times the EC50. Initial treatment of the cells resulted in a period of reduced cell growth and, for some treatments, cell death. Experiments performed on earlier molecules in the series showed that activity was substantially reduced when the NS5A sequences in the genotype 1b subgenomic replicon were replaced with the NS5A sequences from the genotype 1a H77 subgenomic replicon (see Table S3 in the supplemental material). These data led us to focus on sequences in the NS5A region of the resistant replicons. RNA was isolated from cells that grew out of GSK2336805 treatment, and the NS5A gene was analyzed by population sequencing. The amino acid changes resulting from mutations in the nucleotide sequence are shown in Table 3. Variants were found throughout the NS5A-coding sequence for genotype 1b replicons selected at 5× or 10× EC50 for GSK233805 but were limited to one residue in genotype 1b replicons selected at 20× EC50 (Table 3). Both selection conditions (5× and 20× EC50) used for the genotype 1a replicon cells resulted in amino acid changes at only two positions, amino acids 30 and 31, of NS5A (Table 3).
TABLE 3.
Population sequencing results from GSK2336805 resistance selections
| HCV genotype | NS5A amino acid | Amino acid(s) detected with: |
||
|---|---|---|---|---|
| 5× EC50 | 10× EC50 | 20× EC50 | ||
| 1a | Q30 | H | NDa | H |
| L31 | M | ND | M | |
| 1b | L31 | V, W | V | V, W, G |
| N69 | T | |||
| V198 | A | |||
| P223 | S | |||
| I280 | V | |||
| V298 | A | |||
| S364 | P | |||
| S368 | P | |||
ND, not determined.
We followed up this data set by making single point mutations in the NS5A gene to determine which changes were causing resistance. The results of this profiling are shown in Fig. 1 and 2. For genotype 1a, both variants identified in the resistance screen (Q30H and L31M) reduced the potency of GSK2336805. EC50s for the variants were 8.7 and 4.1 nM, respectively, a >150-fold decrease in potency. Compounds that inhibit HCV replication via the NS5B polymerase were used as controls in these experiments. The EC50s for these compounds were not shifted by more than 1.3-fold on either of these mutations in NS5A (see Table S4 in the supplemental material).
FIG 1.
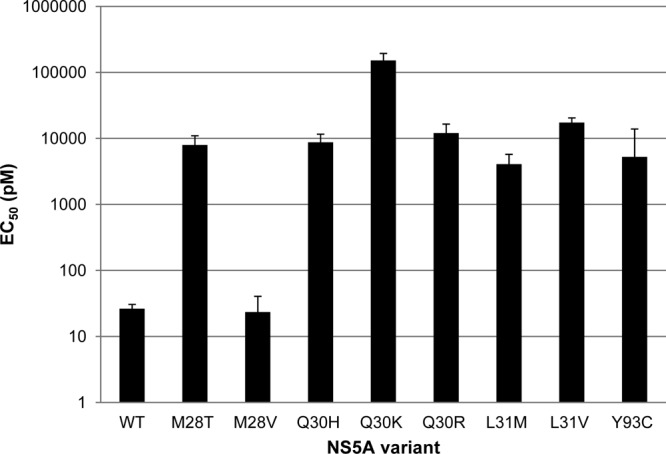
Activity of GSK2336805 on NS5A variants in genotype 1a. GSK2336805 was profiled against a panel of NS5A variants observed in genotype 1a resistance screens with GSK2336805 or published genotype 1a amino acid changes leading to resistance for other NS5A inhibitors. Substitutions were made in the 1a fit replicon as described in Materials and Methods (18). Replicons containing the amino acid changes were tested in transient-transfection assays. The average EC50s are plotted on a log10 scale, and the error bars represent the 95% confidence intervals. Each replicon was tested at least 3 times.
FIG 2.
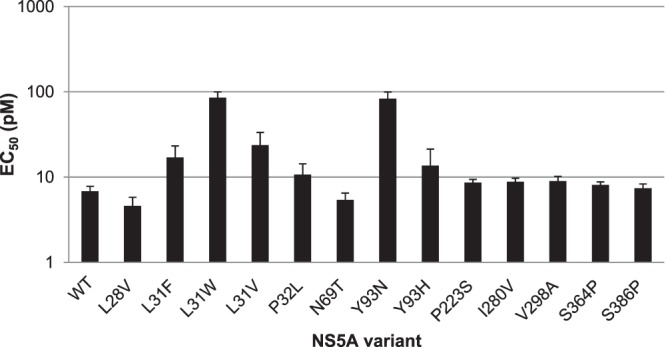
Activity of GSK2336805 on NS5A variants in genotype 1b. GSK2336805 was profiled against a panel of NS5A variants observed in genotype 1b resistance screens with GSK2336805 or published genotype 1b variants leading to resistance for other NS5A inhibitors. Mutations were made in the genotype 1b Con-1 ET replicon as described in Materials and Methods. Variant replicons were tested in transient-transfection assays. The average EC50s are plotted on a log10 scale, and the error bars represent the 95% confidence intervals. Each replicon was tested at least 3 times.
In the genotype 1b replicon variant profiling, there were only three variants tested that altered the potency of GSK2336805 more than 2-fold. When the leucine at position 31 was mutated to tryptophan (L31W) the EC50 was 85.1 pM, a 12.5-fold decrease in potency. Changing the leucine at position 31 to a valine (L31V) resulted in an EC50 of 23.7 pM, a 3.5-fold reduction in potency. The other variants had EC50s between 4.6 and 10.7 pM, a maximum change in potency of 1.6-fold from that of the wild type. These variants do not appear to be sites of primary resistance due to the low impact on GSK2336805 activity, but they may impact the behavior of another variant when present in combination. The potency of control compounds, NS5B polymerase inhibitors, shifted by less than 1.8-fold on any of the changes made in the genotype 1b NS5A sequence (see Table S5 in the supplemental material).
The resistance profiling of GSK2336805 demonstrated that the compound is sensitive to amino acid changes at the same positions in NS5A (amino acids 30 and 31) as another small-molecule NS5A HCV inhibitor, daclatasvir (32). We decided to test several of the variants known to impact daclatasvir potency that were not identified in our resistance screen (32–34). In the genotype 1a replicon, positions 28, 30, 31, 32, and 93 were interrogated. Changing position 28 from methionine to valine had no impact on GSK2336805 potency, but all of the other substitutions tested caused >150-fold reductions in the EC50, with the largest reduction seen for the lysine substitution at position 30 (Fig. 1). This profiling demonstrated that GSK2336805 was susceptible to amino acid substitutions in genotype 1a NS5A arising from daclatasvir selection even though they did not arise in our resistance screens. Interestingly, the Q30R substitution was present in the chimeric replicon containing the NS5A sequence derived from a patient that showed a 40-fold shift in GSK2336805 activity. The activity of GSK2336805 on this single amino acid change is more dramatically shifted than for the chimera, suggesting that the sequence context will impact the potency change. The potency of control compounds, HCV polymerase inhibitors, was shifted less than 1.8-fold on these variants in NS5A (see Table S4 in the supplemental material).
We continued this profiling in the genotype 1b replicon. We examined the impact of changing the leucine at position 31 to a phenylalanine (34). This change caused a <3-fold change in EC50 for GSK2336805 from that of the wild type (Fig. 2). We also altered the proline at position 32 to a leucine and the tyrosine at position 93 to an asparagine or histidine. Less-than-2-fold changes in EC50 were seen for the leucine or histidine variants, but altering position 93 to asparagine reduced GSK2336805 potency 12-fold (Fig. 2). These data showed that GSK2336805 is susceptible to only some of the genotype 1b NS5A variants that impact daclatasvir activity. The EC50s for control compounds, HCV polymerase inhibitors, were shifted less than 1.8-fold on the genotype 1b NS5A variants tested (see Table S5 in the supplemental material).
Cross-resistance profiling.
We wanted to determine if amino acid changes causing resistance to other HCV direct-acting antivirals (DAAs) altered the potency of GSK2336805. We tested a panel of variants in NS3, NS4B, and NS5B that cause resistance to NS3 protease inhibitors, NS4B inhibitors, or NS5B nucleoside or nonnucleoside inhibitors (reviewed in reference 35). Additionally, we tested the potency of GSK2336805 on an amino acid substitution in NS5A that was reported to cause resistance to cyclophilin inhibitors (13). None of the tested amino acid substitutions in NS3, NS4B, or NS5B decreased the activity of GSK2336805 more than 2.6-fold, and neither did the NS5A D320E substitution tested (Table 4). These data showed that GSK2336805 retains activity on variants arising from other HCV DAAs targeting NS3, NS4B, or NS5B or a cyclophilin inhibitor. Control compounds confirmed that the amino acid changes behaved as expected (see Tables S6 to S8 in the supplemental material).
TABLE 4.
Activity of GSK2336805 on resistant variants raised by other HCV DAAsa
| HCV gene | Variant | EC50 (pM) |
Fold shift from WT value | n | |
|---|---|---|---|---|---|
| Mean | 95% CI | ||||
| WT | 6.8 | 5.8–7.8 | 59 | ||
| NS3 | A156T | 3.9 | 2.4–6.3 | 0.6 | 4 |
| NS3 | D168A | 6.2 | 5.1, 7.4b | 0.9 | 2 |
| NS3 | D168V | 6.7 | 6.0, 7.4b | 1.0 | 2 |
| NS3 | R155K | 10.5 | ND | 1.5 | 1 |
| NS4B | H94R | 4.2 | 2.2–7.8 | 0.6 | 6 |
| NS4B | V105L | 11.4 | 7.1–18.2 | 1.7 | 6 |
| NS5A | D320E | 5.5 | 4.0–7.7 | 0.8 | 6 |
| NS5B | C316N | 4.3 | 3.0–6.2 | 0.6 | 12 |
| NS5B | C316Y | 4.4 | 3.1–6.3 | 0.7 | 12 |
| NS5B | C445F | 4.8 | 3.3–7.0 | 0.7 | 12 |
| NS5B | H95Q | 15.9 | 9.3–27.4 | 2.3 | 5 |
| NS5B | M414T | 2.2 | 0.7–4.1c | 0.3 | 3 |
| NS5B | N411S | 17.5 | 11.0–27.7 | 2.6 | 5 |
| NS5B | P495L | 8.3 | 5.1–13.5 | 1.2 | 6 |
| NS5B | P496S | 10.6 | 4.4–25.1 | 1.6 | 6 |
| NS5B | S365T | 6.2 | 2.0–19.0 | 0.9 | 6 |
| NS5B | S368T | 7.6 | 2.7–21.4 | 1.1 | 6 |
| NS5B | Y448H | 3.9 | 2.9–5.3 | 0.6 | 12 |
| NS5B | Y452H | 4.7 | 3.5–6.4 | 0.7 | 12 |
CI, confidence interval; ND, not determined.
EC50s are listed for n = 2.
Range of EC50s is given for n = 3.
Testing the activity of GSK2336805 on chimeric replicons containing NS5A patient sequences from genotype 1.
The H77 and Con-1 ET replicons used in the initial characterization of GSK2336805 represent only a fraction of the diversity present in HCV genotype 1. To determine the breadth of coverage for this genotype, we further characterized the activity of GSK2336805 against chimeric replicons containing sequences derived from HCV-infected patients. The results of the resistance profiling demonstrated that GSK2336805 was sensitive to changes in NS5A, so we focused on diversity across this gene for the chimeric replicons. Alignments were constructed from full-length HCV sequences retrieved from the European HCV database. These were used to generate polymorphism reports and phylogenetic trees for genotypes 1a and 1b. NS5A sequences for genotypes 1a and 1b were chosen for testing from across each subtype using a clustering program to ensure maximal breadth of coverage. The NS5A gene from each of these sequences was synthesized and cloned into the genotype 1b Con-1 ET replicon for testing in a transient-transfection assay. EC50s for GSK2336805 on these chimeric replicons showed a <3-fold shift from those on the genotype 1b Con-1 ET replicon on 7 of 9 genotype 1a and all 10 genotype 1b chimeric replicons tested (Fig. 3). One genotype 1a chimera had an EC50 of 50.1 pM, a 7.4-fold shift from the reference strain, while another genotype 1a chimera had an EC50 of 275 pM, a 40-fold shift in potency (Fig. 3). These data demonstrate that GSK2336805 retains picomolar activity across the sequence diversity found in genotypes 1a and 1b.
FIG 3.
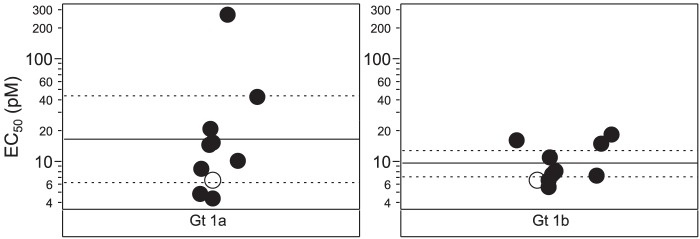
Activity of GSK2336805 on chimeric replicons containing genotype 1a and 1b NS5A patient sequences. GSK2336805 was tested in transient-transfection assays for activity on chimeric replicons containing genotype 1a or 1b patient sequences. The wild-type replicon is the genotype 1b Con-1 ET replicon, the backbone for the chimeric replicons, represented by the open circles. EC50s for each chimeric replicon are represented by black circles. The solid lines represent the mean EC50 for the chimeric replicons, and the dashed lines are the 95% confidence intervals for GSK2336805 activity. Each replicon was tested at least 3 times.
Testing the activity of GSK2336805 on chimeric replicons containing NS5A patient and consensus sequences from genotypes 2 to 6.
HCV isolates exhibit substantial diversity, leading to classification of an isolate into one of six genotypes based on nucleotide sequences (36, 37). The activity of GSK2336805 against other genotypes was assessed using chimeric replicons containing consensus or patient NS5A sequences from genotypes 2 to 6. Full-length NS5A sequences were obtained from the European HCV Database. Alignments and consensus building procedures are described in Materials and Methods. The activity of GSK2336805 was unchanged on chimeric replicons containing consensus sequences from genotypes 4a, 5a, and 6ab and patient sequences from genotypes 4a and 5a (Fig. 4). The chimeric replicon containing the genotype 6a patient sequence had an EC50 of 112 pM for GSK2336805, which represents a slight shift in potency but nonetheless remains at the low picomolar level. Substantial effects on potency were detected when GSK2336805 was tested against replicons containing patient or consensus sequences from genotypes 2a, 2b, 3a, 6cg, and 6hn. EC50s ranged from 3.5 nM to 204 nM, which represents a minimum of a >1,000-fold shift from the EC50 for the genotype 1b Con-1 ET sequence in the genotype 2a background (Fig. 4).
FIG 4.
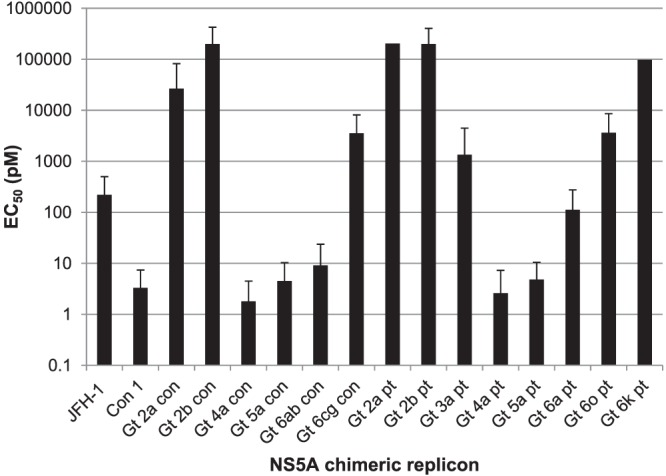
Activity of GSK2336805 on chimeric replicons containing NS5A consensus and patient sequences from HCV genotypes 2 to 6. GSK2336805 was profiled on chimeric replicons containing the NS5A sequences from consensus sequences derived as described in Materials and Methods or from patients. Chimeric replicons were assayed in transient-transfection assays. The average EC50s are plotted on a log10 scale, and the error bars represent the 95% confidence intervals. Each replicon was tested at least three times. Error bars are not included for the chimeric replicons containing the genotype 2a and 6k sequences, as they were run only three times. The EC50s of GSK2336805 on genotype 3a and 6hn consensus replicons were >5,000 pM and are not plotted.
The genotype 2a result was surprising given the potent activity on the genotype 2a JFH-1 replicon. We tested several other compounds from the series on these chimeras and found the loss in potency to be consistent within the series, showing the GSK2336805 result is not an anomaly (data not shown). Sequence analysis showed that several positions were different within the first 100 amino acids when the JFH-1 was compared to the genotype 2a patient and consensus sequences in the chimeric replicon, including a leucine-to-methionine change at position 31 (see Fig. S9 in the supplemental material). This position is important for loss of GSK2336805 activity in genotype 1a and 1b replicons and is the likely cause of the potency reduction in the genotype 2a patient and consensus replicons. These data show that GSK2336805 has multigenotype activity, as it retains picomolar potency on genotype 4a, 5a, and 6ab patient and consensus sequences.
Combination studies.
GSK2336805 has picomolar activity in genotype 1a, 1b, and JFH-1 2a replicons as well as a genotype 2a virus. Although it has impressive activity, published in vitro and in vivo data on other DAAs targeting HCV replication as a monotherapy have demonstrated that resistant RNAs and/or viruses will emerge when the quasispecies population is targeted with a single DAA (38). Thus, GSK2336805 would be used in combination with the current SOC, with other DAAs, or with a combination of other DAAs and components of the SOC.
The ability of GSK2336805 to work in combination with other inhibitors of HCV replication was assessed using the HCV genotype 1b replicon. GSK2336805 was tested in combination with representative protease, polymerase, and NS4B inhibitors as well as cyclosporine, interferon alpha, and ribavirin. Data analysis was performed using the Bliss independence model via the MacSynergy II program. In all combinations tested, GSK2336805 produced strong synergism and insignificant antagonism when combined with another HCV inhibitor (Table 5). The in vitro combination studies performed demonstrate that GSK2336805 is a good candidate for HCV combination therapy with either the SOC, other classes of DAAs, or a combination with the components of the current SOC and other DAAs.
TABLE 5.
Activity of GSK2336805 in combination with other HCV antivirals
| Compound combined with GSK2336805A | Synergy vol | Antagonism vol | MacSynergy II analysis results |
|
|---|---|---|---|---|
| Synergism | Antagonism | |||
| IFN-α | 172.88 | −3.03 | Strong | Insignificant |
| Ribavirin | 102.28 | 0 | Strong | Insignificant |
| NS3 protease inhibitor (BILN 2061) | 110.6 | −14.68 | Strong | Insignificant |
| NS4B inhibitor (GSK2358853)a | 121.49 | −1.49 | Strong | Insignificant |
| GSK2335805A | 289.03 | −4.36 | Strong | Insignificant |
| HCV nucleoside inhibitor (NM-107) | 151.59 | −6.03 | Strong | Insignificant |
| HCV nucleoside inhibitor (MK-0608) | 219.71 | −2.01 | Strong | Insignificant |
| NS5B NNI thumb pocket 1 (Merck NNI) | 273.73 | 0 | Strong | Insignificant |
| NS5B NNI palm site 1 (SB-711845) | 214.65 | −0.5 | Strong | Insignificant |
| NS5B NNI palm site 2 inhibitor (HCV-796) | 103.92 | −3.57 | Strong | Insignificant |
| HCV NS5B inhibitor (GSK2286853)b | 443.54 | 0 | Strong | Insignificant |
| Cyclosporine | 233.68 | −1.34 | Strong | Insignificant |
Compound 1a in reference 22.
Compound D in Fig. S1 in the supplemental material.
DISCUSSION
We have presented data to show that GSK2336805 is an inhibitor of HCV replication with picomolar activity across the standard genotype 1a, 1b, and 2a subgenomic replicons and a chimeric genotype 2a virus with a modest serum shift. We have also shown that variants in the NS5A region of the virus lead to a decrease in activity for GSK2336805. Although the variants identified in genotypes 1a and 1b partially overlap and are in the same region of the protein, the impact of these variants was found to be dramatically different. The genotype 1b resistance profiling showed no changes impacting GSK2336805 activity more than 13-fold, while the genotype 1a resistance variants could reduce activity by more than 5,000-fold. Resistance to DAAs is not unique to GSK2336805 or other similar inhibitors, and the relevance of these changes will be determined through clinical trials (reviewed in reference 35). GSK2336805 exhibits picomolar activity across the diversity of HCV genotype 1 sequences and intergenotypic chimeras with NS5A sequences from genotypes 4, 5, and part of 6.
The NS5A gene is an essential component of the viral replication cycle, but its exact function is unknown. Published data link NS5A activity to both replication of the viral RNA and virus egress (5, 16, 39–42). There are also a significant number of papers demonstrating interactions between NS5A and various cellular and virus proteins (reviewed in reference 43). The numerous publications on NS5A, including two crystal structures of the N-terminal portion of the protein, have failed to fully elucidate the function(s) of the protein (44, 45).
Given the unknown function of NS5A, the exact mechanism of compound inhibition is still under investigation. We have recently published data showing that a compound in the same series as GSK2336805 can change the cellular conformation of the NS5A protein. The proximity of inter- and intramolecular positions of NS5A was disrupted by the presence of compound 1, an active molecule in the GSK2336805 series, while compound 2, an inactive molecule from the same series, did not cause any change (46). These experiments do not provide evidence of direct binding of compound 1 to NS5A, as other cellular factors are present, but they do add experimental data to the resistance profiling results supporting the mechanism of inhibition involving the NS5A protein, either directly or indirectly.
Two sets of data suggest that the impact of the compound is largely on the replication of the RNA. First, the data for the HCVcc virus and stable genotype 2 JFH-1 replicon show that GSK2336805 exhibits similar activity in both systems. The stable subgenomic replicon system represents only the part of the replication cycle where viral RNA is being replicated, while the live virus system includes all aspects of virus replication cycle, including entry, uncoating, RNA replication, assembly, and egress. The data suggest that either the impact of the compound on the NS5A virus replicon activity is the only impact on the virus or the impact on replication occurs at a lower concentration than that on the other potential functions of NS5A. Inhibition of nonreplication functions would be masked if they occurred at a concentration above the replicon EC50. Second, we present data showing that the potency of GSK2336805 is similar in the transient and stable replicons. These two systems differ in one respect: the transient-transfection replicon system requires the successful assembly of a replication complex. The fact that the EC50s for both systems are similar suggests that if the compound is impacting assembly of the replication complex, then the inhibition is equivalent to the concentration required for inhibition of viral RNA synthesis. It is possible that additional inhibition outside RNA replication could be masked in current in vitro systems and is only visualized in the in vivo setting, where it provides additional advantages to this mechanism of inhibition in successfully reducing viral loads in HCV-infected patients. A recently published paper modeled the response of HCV in vitro and in vivo to daclatasvir and predicted that the compound likely works via two different mechanisms, replication and assembly/secretion (47). Efforts to understand the mechanism of action for this class of inhibitors are ongoing.
The combination and cross-resistance data support the inclusion of GSK2336805 in drug regimens either with the current standard of care or with other mechanisms of HCV inhibition currently in development. GSK2336805 has successfully completed both phase 1 and phase 2a clinical trials (48, 49). The addition of protease inhibitors to the current treatment regimen for genotype 1 patients significantly improved SVR rates, but there are still underserved populations that could benefit from a regimen including GSK2336805. Additionally, patients who had relapsed or did not respond to previous HCV therapy showed an increased cure rate with the addition of the protease inhibitors, but it is still not 100%. For example, when telaprevir was combined with PEG-IFN/ribavirin, previous nonresponders had a 29 to 33% SVR rate, which, although it is a substantial improvement from no response to therapy, leaves room for improvement (50). There is also a population of HCV-infected individuals for whom IFN/ribavirin based therapies are contraindicated. These patients highlight a need for more varied therapies that can improve outcomes for all populations. Several combination therapies combining multiple DAAs with the PEG-IFN/ribavirin regimen, as well as therapies attempting to cure patients with an all-DAA regimen to improve outcomes without the significant side effects caused by IFN, are currently in clinical development. GSK2336805 is currently in phase 2b clinical studies to fully elucidate its place as part of an HCV combination therapy.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge all members of the GSK2336805 program team, especially K. Creech and L. H. Kryn for replicon screening, M. Leivers and M. Tallant for chemistry support, and D. Hazen and J. Johnson for virology support. We also thank C. Roberts, A. Spaltenstein, and Z. Hong for their leadership during the GSK2336805 program.
All authors are current or former employees of GlaxoSmithKline.
Footnotes
Published ahead of print 14 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01363-13.
REFERENCES
- 1.Lavanchy D. 2011. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 17:107–115. 10.1111/j.1469-0691.2010.03432.x [DOI] [PubMed] [Google Scholar]
- 2.Kanwal F, Hoang T, Kramer JR, Asch SM, Goetz MB, Zeringue A, Richardson P, El-Serag HB. 2011. Increasing prevalence of HCC and cirrhosis in patients with chronic hepatitis C virus infection. Gastroenterology 140:1182–1188. 10.1053/j.gastro.2010.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244:359–362. 10.1126/science.2523562 [DOI] [PubMed] [Google Scholar]
- 4.Poenisch M, Bartenschlager R. 2010. New insights into structure and replication of the hepatitis C virus and clinical implications. Semin. Liver Dis. 30:333–347. 10.1055/s-0030-1267535 [DOI] [PubMed] [Google Scholar]
- 5.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113. 10.1126/science.285.5424.110 [DOI] [PubMed] [Google Scholar]
- 6.Blight KJ, McKeating JA, Marcotrigiano J, Rice CM. 2003. Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J. Virol. 77:3181–3190. 10.1128/JVI.77.5.3181-3190.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817. 10.1053/j.gastro.2003.09.023 [DOI] [PubMed] [Google Scholar]
- 8.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796. 10.1038/nm1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. 10.1126/science.1114016 [DOI] [PubMed] [Google Scholar]
- 10.Ikeda M, Yi M, Li K, Lemon SM. 2002. Selectable subgenomic and genome-length dicistronic RNAs derived from an infectious molecular clone of the HCV-N strain of hepatitis C virus replicate efficiently in cultured Huh7 cells. J. Virol. 76:2997–3006. 10.1128/JVI.76.6.2997-3006.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poordad F, McCone J, Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, DiNubile MJ, Sniukiene V, Brass CA, Albrecht JK, Bronowicki JP. 2011. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 364:1195–1206. 10.1056/NEJMoa1010494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci P, Flisiak R, George J, Rizzetto M, Shouval D, Sola R, Terg RA, Yoshida EM, Adda N, Bengtsson L, Sankoh AJ, Kieffer TL, George S, Kauffman RS, Zeuzem S. 2011. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 364:2405–2416. 10.1056/NEJMoa1012912 [DOI] [PubMed] [Google Scholar]
- 13.Coelmont L, Hanoulle X, Chatterji U, Berger C, Snoeck J, Bobardt M, Lim P, Vliegen I, Paeshuyse J, Vuagniaux G, Vandamme AM, Bartenschlager R, Gallay P, Lippens G, Neyts J. 2010. DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS One 5:e13687. 10.1371/journal.pone.0013687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Randall G, Panis M, Cooper JD, Tellinghuisen TL, Sukhodolets KE, Pfeffer S, Landthaler M, Landgraf P, Kan S, Lindenbach BD, Chien M, Weir DB, Russo JJ, Ju J, Brownstein MJ, Sheridan R, Sander C, Zavolan M, Tuschl T, Rice CM. 2007. Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. U. S. A. 104:12884–12889. 10.1073/pnas.0704894104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berger KL, Kelly SM, Jordan TX, Tartell MA, Randall G. 2011. Hepatitis C virus stimulates the phosphatidylinositol 4-kinase III alpha-dependent phosphatidylinositol 4-phosphate production that is essential for its replication. J. Virol. 85:8870–8883. 10.1128/JVI.00059-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lohmann V, Hoffmann S, Herian U, Penin F, Bartenschlager R. 2003. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol. 77:3007–3019. 10.1128/JVI.77.5.3007-3019.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van den Hoff MJ, Moorman AF, Lamers WH. 1992. Electroporation in ‘intracellular' buffer increases cell survival. Nucleic Acids Res. 20:2902. 10.1093/nar/20.11.2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Voitenleitner C, Bechtel J, Arfsten A, Hamatake R. 2012. Hepatitis C genotype 1a replicon improved through introduction of fitness mutations. Biotechniques 52:273–275. 10.2144/000113841 [DOI] [PubMed] [Google Scholar]
- 19.Blackham S, Baillie A, Al-Hababi F, Remlinger K, You S, Hamatake R, McGarvey MJ. 2010. Gene expression profiling indicates the roles of host oxidative stress, apoptosis, lipid metabolism, and intracellular transport genes in the replication of hepatitis C virus. J. Virol. 84:5404–5414. 10.1128/JVI.02529-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamarre D, Anderson PC, Bailey M, Beaulieu P, Bolger G, Bonneau P, Bos M, Cameron DR, Cartier M, Cordingley MG, Faucher AM, Goudreau N, Kawai SH, Kukolj G, Lagace L, Laplante SR, Narjes H, Poupart MA, Rancourt J, Sentjens RE, St GR, Simoneau B, Steinmann G, Thibeault D, Tsantrizos YS, Weldon SM, Yong CL, Llinas-Brunet M. 2003. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 426:186–189. 10.1038/nature02099 [DOI] [PubMed] [Google Scholar]
- 22.Miller JF, Chong PY, Shotwell JB, Catalano JG, Tai VW, Fang J, Banka AL, Roberts CD, Youngman M, Zhang H, Xiong Z, Mathis A, Pouliot JJ, Hamatake RK, Price DJ, Seal JW, III, Stroup LL, Creech KL, Carballo LH, Todd D, Spaltenstein A, Furst S, Hong Z, Peat AJ. 1 April 2013. Hepatitis C replication inhibitors that target the viral NS4B protein. J. Med. Chem. 10.1021/jm400125h [DOI] [PubMed] [Google Scholar]
- 23.Pierra C, Benzaria S, Amador A, Moussa A, Mathieu S, Storer R, Gosselin G. 2005. Nm 283, an efficient prodrug of the potent anti-HCV agent 2′-C-methylcytidine. Nucleosides Nucleotides Nucleic Acids 24:767–770. 10.1081/NCN-200060112 [DOI] [PubMed] [Google Scholar]
- 24.Carroll SS, Ludmerer S, Handt L, Koeplinger K, Zhang NR, Graham D, Davies ME, MacCoss M, Hazuda D, Olsen DB. 2009. Robust antiviral efficacy upon administration of a nucleoside analog to hepatitis C virus-infected chimpanzees. Antimicrob. Agents Chemother. 53:926–934. 10.1128/AAC.01032-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giuliano C, Fiore F, Di MA, Padron VJ, Bishop A, Bonelli F, Gonzalez-Paz O, Marcucci I, Harper S, Narjes F, Pacini B, Monteagudo E, Migliaccio G, Rowley M, Laufer R. 2005. Preclinical pharmacokinetics and metabolism of a potent non-nucleoside inhibitor of the hepatitis C virus NS5B polymerase. Xenobiotica 35:1035–1054. 10.1080/00498250500356548 [DOI] [PubMed] [Google Scholar]
- 26.Howe AY, Cheng H, Johann S, Mullen S, Chunduru SK, Young DC, Bard J, Chopra R, Krishnamurthy G, Mansour T, O'Connell J. 2008. Molecular mechanism of hepatitis C virus replicon variants with reduced susceptibility to a benzofuran inhibitor, HCV-796. Antimicrob. Agents Chemother. 52:3327–3338. 10.1128/AAC.00238-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw AN, Tedesco R, Bambal R, Chai D, Concha NO, Darcy MG, Dhanak D, Duffy KJ, Fitch DM, Gates A, Johnston VK, Keenan RM, Lin-Goerke J, Liu N, Sarisky RT, Wiggall KJ, Zimmerman MN. 2009. Substituted benzothiadizine inhibitors of hepatitis C virus polymerase. Bioorg. Med. Chem. Lett. 19:4350–4353. 10.1016/j.bmcl.2009.05.091 [DOI] [PubMed] [Google Scholar]
- 28.Prichard MN, Shipman C., Jr 1990. A three-dimensional model to analyze drug-drug interactions. Antiviral Res. 14:181–205. 10.1016/0166-3542(90)90001-N [DOI] [PubMed] [Google Scholar]
- 29.Bliss CI. 1939. The toxicity of poisons applied jointly. Ann. Appl. Biol. 26:585–615. 10.1111/j.1744-7348.1939.tb06990.x [DOI] [Google Scholar]
- 30.Buckwold VE, Wei J, Wenzel-Mathers M, Russell J. 2003. Synergistic in vitro interactions between alpha interferon and ribavirin against bovine viral diarrhea virus and yellow fever virus as surrogate models of hepatitis C virus replication. Antimicrob. Agents Chemother. 47:2293–2298. 10.1128/AAC.47.7.2293-2298.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Powdrill MH, Tchesnokov EP, Kozak RA, Russell RS, Martin R, Svarovskaia ES, Mo H, Kouyos RD, Gotte M. 2011. Contribution of a mutational bias in hepatitis C virus replication to the genetic barrier in the development of drug resistance. Proc. Natl. Acad. Sci. U. S. A. 108:20509–20513. 10.1073/pnas.1105797108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O'Boyle DR, Lemm JA, Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA, Hamann LG. 2010. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465:96–100. 10.1038/nature08960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fridell RA, Wang C, Sun JH, O'Boyle DR, Nower P, Valera L, Qiu D, Roberts S, Huang X, Kienzle B, Bifano M, Nettles RE, Gao M. 2011. Genotypic and phenotypic analysis of variants resistant to hepatitis C virus nonstructural protein 5A replication complex inhibitor BMS-790052 in humans: in vitro and in vivo correlations. Hepatology 54:1924–1935. 10.1002/hep.24594 [DOI] [PubMed] [Google Scholar]
- 34.Fridell RA, Qiu D, Wang C, Valera L, Gao M. 2010. Resistance analysis of the hepatitis C virus NS5A inhibitor BMS-790052 in an in vitro replicon system. Antimicrob. Agents Chemother. 54:3641–3650. 10.1128/AAC.00556-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aloia AL, Locarnini S, Beard MR. 2012. Antiviral resistance and direct-acting antiviral agents for HCV. Antivir. Ther. 17:1147–1162. 10.3851/IMP2426 [DOI] [PubMed] [Google Scholar]
- 36.Zein NN. 2000. Clinical significance of hepatitis C virus genotypes. Clin. Microbiol. Rev. 13:223–235. 10.1128/CMR.13.2.223-235.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simmonds P, Holmes EC, Cha TA, Chan SW, McOmish F, Irvine B, Beall E, Yap PL, Kolberg J, Urdea MS. 1993. Classification of hepatitis C virus into six major genotypes and a series of subtypes by phylogenetic analysis of the NS-5 region. J. Gen. Virol. 74:2391–2399. 10.1099/0022-1317-74-11-2391 [DOI] [PubMed] [Google Scholar]
- 38.Sarrazin C, Zeuzem S. 2010. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 138:447–462. 10.1053/j.gastro.2009.11.055 [DOI] [PubMed] [Google Scholar]
- 39.Tellinghuisen TL, Foss KL, Treadaway JC, Rice CM. 2008. Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J. Virol. 82:1073–1083. 10.1128/JVI.00328-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tellinghuisen TL, Foss KL, Treadaway J. 2008. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 4:e1000032. 10.1371/journal.ppat.1000032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hughes M, Griffin S, Harris M. 2009. Domain III of NS5A contributes to both RNA replication and assembly of hepatitis C virus particles. J. Gen. Virol. 90:1329–1334. 10.1099/vir.0.009332-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blight KJ, Kolykhalov AA, Rice CM. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1974. 10.1126/science.290.5498.1972 [DOI] [PubMed] [Google Scholar]
- 43.Cordek DG, Bechtel JT, Maynard AT, Kazmierski WM, Cameron CE. 2011. Targeting the NS5A protein of HCV: an emerging option. Drugs Future 36:691–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Love RA, Brodsky O, Hickey MJ, Wells PA, Cronin CN. 2009. Crystal structure of a novel dimeric form of NS5A domain I protein from hepatitis C virus. J. Virol. 83:4395–4403. 10.1128/JVI.02352-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tellinghuisen TL, Marcotrigiano J, Rice CM. 2005. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 435:374–379. 10.1038/nature03580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhattacharya D, Ansari IU, Hamatake R, Walker J, Kazmierski W, Striker R. 30 August 2013. Pharmacologic disruption of hepatitis C non-structural 5A (NS5A) intra and intermolecular conformations. J. Gen. Virol. 10.1099/vir.0.054569-0 [DOI] [PubMed] [Google Scholar]
- 47.Guedj J, Dahari H, Rong L, Sansone ND, Nettles RE, Cotler SJ, Layden TJ, Uprichard SL, Perelson AS. 2013. Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Proc. Natl. Acad. Sci. U. S. A. 110:3991–3996. 10.1073/pnas.1203110110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilfret D, Walker J, Adkison K, Jones L, Lou Y, Gan J, Castellino S, Moseley C, Horton J, SMde Culp A, Goljer I, Spreen W. 2013. Safety, tolerability, pharmacokinetics, and antiviral activity of GSK2336805, an inhibitor of hepatitis C virus (HCV) NS5A, in healthy subjects and subjects chronically infected with HCV genotype 1. Antimicrob. Agents Chemother. 57:5037–5044. 10.1128/AAC.00910-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gardner S, Cutrell A, Elko-Simms C, Adkison K, Hamatake R, Walker J, Rodriguez-Torres M, Hong Z. 25 September 2013. A double-blind, randomized, placebo-controlled study to assess the safety, antiviral activity, and pharmacokinetics of GSK2336805 when given as monotherapy and in combination with peginterferon alfa-2a and ribavirin in hepatitis C virus genotype 1-infected treatment-naive subjects. Liver Int. 10.1111/liv.12334 [DOI] [PubMed] [Google Scholar]
- 50.Zeuzem S, Andreone P, Pol S, Lawitz E, Diago M, Roberts S, Focaccia R, Younossi Z, Foster GR, Horban A, Ferenci P, Nevens F, Mullhaupt B, Pockros P, Terg R, Shouval D, HBvan Weiland O, Van HR, De MS, Luo D, Boogaerts G, Polo R, Picchio G, Beumont M. 2011. Telaprevir for retreatment of HCV infection. N. Engl. J. Med. 364:2417–2428. 10.1056/NEJMoa1013086 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.