

Abstract
The retinoblastoma tumor-suppressor protein (pRb) is known to induce growth arrest and cellular differentiation. The molecular determinants of pRb function include protein–protein interactions and post-translational modifications such as phosphorylation. Recently, the co-activator p300 was found to acetylate pRb. The biological significance of pRb acetylation, however, remains unclear. In the present study, we provide evidence that pRb undergoes acetylation upon cellular differentiation, including skeletal myogenesis. In addition to p300, the p300-Associated Factor (P/CAF) can mediate pRb acetylation as pRb interacts directly with the acetyltransferase domain of P/CAF in vitro and can associate with P/CAF in differentiated cells. Significantly, by using a C terminal acetylation-impaired mutant of pRb, we reveal that acetylation does not affect pRb-dependent growth arrest or the repression of E2F transcriptional activity. Instead, acetylation is required for pRb-mediated terminal cell cycle exit and the induction of late myogenic gene expression. Based on these results, we propose that acetylation regulates the differentiation-specific function(s) of pRb.
Keywords: acetylation, differentiation, P/CAF, retinoblastoma protein
Introduction
The retinoblastoma (Rb) gene family consists of three members, Rb, p107 and p130, known to encode ‘pocket' proteins that regulate the mammalian cell cycle. More specifically, pRb family members have overlapping roles in promoting cellular growth arrest. Targeted inactivation of all the three Rb-related genes in embryonic stem cells causes deregulated G1/S transition, abrogation of G1 arrest and cellular immortalization (Dannenberg et al, 2000; Sage et al, 2000). The underlying mechanism of these observations involves the common ability of pocket proteins to interact with the E2F family of transcription factors. This is exemplified by the activity of pRb, which, once in a hypophosphorylated state, can bind to E2F-1 or E2F-3 and repress E2F-induced expression of genes normally required for cell cycle progression (Dyson, 1998; Harbour and Dean, 2000).
Despite the apparent similarities, multiple studies have now revealed the biochemical and biological differences that distinguish pRb from p107 and p130 (Classon and Dyson, 2001). Most noteworthy is the fact that pRb is the only pocket protein known to exhibit features of a bona-fide tumor suppressor. Accordingly, Rb heterozygous mice are predisposed to the onset of pituitary and thyroid cancer (Clarke et al, 1992; Jacks et al, 1992), and deregulation of the Rb signaling pathway or inactivation of Rb itself is a hallmark of nearly all human tumors (Hanahan and Weinberg, 2000). Furthermore, genetic experiments have demonstrated a unique role for Rb in metazoan development and cellular differentiation. Germline deletion of Rb in mice, for instance, results in embryonic lethality between 13 and 15 days of gestation (Clarke et al, 1992; Jacks et al, 1992; Lee et al, 1992), whereas p107−/− or p130−/− mice are seemingly viable (Cobrinik et al, 1996; Lee et al, 1996). More recent reports, however, suggest that the penetrance of p107/p130 null phenotypes may depend on the genetic background (LeCouter et al, 1998a, 1998b). Nonetheless, Rb−/− animals exhibit distinct developmental abnormalities, with deregulated proliferation, cell death and defects in differentiation seen in multiple embryonic tissues. Although a compound E2F-1 deletion extends the survival of Rb−/− animals, these mice eventually die of disruptions in erythroid, pulmonary and muscular development (Yamasaki et al, 1998). Consequently, important function(s) of the pRb tumor suppressor are not restricted to the regulation of E2F activity during cell cycle progression.
In vitro tissue culture models also support the notion that pRb possesses a unique role in mediating differentiation. This is best demonstrated by the requirement for Rb during skeletal myogenesis (Gu et al, 1993; Schneider et al, 1994; Zacksenhaus et al, 1996). Three coordinated yet distinct biological events are known to occur during muscle differentiation. The first involves an initial growth arrest linked to the upregulation of the cell cycle inhibitor p21cip1 and repression of E2F-regulated proliferative genes. This early phase of the myogenic program does not seem to necessitate pRb, as other pocket proteins can substitute for loss of Rb in inducing growth arrest and Rb−/− myoblasts are capable of expressing early markers of differentiation (Schneider et al, 1994; Novitch et al, 1996). Following this acute growth arrest, a more permanent cell cycle withdrawal is considered necessary for the later stages of differentiation. This then renders myotubes conducive to the expression of late markers including the myosin heavy chain (MyHC). Rb-deficient cells are incapable of undergoing terminal arrest, exhibit delayed expression of late differentiation markers and can re-enter the cell cycle. Significantly, the aberrant myogenic phenotype associated with Rb disruption does not occur in cells lacking p107 or p130 (Novitch et al, 1996). Thus, pRb fills a more specific role throughout the later phases of myogenesis. Consistent with animal models, the ability of pRb to promote differentiation is independent of acute G1/S block and does not require stable binding to E2F (Sellers et al, 1998). Instead, preliminary efforts led to the idea that pRb can potentiate the muscle-specific transcription factor MyoD (Gu et al, 1993). Although several studies have confirmed the ability of pRb to stimulate MyoD-dependent transcription, this cooperation is probably of an indirect nature, as the association of pRb with MyoD in vivo remains controversial (Li et al, 2000). Alternative models propose that pRb binding to inhibitors of MyoD, such as the E1A-like inhibitor of differentiation-1 (EID-1) or histone deacetylases (HDACs), leads to ‘de-repression' of MyoD activity (MacLellan et al, 2000; Miyake et al, 2000; Puri et al, 2001). Multiple biochemical factors are likely to contribute to the overall functions of pRb required for differentiation.
In addition to pRb, other proteins are known to cooperate with MyoD in promoting muscle differentiation. Among these are the transcriptional co-activators CREB-binding protein (CBP), p300 and the p300-associated factor (P/CAF), which possess intrinsic acetyltransferase activity (Bannister and Kouzarides, 1996; Ogryzko et al, 1996; Yang et al, 1996) and are required for myogenesis (Puri et al, 1997; Polesskaya et al, 2001b). p300 and CBP are in fact known to regulate the activity of numerous transcription factors (Vo and Goodman, 2001), and possess conserved functions in cell growth, transformation and development (Goodman and Smolik, 2000). P/CAF has likewise been linked to differentiation and tumorigenesis (Yang et al, 1996; Schiltz and Nakatani, 2000). Current models propose that p300/CBP and P/CAF can form a multimeric complex with MyoD, which recruits these co-activators to muscle-specific promoters (Eckner et al, 1996; Yuan et al, 1996; Puri et al, 1997; Sartorelli et al, 1997). P/CAF and p300/CBP are then believed to hyperacetylate the surrounding nucleosomes, thus increasing the accessibility of additional transcription factors to MyoD target promoters (McKinsey et al, 2001). Interestingly, besides acetylating histone tails, p300/CBP and P/CAF can acetylate MyoD itself at conserved lysine residues (Sartorelli et al, 1999; Polesskaya et al, 2000; Polesskaya and Harel-Bellan, 2001). The acetylation of MyoD is postulated to increase DNA binding and is important for MyoD activity. Remarkably, p300/CBP and P/CAF have now been demonstrated to acetylate a wide variety of non-histone proteins, including many DNA-binding proteins, also general transcription factors, cytoplasmic proteins and tumor suppressors (Kouzarides, 2000).
Recently, the co-activator p300 was shown to acetylate pRb in vitro (Chan et al, 2001). The mechanism and biological relevance of pRb acetylation, however, remain unclear. In the present study, we provide evidence that the differentiation-specific functions of pRb are modulated by acetylation.
Results
pRb is acetylated upon cellular differentiation
The aim of the present study was to determine the importance of pRb acetylation during cellular differentiation. To this end, the acetylation state of pRb was examined in various differentiation cell culture systems. Murine C2C12 myoblasts are induced to differentiate into myotubes by growth to confluence and depletion of serum (DM). Using this system, expression of the late myogenic marker, MyHC, was detected by 36 h (Figure 1A, left). Acetylation of pRb was examined via immunoprecipitation of C2C12 nuclear extracts using either anti-pRb or control IgG antibody, followed by immunoblotting with an antibody that detects acetylated lysine residues (Ac-K). As shown in Figure 1A (right panel), acetylated pRb was more prevalent in differentiated (DM) extracts when compared to undifferentiated cells (GM). When the same blot was stripped and re-probed for pRb, a predominance of the active, hypophosphorylated (faster migrating) form of pRb was detected in differentiated myotubes. The acetylation state of pRb was also assessed in human foreskin keratinocytes (HFKs) that undergo cellular differentiation upon suspension in methylcellulose. Significantly, pRb became acetylated as the HFK differentiation marker involucrin was induced (Figure 1B). Taken together, these results demonstrate that pRb acetylation occurs upon cellular differentiation and that this regulatory process is conserved.
Figure 1.

pRb is acetylated upon cellular differentiation. (A) Asynchronous C2C12 cells were grown in 20% FCS and harvested at low confluence (GM), or grown to high confluence and incubated in 1% horse serum (DM). Samples were harvested following 18 and 36 h in DM. C2C12 differentiation was confirmed by immunoblotting for MyHC from whole-cell lysates (left). In the right panel, acetylation of pRb was detected by immunoprecipitating nuclear-enriched extracts with IgG control or an anti-pRb agarose-conjugated antibody. Immunoprecipitated samples were run on an SDS–PAGE gel, transferred to nitrocellulose and probed with anti-acetylated lysine (Ac-K) antibody. Blots were stripped and re-probed using anti-pRb to confirm the presence of an overlapping pRb band. (B) Asynchronous primary HFKs were either harvested as cycling (cyc) or suspended in methylcellulose. Differentiated cells (diff) were collected after 24 h in methylcellulose, and differentiation-induced expression of involucrin was confirmed by RT–PCR (left panel). The acetylation state of pRb was detected as in (A).
Myogenesis induces pRb to associate with P/CAF
As P/CAF is necessary for both muscle and keratinocyte differentiation (Puri et al, 1997; Kawabata et al, 2002), we examined whether pRb can form a complex with P/CAF. As demonstrated in Figure 2B, in vitro translated (IVT) P/CAF bound to the pRb large pocket (pRbLp). Deletion mutations at exons 21 and 22 (pRbLpΔ21, pRbLpΔ22) in the pocket region did not noticeably affect binding to P/CAF. The pRbA (aa 379–610, Figure 2A) domain alone did not pull down P/CAF, while the B (aa 612–767) and C (aa 792–928) regions of pRb interacted with P/CAF, albeit less efficiently when used individually. To confirm that pRb binds directly to P/CAF, the His-tagged HAT domain of P/CAF (aa 353–658, Figure 2A) was purified from bacteria and used in pull-down assays with full-length recombinant pRb (GST-pRbFl). This HAT domain was sufficient to interact directly with pRb (Figure 2C), and was as potent as the association of P/CAF with p53, a well-known in vitro and in vivo binding partner of P/CAF (Liu et al, 1999). Next, we used a recombinant P/CAF fragment lacking its first 351 amino acids (ΔN-P/CAF) in an attempt to co-purify endogenous pRb from differentiated C2C12 extract. ΔN-P/CAF does not interact with p300 (Reid et al, 1998), yet readily precipitated endogenous pRb (Figure 2D). Thus, P/CAF and pRb might form a complex in cells independently of p300. Having established that pRb and P/CAF interact in vitro, we attempted to determine whether such a complex could be detected in vivo. As pRb levels increase upon myogenesis (Figure 1A), GM and DM nuclear lysates were standardized for equal pRb levels before being immunoprecipitated. Consequently, pRb was found to co-precipitate with endogenous P/CAF and this association was significantly increased in differentiated cells (Figure 2E). Furthermore, endogenous P/CAF binding correlated with the hypophosphorylated and acetylated form of pRb (Figure 2E, lower panels). Similar observations were obtained in differentiating HFKs (data not shown). Thus, cellular differentiation induces P/CAF to associate with pRb.
Figure 2.
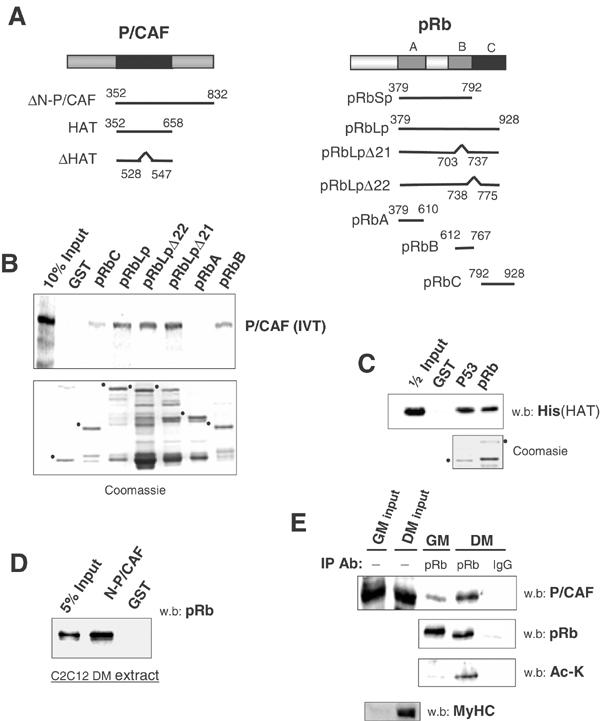
pRb associates with P/CAF. (A) Schematic diagram of pRb and P/CAF fragments used in this study. (B) In vitro translated 35S-labeled full-length P/CAF was incubated with the indicated GST-pRb fragments in a standard pull-down assay. Samples were resolved via SDS–PAGE gel; bound P/CAF was analyzed using phosphoimaging, while appropriate GST loading was confirmed via coomassie staining of a replica gel (bottom). Dots designate the running positions of GST proteins used. (C) Full-length GST-pRb or GST-p53 were incubated with the His-tagged HAT domain of P/CAF in a pull-down experiment and analyzed via Western blot. (D) GST-ΔN-P/CAF (Δ1–351) was used to purify endogenous pRb from differentiated C2C12 extracts. (E) GM or DM C2C12 extracts were standardized for pRb levels, immunoprecipitated as in Figure 1 and subjected to Western blot analysis for endogenous P/CAF. Samples immunoprecipitated in parallel were blotted with anti-Ac-K. The Ac-K blot was stripped and re-probed with anti-pRb.
P/CAF mediates acetylation of pRb
To assess whether P/CAF could also directly catalyze the acetylation of pRb, we used GST-pRbLp as a putative substrate for the HAT domain of P/CAF in an acetyltransferase assay. In addition, acetylation of pRbLp was compared to that of the small pocket (pRbSp: aa 379–792), which lacks the C terminus of pRb. Recombinant P/CAF was able to acetylate specifically the large pocket of pRb up to five-fold over background levels, as detected via Western blot (Figure 3A). Conversely, the small pocket of pRb was not markedly acetylated, even though it was capable of binding to P/CAF. We extrapolate from this result that the C terminus of pRb contains a major site(s) targeted by P/CAF for acetylation. Importantly, P/CAF could also enhance pRb acetylation in differentiating C2C12 cells (Figure 3B, lane 3). When an inactive version of P/CAF lacking its acetyltransferase domain (Flag-P/CAFΔHAT) was co-transfected along with HA-pRb, pRb acetylation was reduced, potentially due to a lower capacity of P/CAFΔHAT to associate with pRb (Figure 3B, lane 4). Consistent with a role for p300 in pRb acetylation, overexpression of p300 in C2C12 cells also increased detectable levels of acetylated pRb (Figure 3B, lane 5). Interestingly, co-transfection of P/CAF and p300 along with pRb resulted in an overall increase in pRb acetylation compared to samples transduced with either co-activator alone (Figure 3B, last lane). In sum, we conclude that P/CAF is a mediator of pRb acetylation, and that it can cooperate with p300 in fulfilling this activity.
Figure 3.
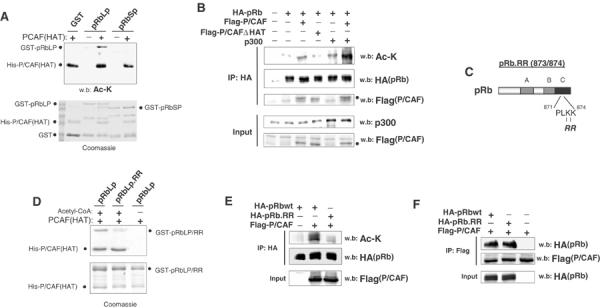
P/CAF mediates acetylation of pRb. (A) GST-pRbLp or GST-pRbSp were incubated with His-P/CAF(HAT) in an acetyltransferase assay with cold acetyl-CoA. Reaction products were separated via SDS–PAGE. Upper panel: Ac-K Western blot. Lower panel: coomassie-stained replica gel. (B) C2C12 cells were transfected with the indicated constructs before incubation in DM for 24 h. Lysates were immunoprecipitated with anti-HA(pRb) and subjected to Western blot analysis for the presence of acetylated pRb using anti-Ac-K. Dots indicate the presence of Flag-P/CAF or Flag-P/CAFΔHAT. (C) Schematic diagram of the pRb.RR mutant, in which lysines 873/874 in the C terminus have been mutated to non-acetylatable arginine residues. (D) GST-pRbLp or GST-pRbLp.RR were incubated with His-P/CAF(HAT) and radiolabeled 14C-acetyl-CoA as previously mentioned. (E) COS-1 cells were co-transfected with Flag-P/CAF and either wild-type pRb or the pRb.RR mutant. COS-1 cells were used here as they do not undergo differentiation and provide more limiting conditions for acetylation in cells. Acetylation of pRb or pRb.RR was determined as in (B). (F) Cells were transfected as in (E). Lysates were immunoprecipitated with anti-Flag (P/CAF) and blotted for HA (pRb/pRb.RR).
Previously, p300 was found to acetylate the C terminus of pRb in vitro at lysine residues 873/874 (Chan et al, 2001). As our results demonstrate that P/CAF equally acetylates the C terminus of pRb in vitro, and that P/CAF cooperates with p300 in acetylating pRb in cells, we reasoned that amino acids 873/874 of pRb might be targeted by P/CAF as well. To verify this hypothesis, lysine residues 873/874 of pRb were mutated to arginine (RR), a change that preserves the overall charge, but inhibits acetylation of the sites in question. The resulting mutant is referred to as pRb.RR (Figure 3C). When used in vitro within the context of pRbLP, the RR mutant displayed a reduced capacity for P/CAF-mediated acetylation (Figure 3D). This was confirmed in COS-1 cells that were co-transfected with Flag-P/CAF and full-length pRb or pRb.RR (Figure 3E). Moreover, this decrease in acetylation was not caused by defects in P/CAF binding, as pRb.RR co-precipitates with Flag-P/CAF (Figure 3F). We note that the residual acetylation of pRb.RR is likely due to the presence of additional unidentified acetylated sites. Nevertheless, we conclude that lysine residues 873/874 are important sites of pRb acetylation.
pRb acetylation is required for permanent cell cycle withdrawal and differentiation-specific gene expression
To identify which biological function(s) requires pRb acetylation, the acetylation-impaired mutant pRb.RR was expressed in various Rb-deficient cell lines. Consistent with previous experiments, the expression of either wild-type pRb or pRb.RR in Rb−/− SAOS-2 cells resulted in G1/S growth arrest (data not shown) (Chan et al, 2001), demonstrating that pRb acetylation is not required for acute cell cycle arrest. Following these results, the requirement for pRb acetylation was more specifically examined within the context of cellular differentiation. As the genetic requirement for Rb during muscle development has been firmly established, skeletal myogenesis was used as our model system. Moreover, Rb−/− 3T3 fibroblasts were employed because ectopic expression of MyoD in normal 3T3 cells can induce myogenesis, while MyoD-Rb−/− cells do not differentiate properly and are incapable of undergoing permanent cell cycle withdrawal (Novitch et al, 1996). 3T3 cells were infected with amphotrophic retrovirus encoding MyoD or an empty vector to create stable cell lines (MyoD-Rb−/− or pBabe-Rb−/−) that were then transfected with pRb or the pRb.RR mutant. Finally, subconfluent cells were either starved in DM media for 24 h (DM) or starved for 48 h before being re-stimulated with 10% serum for 18 h (serum).
When MyoD was stably expressed alone in an Rb−/− background, cells failed to undergo growth arrest upon incubation in DM, with approximately 15% of GFP-positive cells maintaining DNA synthesis as measured by BrdU incorporation (Figure 4A). This is in agreement with the fact that Rb−/− 3T3 fibroblasts are deficient for growth arrest upon serum starvation (Classon et al, 2000) and that MyoD-induced arrest requires Rb (Peschiaroli et al, 2002). Predictably, these samples had a higher proliferative potential when re-incubated in serum-containing media. Expression of pRb in MyoD-Rb−/− fibroblasts rescued the arrest mediated by growth factor depletion and further rendered transduced cells refractory to mitogenic stimulation (Figure 4A), the latter being a hallmark of cells committed towards differentiation. When pRb.RR was introduced into a MyoD-Rb−/− background, cells initially arrested indistinguishably from wild-type pRb when starved. Importantly though, 15% of pRb.RR transfected cells re-entered the cell cycle upon addition of serum, indicating that cells expressing the acetylation defective mutant maintain their proliferative potential. Similar experiments were performed with CC42 muscle precursor cells, which do not exhibit terminal growth arrest upon differentiation due to impaired pRb function (Schneider et al, 1994; Chen and Wang, 2000). Stable expression of pRb or pRb.RR in CC42 myotubes confirmed that the acetylation defective mutant could not establish a growth refractory state (Figure 4B). Moreover, while wild-type pRb retained a more hypophosphorylated (p) state following serum re-stimulation, pRb.RR was predominantly hyperphosphorylated (pp), consistent with its inability (Figure 4C) to establish permanent cell cycle withdrawal.
Figure 4.

Acetylation of pRb at lysine residues 873/874 is required for permanent cell cycle withdrawal. (A) Rb−/− fibroblasts were infected with amphotrophic retrovirus expressing MyoD or empty vector. Following selection, stable control Rb−/− or MyoD-Rb−/− cell lines were transfected with pRb.wt, pRb.RR or vector control. A GFP construct was included in all samples at 1/10 the total amount of DNA. Cells were then either starved in DM for 24 h or starved for 48 h before being re-stimulated with 10% serum, labeled with BrdU and fixed for immunostaining. Proliferative cells incorporating BrdU were identified under fluorescent microscopy and scored as a percentage of BrdU-GFP double-positive cells over the total number of GFP transfected cells. (B) CC42 myoblasts were co-transfected with pRb or pRb.RR along with a puromycin-expressing vector. Stable transformants were isolated following selection, incubated in GM (asynchronous cycling), DM (for 72 h) or DM followed by re-stimulation in 20% serum for 18 h. BrdU-incorporating cells were analyzed via flow cytometric analysis. (C) Immunoblots detecting HA-pRb from stable CC42 lines in (B).
As acetylation of pRb is necessary to cooperate with MyoD in establishing permanent growth arrest, we hypothesized that pRb acetylation might also affect other MyoD-dependent functions, such as differentiation-specific gene expression. To test this, MyoD-Rb−/− lines were transfected as in Figure 4 and the steady-state levels of myogenic markers were examined. As previously demonstrated, pRb had little or no effect on MyoD-dependent expression of the early markers of differentiation myogenin (Figure 5A, left) and p21cip1 (right), while both MyoD and pRb were required for optimal induction of the late marker MyHC (Figure 5A). When pRb.RR was expressed, no defects were seen on myogenin or p21cip1 expression. However, induction of MyHC was reduced compared to wild-type pRb/MyoD-expressing cells, suggesting a failure of the pRb.RR mutant to stimulate MyoD activity fully. Finally, immunofluorescent microscopy revealed that even at later time points (DM=72 h), cells expressing pRb.RR displayed a more diffuse pattern of MyHC expression (Figure 5B) as well as an overall lower percentage of MyHC-positive cells compared to wild-type pRb (Figure 5C). We conclude from these results that acetylation of pRb at residues 873/874 is dispensable for the early stages of differentiation, including acute cell cycle arrest and expression of early markers. More importantly, pRb acetylation is required to establish permanent cell cycle withdrawal and induction of late differentiation gene expression.
Figure 5.
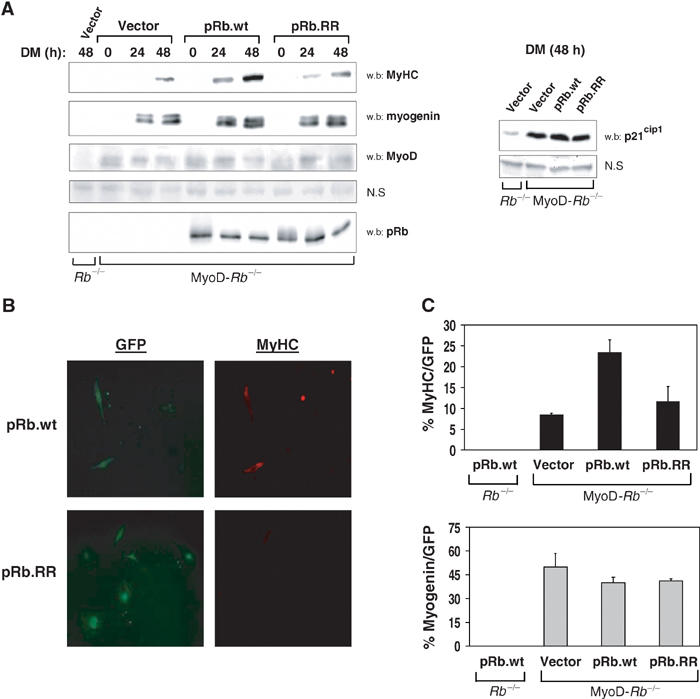
Acetylation of pRb at lysine residues 873/874 is required for optimal expression of late differentiation markers. (A) Control Rb−/− or MyoD-Rb−/− cells were transfected as in Figure 4, grown to confluence and starved in DM for the indicated times. Steady-state levels of MyHC, myogenin, p21cip1, MyoD and pRb were assessed via Western blot. MyoD appears as a doublet, previously described as different phosphorylated forms. Endogenous MyoD was detectable upon overexposure. Nonspecific (NS) bands serve as loading controls. (B) MyoD-Rb−/− samples were transfected with pRb or pRb.RR along with GFP and fixed after 72 h in DM for MyHC and myogenin immunostaining. Depicted are representative fields (similar exposure times) of GFP and MyHC-positive cells. (C) Quantitation of MyHC/GFP and myogenin/GFP-positive cells following 72 h in DM.
Mutation of pRb-acetylated residues 873/874 does not affect E2F transcriptional repression, but results in impaired MyoD-dependent transactivation
The molecular properties of pRb can be categorized into two distinct activities: (1) pRb transcriptional repression of cell cycle-regulated genes and (2) its ability to act as a differentiation-specific co-activator. To determine whether these activities are regulated by acetylation, the pRb.RR mutant was used in transcriptional reporter assays. In the first set of experiments, the ability of pRb.RR to repress E2F-1 transcription was examined. Rb−/− 3T3 cells were transfected with Gal4-E2F-1, a Gal4 reporter plasmid and pRb or the indicated mutants. Luciferase activity was measured following 24 h of serum starvation. The pocket mutant pRbΔ22, which does not induce cell cycle arrest or muscle-specific gene expression (Sellers et al, 1998), was included as a negative control and is unable to repress E2F transcription when compared to wild-type pRb (Figure 6A). In contrast, pRb.RR was still capable of significantly repressing E2F activity. Furthermore, co-transfection of P/CAF with wild-type pRb did not alter E2F repression (data not shown). In Figure 6B, pRb-mediated co-activation was analyzed using a reporter construct responsive to MyoD activation. Rb-negative cell lines were transfected with 4RE-luc, MyoD and limiting amounts of pRb or the indicated mutants, followed by incubation in DM. Interestingly, while pRb was found to stimulate MyoD activity, pRb.RR was impaired in co-activating MyoD-dependent transcription (Figure 6B). These results were confirmed by examining the endogenous levels of cyclin A, a marker of proliferation that is repressed by pRb, and MyHC that is activated by MyoD (Figure 6C). Acetylation is thus required for pRb to function as a co-activator, but dispensable for transcriptional repression.
Figure 6.

Mutation of pRb-acetylated residues 873/874 does not affect E2F transcriptional repression, but results in impaired MyoD-dependent transactivation. (A) Rb−/− 3T3 cells were transfected with plasmids encoding the E2F-1 transactivating domain fused to Gal-4 (Gal4-E2F), pRb or the indicated pRb mutants, and a Gal-4 luc reporter. Following transfection, cells were starved for 24 h before luciferase activity was measured and standardized. (B) The co-activating potential of pRb was assessed by transfecting Rb−/− 3T3s with pRb, pRb.RR or pRbΔ22, along with MyoD and the MyoD-responsive reporter 4RE-luc. Transfected cells were incubated in DM for 24 h prior to measurements of luciferase activity. A representative of three independent experiments is shown in both (A) and (B). (C) Samples transiently expressing MyoD and pRb or pRb mutants were differentiated for 32 h and the levels of the indicated proteins were examined via Western blot.
pRb acetylation increases MDM2 binding and degradation of the inhibitor of differentiation EID-1
Two nonexclusive models have been proposed to explain how pRb can activate transcription during differentiation. Both hypotheses imply that pRb indirectly potentiates MyoD activity by associating with inhibitors of MyoD. It has been postulated, for instance, that the interaction between pRb and HDACs sequesters deacetylase activity from MyoD, thus alleviating HDAC-mediated repression of muscle-specific genes (Puri et al, 2001). Alternatively, a recently cloned repressor of MyoD transcription, the E1A-like inhibitor of differentiation (EID-1), can associate with pRb and is degraded upon terminal growth arrest/myogenesis (MacLellan et al, 2000; Miyake et al, 2000). The degradation of EID-1 requires the concomitant association of pRb to the ubiquitin ligase MDM2, which then targets EID-1 for proteosomal destruction (Miyake et al, 2000). It is within the context of these models that we examined the mechanism by which acetylation affects pRb.
To accomplish this, wild-type HA-tagged pRb or HA-pRb.RR was expressed in C2C12 cells and immunoprecipitated following the induction of myogenesis for 18 h. In the case of EID-1, a construct encoding a T7-tagged version of EID-1 was transfected with pRb, because antibodies against endogenous murine EID-1 are not readily available. Samples immunoprecipitated in parallel revealed that wild-type pRb co-precipitated MDM2, HDAC-1 and T7-EID-1 in myotubes (Figure 7A). Interestingly, mutations of pRb lysine residues 873/874 to arginine reduced the efficiency of MDM2 binding without affecting the amount of pRb-associated HDAC-1 or EID-1. This is consistent with HDAC and EID-1 interactions occurring through the pocket region of pRb (Brehm et al, 1998; Magnaghi-Jaulin et al, 1998; Miyake et al, 2000), whereas MDM2 association takes place in the C terminus (Xiao et al, 1995). Also, overexpression of P/CAF with p300 in vivo can increase the affinity of pRb for MDM2 (data not shown). Finally, during C2C12 differentiation, MDM2 associated with endogenous pRb following either simultaneous or delayed kinetics relative to P/CAF binding and pRb acetylation (Figure 7B). To confirm the functional output of reduced MDM2 binding, we examined the ability of pRb.RR to initiate EID-1 degradation. C2C12 cells were transfected and differentiated for 24 h as before and de novo protein synthesis was inhibited by pre-incubating samples in cyclohexamide. Analysis of whole-cell lysates demonstrated that, even though pRb.RR can bind to EID-1, it was not able to promote EID-1 degradation as rapidly as wild-type pRb (Figure 7C and D), presumably due to its impaired recruitment of MDM2 and deficient EID-1 ubiquitination. As such, the modulation of pRb-MDM2 binding via acetylation has subsequent effects on the degradation of EID-1 and differentiation-specific gene expression.
Figure 7.
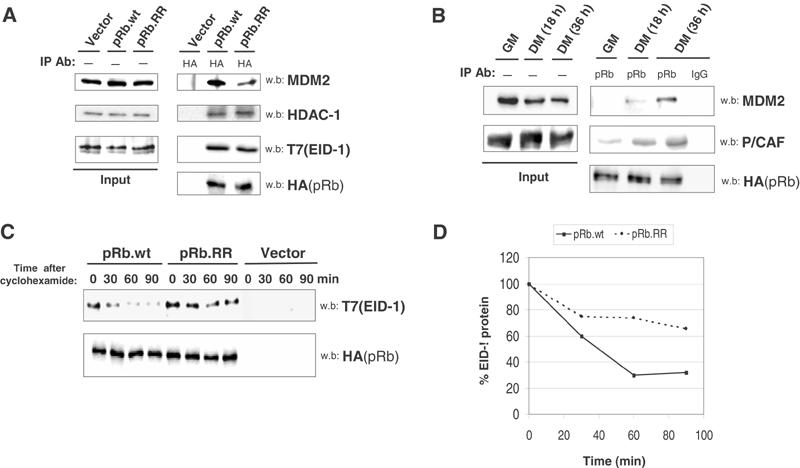
pRb acetylation correlates with binding to MDM2 and degradation of EID-1. (A) C2C12 cells were transfected with HA-pRb or HA-pRb.RR and incubated in DM for 24 h. Nuclear lysates were immunoprecipitated using anti-HA antibody, subjected to SDS–PAGE and Western blot analysis for HA-pRb-associated MDM2, HDAC-1 and EID-1 proteins. In the case of EID-1, a plasmid encoding T7-tagged EID-1 was co-transfected along with the various pRb mutants and detected by immunoblotting with anti-T7 antibody. (B) C2C12 cells were differentiated for the indicated amount of times and IP/Western blot analysis was performed as in Figure 1. Simultaneous sets of endogenous pRb IPs were blotted for associated MDM2 or P/CAF. (C) C2C12 myoblasts were co-transfected with empty vector, HA-pRb plus T7-EID-1 or HA-pRb.RR with T7-EID-1. Cells were maintained in DM for 24 h, followed by addition of 30 μg/ml cyclohexamide for 1 h. After this treatment (designated as T=0), whole-cell lysates were collected at the indicated chase time points. EID-1 and pRb protein stability was assessed via Western blot. (D) Quantitation of (C).
Discussion
The retinoblastoma protein (pRb) is the prototypical tumor suppressor and its molecular functions have been the subject of numerous studies. In the past decade, pRb has been shown to interact with over 100 cellular proteins (Classon and Dyson, 2001), involved in multiple biological processes, including cell cycle control, development and differentiation. Nonetheless, the physiological significance of most of these complexes remains elusive, and it has become important to ascertain the regulatory processes that coordinate pRb function with such a wide variety of biochemical targets. One potential mechanism is via covalent modification. It is a well-known fact that pRb can be phosphorylated during various stages of the cell cycle and that this in turn regulates the association/disassociation of pRb with proteins such as E2F (Buchkovich et al, 1989; Chen et al, 1989; Dynlacht et al, 1994). The results presented herein indicate that modification by acetylation may also serve as a method of controlling pRb functions, particularly during cellular differentiation. Indeed, we demonstrate that acetylation of pRb regulates its ability to cooperate with the tissue-specific transcription factor MyoD to induce cellular differentiation (Figures 4 and 5). Moreover, as pRb is acetylated during monocyte (Chan et al, 2001), keratinocyte and muscle differentiation (Figure 1), this regulatory process is likely to be conserved.
Although pRb and the co-activators p300/CBP and P/CAF all possess necessary roles in driving muscle differentiation (Novitch et al, 1996; Puri et al, 1997; Polesskaya et al, 2001b), the mechanism by which their functions are coordinated has been for the most part unknown. Our study provides a potential link between these acetyltransferases and pRb, by demonstrating that pRb acetylation is required for the establishment of permanent cell cycle withdrawal and expression of late differentiation markers. Furthermore, P/CAF may be an important regulator of these pRb-specific functions because it can interact directly with pRb (Figure 2) and promotes the latter's acetylation both in vitro and in vivo (Figure 3). Despite recent results demonstrating that p300 can also acetylate pRb in vitro (Chan et al, 2001), p300 is generally not believed to interact with pRb directly in vivo, and it has been suggested that additional cellular proteins promote the formation of such a complex in cells. Given the fact that P/CAF is found in complex with p300 (Yang et al, 1996) and that it can cooperate with p300 in acetylating pRb during myogenesis (Figure 3C), P/CAF could recruit p300 to pRb upon cellular differentiation. Alternatively, acetylation by one co-activator might promote the subsequent association of pRb with the other, and induce further acetylation. These models have been proposed to explain the cooperative nature of MyoD acetylation by p300 and P/CAF (Sartorelli et al, 1999; Polesskaya et al, 2001a), and warrant further investigation with regard to pRb.
It is interesting to note that acetylation of pRb is targeted to its C-terminal region, at lysine residues 873/874 (Figure 3). Although there is as of yet no identified consensus site for acetylation, the sequence encompassing pRb lysine residues 873/874 (PLKK) (Figure 3C) is homologous to P/CAF and p300 target sites found in other acetylated proteins (data not shown). This finding is also of significance as the pRb C domain is less conserved among the different pRb family members, and we were unable to locate sequences homologous to the one encompassing lysines 873/874 in p107 or p130 (data not shown). Our observations would suggest that acetylation at these discrete sites modulates functions particular to pRb, such as those performed during differentiation. Nonetheless, we do not rule out the possibility of all pocket proteins being modified by acetylation at other sites and/or under different biological conditions. A more systematic mapping of putative acetylation residues within pRb and its family members p107/p130 will be necessary in the future.
In spite of these considerations, the idea that acetylation regulates the differentiation-specific properties of pRb is supported by experiments in which we utilized the acetylation-impaired mutant pRb.RR. Inhibition of pRb acetylation at residues 873/874 was not seen to affect repression of E2F transcription (Figure 6A), a function shared by all pocket proteins. Instead, acetylation at these sites is required for pRb to act as a co-activator of differentiation-specific transcription and stimulate MyoD activity (Figure 6B). Altogether, the fact that pRb acetylation does not affect E2F repression is consistent with acetylation being dispensable for pRb-induced acute cell cycle arrest (Figure 4). Conversely, as pRb is known to cooperate with MyoD in maintaining cell cycle exit and regulating MyHC expression, the disrupted transactivating potential of pRb.RR correlates with its inability to establish terminal arrest (Figure 4B) and induce elevated MyHC levels (Figure 5) during myogenesis.
A mechanistic explanation for this phenotype is provided when acetylation was revealed to increase the affinity of pRb for MDM2 (Figure 7A), the latter being a ubiquitin ligase known to interact with the C terminus of pRb (Xiao et al, 1995). How acetylation might affect protein–protein interactions in this domain remains unclear, but may involve alterations in charge and/or structure. Although MDM2 itself is not generally considered a differentiation-specific factor, it has been shown to ubiquitinate and initiate the degradation of EID-1, an inhibitor of differentiation that forms a trimeric complex with pRb and MDM2 (Miyake et al, 2000). EID-1 is normally degraded upon myogenesis and it ensues that inhibition of pRb acetylation leads to stabilization of EID-1 (Figure 7C), which may prolong the repression of differentiation-specific genes. In this regard, the acetylation-impaired mutant pRb.RR behaves identically to C-terminal deletion fragments of pRb incapable of binding MDM2 (Miyake et al, 2000). Moreover, our study is consistent with the discovery that the pRb C terminus is necessary for myogenic conversion (Guo et al, 2003), and highlights the contribution of this domain to the overall function of pRb. Therefore, it will be interesting to examine the effects of pRb acetylation on its association with other putative C-terminal binding proteins.
In summary, our data lend credence to the idea that discrete residues in the pRb C terminus are targeted for acetylation upon differentiation, subsequently regulating protein–protein interactions in this latter region of pRb. We provide evidence that acetylation of pRb at residues 873/874 increases its affinity for MDM2 and promotes the degradation of EID-1, coinciding with permanent cell cycle exit and de-repression of MyoD-dependent transcription. Based on these results, pRb acetylation may serve as a molecular ‘switch', which selectively engages the functions of pRb required for cellular differentiation.
Materials and methods
Plasmids
pCX and pGEM plasmids encoding fragments of P/CAF, Gal4-E2F-1 (E2F-1 transactivating domain fused to the Gal4 DNA-binding region) as well as Gal4-luc reporter constructs were obtained from T Kouzarides (Martinez-Balbas et al, 2000). Plasmid of His-P/CAF HAT was generated by cloning the HAT domain of P/CAF into pET28A. pGEX-pRb plasmids were described elsewhere (Nead et al, 1998). pCDNA-p300 was obtained from S Grossman (Grossman et al, 1998); pSG5L-HA-Rb, pSG5l-HA-RbΔ22 and pSG5L-T7-EID-1 were provided by W Kaelin (Miyake et al, 2000). The pSG5L-HA-Rb.RR mutant was generated by converting aa 873/874 from KK → RR, via Quick Change site-directed mutagenesis (Stratagene). pCSA-MyoD and 4RE-luc were gifts from Y Sun.
Cell culture and differentiation
Primary HFKs were harvested from neonatal foreskins and grown as previously described (Westbrook et al, 2002). Differentiation of HFKs was induced by re-suspending approximately 2 × 106 cells into 10 ml of methylcellulose (Ruesch and Laimins, 1998) for 24 h. COS-1 cells were grown in DMEM supplemented with 10% fetal calf serum (FCS). C2C12 and pRb defective CC42 murine myoblast stable lines were maintained in DMEM with 20% FCS. For differentiation, cells were grown to confluence and then incubated in differentiation media containing DMEM and 1% horse serum (DM). Rb−/− 3T3 fibroblasts were provided by Classon (Classon et al, 2000), cultured in DMEM supplemented with 10% cosmic calf serum (CCS, Hyclone) and infected with pBabe-MyoD amphotrophic retrovirus (Novitch et al, 1996) to generate stable cell lines. For myogenic conversion of MyoD-Rb−/− 3T3 lines, cells were first transfected with Rb and incubated in DM 24 h later at subconfluence.
Transfections, reporter assays and semiquantitative RT–PCR
Subconfluent Rb−/− 3T3 fibroblasts were transfected with lipofectamine 2000 (GIBCO-BRL). Following a 4-h incubation period, cells were maintained in fresh media for 24 h. After this point, transfected cells reached 75% confluence and were incubated for an additional 24–72 h in DM. Luciferase activity was measured and standardized as previously described (Nead et al, 1998). Primer sequences and RT–PCR conditions are available upon request.
Immunofluorescence and flow cytometry
Immunostaining of muscle-specific markers and bromodeoxyuridine (BrdU)-labeled cells, as well as flow cytometric analysis, was performed following established methods (Chen and Wang, 2000; Westbrook et al, 2002).
Glutathione S-transferase (GST) pull-down and acetyltransferase assay
GST and in vitro translated proteins were prepared as mentioned (Nead et al, 1998). His-tagged fusion proteins were purified from pET-28A expressing Rosetta (Novagen) bacterial strain, using nickel columns (Qiagen) following the manufacturer's recommendations. GST pull-down assays and recombinant protein–protein interactions were carried out following standard methods (Huang and McCance, 2002). In vitro acetylation was carried out following a modified protocol (Bannister and Kouzarides, 1996). Reactions were conducted in 30 μl with either 10 nM acetyl-CoA or 90 ρmol of 14C-radiolabeled acetyl-CoA (50 mCi/mM, Amersham Pharmacia Biotech) at 30°C for 45 min. Typically, 8 pmol of GST-pRb and 3 pmol of His-tagged P/CAF-HAT were used per reaction. Cold samples were resolved by SDS–PAGE gel and transferred to nitrocellulose for Western blot analysis using anti-acetyl-lysine antibody. Radiolabeled samples were transferred to PVDF and exposed to film. A replica gel was stained with coomassie to ensure proper loading.
Immunoprecipitations and Western blot
The antibodies used include those against: pRb (IF8AC, Santa-Cruz), pRb (G3-245, Pharmacia), P/CAF (from Y Nakatani), p300 (C-20, Santa-Cruz), p21 (F-5, Santa-Cruz), MDM2 (H-221, Santa-Cruz), MyoD (M-318, Santa-Cruz), T7 (Novagen), HDAC-1 (H-51, Santa-Cruz), MyHC (MY-32, Sigma), Myogenin (F5D, Santa-Cruz) HA (3F10, Roche), Flag (M2, Sigma). Alexa Fluor 594 conjugated anti-BrdU and Alexa Fluor 594 (Molecular Probes), monoclonal and polyclonal anti-acetyl-lysine (Cell Signaling) antibodies were also used. Whole-cell extracts were obtained by lysing cell pellets in 50 mM Tris (pH 7.5), 150 mM NaCl, 1 mM DTT, 5 mM EDTA, 1% Triton-X and 1:100 dilution of protease inhibitor cocktail (Sigma P8340). Immunoprecipitations were performed on nuclear lysates extracted, based on a modified protocol (Dignam et al, 1983). In all, 2–5 mg of pre-cleared protein extract was immunoprecipitated with the indicated antibodies at 4°C overnight (20 mM HEPES (pH 7.9), 100 mM KCl, 1.5 mM MgCl2, 1% NP-40, 10% glycerol, protease inhibitor cocktail and 10 μM TSA). Immunoprecipitates were resolved via SDS–PAGE and transferred onto nitrocellulose membranes. Protein detection was achieved by Western blot analysis.
Acknowledgments
We are indebted to Tony Kouzarides, Yoshihiro Nakatani, Marie Classon and Yin Sun for providing us with various plasmids, antibodies and cell lines. We also thank Ross LaRossa for assistance with the site-directed mutagenesis, and Thomas Westbrook and JiYong Zhao for review of the manuscript. This work was funded by a grant from the National Institute of Allergy and Infectious Disease, AI 30798 to DJM.
References
- Bannister AJ, Kouzarides T (1996) The CBP co-activator is a histone acetyltransferase. Nature 384: 641–643 [DOI] [PubMed] [Google Scholar]
- Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T (1998) Retinoblastoma protein recruits histone deacetylase to repress transcription [see comments]. Nature 391: 597–601 [DOI] [PubMed] [Google Scholar]
- Buchkovich K, Duffy LA, Harlow E (1989) The retinoblastoma protein is phosphorylated during specific phases of the cell cycle. Cell 58: 1097–1105 [DOI] [PubMed] [Google Scholar]
- Chan HM, Krstic-Demonacos M, Smith L, Demonacos C, La Thangue NB (2001) Acetylation control of the retinoblastoma tumour-suppressor protein. Nat Cell Biol 3: 667–674 [DOI] [PubMed] [Google Scholar]
- Chen PL, Scully P, Shew JY, Wang JY, Lee WH (1989) Phosphorylation of the retinoblastoma gene product is modulated during the cell cycle and cellular differentiation. Cell 58: 1193–1198 [DOI] [PubMed] [Google Scholar]
- Chen TT, Wang JY (2000) Establishment of irreversible growth arrest in myogenic differentiation requires the RB LXCXE-binding function. Mol Cell Biol 20: 5571–5580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AR, Maandag ER, van Roon M, van der Lugt NM, van der Valk M, Hooper ML, Berns A, te Riele H (1992) Requirement for a functional Rb-1 gene in murine development. Nature 359: 328–330 [DOI] [PubMed] [Google Scholar]
- Classon M, Dyson N (2001) p107 and p130: versatile proteins with interesting pockets. Exp Cell Res 264: 135–147 [DOI] [PubMed] [Google Scholar]
- Classon M, Salama S, Gorka C, Mulloy R, Braun P, Harlow E (2000) Combinatorial roles for pRB, p107, and p130 in E2F-mediated cell cycle control. Proc Natl Acad Sci USA 97: 10820–10825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobrinik D, Lee MH, Hannon G, Mulligan G, Bronson RT, Dyson N, Harlow E, Beach D, Weinberg RA, Jacks T (1996) Shared role of the pRB-related p130 and p107 proteins in limb development. Genes Dev 10: 1633–1644 [DOI] [PubMed] [Google Scholar]
- Dannenberg JH, van Rossum A, Schuijff L, te Riele H (2000) Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev 14: 3051–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignam J.D, Martin PL, Shastry BS, Roeder RG (1983) Eukaryotic gene transcription with purified components. Methods Enzymol 101: 582–598 [DOI] [PubMed] [Google Scholar]
- Dynlacht BD, Flores O, Lees JA, Harlow E (1994) Differential regulation of E2F transactivation by cyclin/cdk2 complexes. Genes Dev 8: 1772–1786 [DOI] [PubMed] [Google Scholar]
- Dyson N (1998) The regulation of E2F by pRB-family proteins. Genes Dev 12: 2245–2262 [DOI] [PubMed] [Google Scholar]
- Eckner R, Yao TP, Oldread E, Livingston DM (1996) Interaction and functional collaboration of p300/CBP and bHLH proteins in muscle and B-cell differentiation. Genes Dev 10: 2478–2490 [DOI] [PubMed] [Google Scholar]
- Goodman RH, Smolik S (2000) CBP/p300 in cell growth, transformation, and development. Genes Dev 14: 1553–1577 [PubMed] [Google Scholar]
- Grossman SR, Perez M, Kung AL, Joseph M, Mansur C, Xiao ZX, Kumar S, Howley PM, Livingston DM (1998) p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol Cell 2: 405–415 [DOI] [PubMed] [Google Scholar]
- Gu W, Schneider JW, Condorelli G, Kaushal S, Mahdavi V, Nadal-Ginard B (1993) Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell 72: 309–324 [DOI] [PubMed] [Google Scholar]
- Guo CS, Degnin C, Fiddler TA, Stauffer D, Thayer MJ (2003) Regulation of MyoD activity and muscle cell differentiation by MDM2, pRb, and Sp1. J Biol Chem 278: 22615–22622 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100: 57–70 [DOI] [PubMed] [Google Scholar]
- Harbour JW, Dean DC (2000) The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev 14: 2393–2409 [DOI] [PubMed] [Google Scholar]
- Huang SM, McCance DJ (2002) Down regulation of the interleukin-8 promoter by human papillomavirus type 16 E6 and E7 through effects on CREB binding protein/p300 and P/CAF. J Virol 76: 8710–8721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA (1992) Effects of an Rb mutation in the mouse. Nature 359: 295–300 [DOI] [PubMed] [Google Scholar]
- Kawabata H, Kawahara K, Kanekura T, Araya N, Daitoku H, Hatta M, Miura N, Fukamizu A, Kanzaki T, Maruyama I, Nakajima T (2002) Possible role of transcriptional coactivator P/CAF and nuclear acetylation in calcium-induced keratinocyte differentiation. J Biol Chem 277: 8099–8105 [DOI] [PubMed] [Google Scholar]
- Kouzarides T (2000) Acetylation: a regulatory modification to rival phosphorylation? EMBO J 19: 1176–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeCouter JE, Kablar B, Hardy WR, Ying C, Megeney LA, May LL, Rudnicki MA (1998a) Strain-dependent myeloid hyperplasia, growth deficiency, and accelerated cell cycle in mice lacking the Rb-related p107 gene. Mol Cell Biol 18: 7455–7465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeCouter JE, Kablar B, Whyte PF, Ying C, Rudnicki MA (1998b) Strain-dependent embryonic lethality in mice lacking the retinoblastoma-related p130 gene. Development 125: 4669–4679 [DOI] [PubMed] [Google Scholar]
- Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A (1992) Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 359: 288–294 [DOI] [PubMed] [Google Scholar]
- Lee MH, Williams BO, Mulligan G, Mukai S, Bronson RT, Dyson N, Harlow E, Jacks T (1996) Targeted disruption of p107: functional overlap between p107 and Rb. Genes Dev 10: 1621–1632 [DOI] [PubMed] [Google Scholar]
- Li FQ, Coonrod A, Horwitz M (2000) Selection of a dominant negative retinoblastoma protein (RB) inhibiting satellite myoblast differentiation implies an indirect interaction between MyoD and RB. Mol Cell Biol 20: 5129–5139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Scolnick DM, Trievel RC, Zhang HB, Marmorstein R, Halazonetis TD, Berger SL (1999) p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol Cell Biol 19: 1202–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLellan WR, Xiao G, Abdellatif M, Schneider MD (2000) A novel Rb- and p300-binding protein inhibits transactivation by MyoD. Mol Cell Biol 20: 8903–8915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP, Troalen F, Trouche D, Harel-Bellan A (1998) Retinoblastoma protein represses transcription by recruiting a histone deacetylase [see comments]. Nature 391: 601–605 [DOI] [PubMed] [Google Scholar]
- Martinez-Balbas MA, Bauer UM, Nielsen SJ, Brehm A, Kouzarides T (2000) Regulation of E2F1 activity by acetylation. EMBO J 19: 662–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN (2001) Control of muscle development by dueling HATs and HDACs. Curr Opin Genet Dev 11: 497–504 [DOI] [PubMed] [Google Scholar]
- Miyake S, Sellers WR, Safran M, Li X, Zhao W, Grossman SR, Gan J, DeCaprio JA, Adams PD, Kaelin WG Jr (2000) Cells degrade a novel inhibitor of differentiation with E1A-like properties upon exiting the cell cycle. Mol Cell Biol 20: 8889–8902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nead MA, Baglia LA, Antinore MJ, Ludlow JW, McCance DJ (1998) Rb binds c-Jun and activates transcription. EMBO J 17: 2342–2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novitch BG, Mulligan GJ, Jacks T, Lassar AB (1996) Skeletal muscle cells lacking the retinoblastoma protein display defects in muscle gene expression and accumulate in S and G2 phases of the cell cycle. J Cell Biol 135: 441–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87: 953–959 [DOI] [PubMed] [Google Scholar]
- Peschiaroli A, Figliola R, Coltella L, Strom A, Valentini A, D'Agnano I, Maione R (2002) MyoD induces apoptosis in the absence of RB function through a p21(WAF1)-dependent re-localization of cyclin/cdk complexes to the nucleus. Oncogene 21: 8114–8127 [DOI] [PubMed] [Google Scholar]
- Polesskaya A, Duquet A, Naguibneva I, Weise C, Vervisch A, Bengal E, Hucho F, Robin P, Harel-Bellan A (2000) CREB-binding protein/p300 activates MyoD by acetylation. J Biol Chem 275: 34359–34364 [DOI] [PubMed] [Google Scholar]
- Polesskaya A, Harel-Bellan A (2001) Acetylation of MyoD by p300 requires more than its histone acetyltransferase domain. J Biol Chem 276: 44502–44503 [DOI] [PubMed] [Google Scholar]
- Polesskaya A, Naguibneva I, Duquet A, Bengal E, Robin P, Harel-Bellan A (2001a) Interaction between acetylated MyoD and the bromodomain of CBP and/or p300. Mol Cell Biol 21: 5312–5320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polesskaya A, Naguibneva I, Fritsch L, Duquet A, Ait-Si-Ali S, Robin P, Vervisch A, Pritchard LL, Cole P, Harel-Bellan A (2001b) CBP/p300 and muscle differentiation: no HAT, no muscle. EMBO J 20: 6816–6825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri P.L, Iezzi S, Stiegler P, Chen TT, Schiltz RL, Muscat GE, Giordano A, Kedes L, Wang JY, Sartorelli V (2001) Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol Cell 8: 885–897 [DOI] [PubMed] [Google Scholar]
- Puri PL, Sartorelli V, Yang XJ, Hamamori Y, Ogryzko VV, Howard BH, Kedes L, Wang JY, Graessmann A, Nakatani Y, Levrero M (1997) Differential roles of p300 and PCAF acetyltransferases in muscle differentiation. Mol Cell 1: 35–45 [DOI] [PubMed] [Google Scholar]
- Reid JL, Bannister AJ, Zegerman P, Martinez-Balbas MA, Kouzarides T (1998) E1A directly binds and regulates the P/CAF acetyltransferase. EMBO J 17: 4469–4477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruesch MN, Laimins LA (1998) Human papillomavirus oncoproteins alter differentiation-dependent cell cycle exit on suspension in semisolid medium. Virology 250: 19–29 [DOI] [PubMed] [Google Scholar]
- Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, Theodorou E, Jacks T (2000) Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev 14: 3037–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorelli V, Huang J, Hamamori Y, Kedes L (1997) Molecular mechanisms of myogenic coactivation by p300: direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol Cell Biol 17: 1010–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorelli V, Puri PL, Hamamori Y, Ogryzko V, Chung G, Nakatani Y, Wang JY, Kedes L (1999) Acetylation of MyoD directed by PCAF is necessary for the execution of the muscle program. Mol Cell 4: 725–734 [DOI] [PubMed] [Google Scholar]
- Schiltz RL, Nakatani Y (2000) The PCAF acetylase complex as a potential tumor suppressor. Biochem Biophys Acta 1437: M37–M53 [DOI] [PubMed] [Google Scholar]
- Schneider JW, Gu W, Zhu L, Mahdavi V, Nadal-Ginard B (1994) Reversal of terminal differentiation mediated by p107 in Rb−/− muscle cells. Science 264: 1467–1471 [DOI] [PubMed] [Google Scholar]
- Sellers WR, Novitch BG, Miyake S, Heith A, Otterson GA, Kaye FJ, Lassar AB, Kaelin WG Jr (1998) Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor cell growth. Genes Dev 12: 95–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo N, Goodman RH (2001) CREB-binding protein and p300 in transcriptional regulation. J Biol Chem 276: 13505–13508 [DOI] [PubMed] [Google Scholar]
- Westbrook TF, Nguyen DX, Thrash BR, McCance DJ (2002) E7 abolishes raf-induced arrest via mislocalization of p21(Cip1). Mol Cell Biol 22: 7041–7052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao ZX, Chen J, Levine AJ, Modjtahedi N, Xing J, Sellers WR, Livingston DM (1995) Interaction between the retinoblastoma protein and the oncoprotein MDM2. Nature 375: 694–698 [DOI] [PubMed] [Google Scholar]
- Yamasaki L, Bronson R, Williams BO, Dyson NJ, Harlow E, Jacks T (1998) Loss of E2F-1 reduces tumorigenesis and extends the lifespan of Rb1(+/−)mice. Nat Genet 18: 360–364 [DOI] [PubMed] [Google Scholar]
- Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y (1996) A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature 382: 319–324 [DOI] [PubMed] [Google Scholar]
- Yuan W, Condorelli G, Caruso M, Felsani A, Giordano A (1996) Human p300 protein is a coactivator for the transcription factor MyoD. J Biol Chem 271: 9009–9013 [DOI] [PubMed] [Google Scholar]
- Zacksenhaus E, Jiang Z, Chung D, Marth JD, Phillips RA, Gallie BL (1996) pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes Dev 10: 3051–3064 [DOI] [PubMed] [Google Scholar]